

Abstract
Vascular inflammation is associated with and in large part driven by changes in the leukocyte compartment of the vessel wall. Here, we focus on monocyte influx during atherosclerosis, the most common form of vascular inflammation. Although the arterial wall contains a large number of resident macrophages and some resident dendritic cells, atherosclerosis drives a rapid influx of inflammatory monocytes (Ly-6C+ in mice) and other monocytes (Ly-6C− in mice, also known as patrolling monocytes). Once in the vessel wall, Ly-6C+ monocytes differentiate to a phenotype consistent with inflammatory macrophages and inflammatory dendritic cells. The phenotype of these cells is modulated by lipid uptake, Toll-like receptor ligands, hematopoietic growth factors, cytokines and chemokines. In addition to newly recruited macrophages, it is likely that resident macrophages also change their phenotype. Monocyte-derived inflammatory macrophages have a short half-life. After undergoing apoptosis, they may be taken up by surrounding macrophages or, if the phagocytic capacity is overwhelmed, can undergo secondary necrosis, a key event in forming the necrotic core of atherosclerotic lesions. In this review, we discuss these and other processes associated with monocytic cell dynamics in the vascular wall and their role in the initiation and progression of atherosclerosis.
Introduction
Monocytes, macrophages and dendritic cells are key cells in the initiation and progression of atherosclerosis. Here, we review the available evidence on how monocytes reach atherosclerotic lesions, how they differentiate into inflammatory macrophages, the possible relation between resident and inflammatory macrophages, and the fate of these cell types as the atherosclerotic lesions progress. We also refer the reader to several excellent previous reviews on the subject 1-6. Because of space constraints, we will consider dendritic cells only insofar as they are monocyte-derived. Reviews on dendritic cells in atherosclerosis can be found elsewhere 7-9.
The healthy mouse aorta contains a significant number of Mac-1+ cells, most of which are resident macrophages 10-12. In addition to CD11b (the α chain of the Mac-1 integrin), these cells also express the phagocyte marker CD68 and the macrophage marker F4/80. Although some macrophages in lymphoid tissues like the spleen derive from blood monocytes 13-15, the origin and lineage of resident vascular macrophages is unknown. Microglia 16 and Langerhans cells 17 have been shown to proliferate in situ. Since there was no evidence of microglia progenitor recruitment from the circulation 16, microglia cells may arise from embryonic precursor cells. It is possible that resident vascular macrophages share a similar nature, although this has not been investigated.
Monocytes
Under conditions of atherosclerosis, monocytes are rapidly recruited into the vessel wall. Mouse monocytes develop from a bone marrow precursor cell, the monocyte-dendritic cell precursor (MDP) 18. MDPs give rise to all blood monocytes and the common dendritic cell precursor (CDP) 19, but MDPs do not give rise to granulocytes. Under conditions of acute inflammation, hematopoietic progenitor cells can differentiate to dendritic cells outside the bone marrow 20. The growth factor M-CSF and its receptor CD115 are critical for MDP differentiation into monocytes 21, 22. The cell fate decisions that occur in the bone marrow for monocyte differentiation require a number of specific transcription factors. Expression of PU.1, a member of the Ets transcription factor family, is induced during early myeloid differentiation and is high in mature monocytes and granulocytes 23, 24. Studies of mice in which PU.1 has been deleted show a complete defect in production of monocytes, granulocytes and B and T lymphocytes. These mice are either embryonic lethal or die from sepsis shortly after birth 25, 26. PU.1 stimulates the Egr family of transcription factors that play a role in monocyte development 27, 28. Interferon regulatory factor-8 (IRF-8) also known as interferon consensus sequence binding protein or ICSBP, is a member of the IRF family of transcription factors. Studies with IRF8 knockout mice (Irf8−/−) have demonstrated that IRF8 inhibits granulocyte differentiation, while promoting monocyte proliferation from progenitor bone marrow cells 29, 30. JunB is a component of the AP-1 transcriptional complex, and junB is highly expressed in granulocytes and in myeloid precursors 31, 32. JunB is a negative regulator of cell proliferation through inhibition of cyclin D1 and activation of the Cdk inhibitor p16 33, 34. Moreover, several studies have shown that mice lacking myeloid expression of junB develop a myeloproliferative disorder similar to chronic myeloid leukemia 35, 36. JunB expression was reduced in bone marrow of Nr4a1−/−Nr4a3−/− knockout mice 37. Taken together, these data suggest that junB is an essential component of myeloid differentiation, and that the orphan nuclear receptors Nr4a1 and/or Nr4a3 may regulate junB expression and monocyte differentiation in bone marrow. Other transcription factors, including KLF4 38 and the Maf family 39 have also been implicated in monocyte development and differentiation. Once mouse monocytes egress from the bone marrow, they circulate in the blood with a half-life of about 17 hours 40-42. Many of these measurements were made before monocyte subsets were known, and the circulatory half-life may well differ for different monocyte subsets. Mature monocytes reside in the subcapsular red pulp of the spleen, where they are rapidly deployed in response to inflammatory signals 15. These spleen monocytes show a gene expression profile that is very similar to that of blood monocytes.
In mice and humans, several monocyte subsets have been described. Originally, Ly-6C+ monocytes were described as “inflammatory” and Ly-6C− as “resident” 43. The Ly-6C+ monocytes also express the MCP-1 receptor CCR2, the adhesion molecule L-selectin, and low levels of the chemokine receptor CX3CR1. Conversely, Ly-6C− monocytes express high levels of CX3CR1 and of the αLβ2 integrin LFA-1 43, 44. In humans, CD14high and CD14+CD16+ monocytes were described 45. More recently, a CD14dim human monocyte subset was found to be the subset containing the population that patrols blood vessels 46. Human CD14high monocytes also express CCR2, L-selectin and the Fc receptor CD64. Among human monocytes, CD14+CD16+ cells lack CCR2, but express CD32 and higher levels of MHC-II. The differential function of CD14+CD16+ and CD14dim monocytes was recently described 46. Since the definition of the monocyte subsets is currently based on surface markers, it is unclear and controversial whether they can interconvert 43, 47. Lineage tracking studies will be needed to understand the lineage relationship between these monocyte subsets and their differentiation into resident and inflammatory vascular macrophages.
Circulating monocytes of patients with cardiovascular disease display high levels of surface receptors that may be involved in inflammatory responses. Monocyte TLR4 expression has been reported to increase in patients with coronary arteriosclerosis and acute coronary syndrome 48-50. TLR4 is a signaling co-receptor of CD14. Both CD14+CD16+ and CD14highCD16− monocytes secrete proinflammatory cytokines in response to LPS, the bacterial TLR4 ligand. CD14dim monocytes do not respond to LPS, but respond to viruses and endogenous nucleic acids via TLR7 and TLR8 46. In asymptomatic hypercholesterolemic subjects, TLR4 is expressed on CD14+CD16+ and at lower levels on CD14highCD16− monocytes 51. However, it is unclear whether expansion of TLR4-positive monocytes in patients with clinical manifestations of atherosclerosis is associated with the CD14+CD16+ or the CD14highCD16− population.
At sites of inflammation, monocytes are recruited to lymphoid and non-lymphoid tissues 52, where they phagocytose microbes, apoptotic cells and host-derived damage-associated molecules and complexes, such as oxidized or otherwise modified low density lipoprotein (LDL) 53, 54. These monocyte-derived cells have variably been described as inflammatory dendritic cells 55, 56 or inflammatory macrophages. In the context of atherosclerosis, these cells can give rise to foam cells and initiate formation of fatty streaks 57, but resident dendritic cells have also been shown to produce foam cells 56. Interestingly, increased numbers of inflammatory macrophages and DCs are found even in the normal mouse aorta at sites that are predisposed to develop atherosclerotic lesions 55. It is presently unclear to what extent these cells differentiate (lineage commitment) or whether they show plasticity (phenotypic changes in response to their environment) 58.
Ly-6C+ monocytes are known to give rise to CD11b+CD11c+ inflammatory macrophages in the intestinal lamina propria 59, 60. In a model of Listeria monocytogenes infection, monocyte-derived cells have been described as TNF and iNOS-producing Tip-DCs 61. These cells express high levels of MHC-II, CD80 and CD86, and are efficient at presenting antigens 62. They phagocytose microbes, promote inflammation by secreting cytokines and degrade tissue by proteolytic enzymes including matrix metalloproteinases. Although Tip-DCs migrate to secondary lymphoid organs in listeria-infected mice 61, their migration in atherosclerosis has not been studied. These Ly-6C+ monocyte-derived cells have been compared to M1 (see below) macrophages 1, but it is unclear whether these cells fully recapitulate the phenotype of classical M1 macrophages 63, 64, which were originally defined based on human blood monocyte-derived macrophages grown in vitro in the presence of M-CSF, IFN-γ and LPS 65. It is presently not clear whether Tip-DCs and inflammatory macrophages are distinct or overlapping subsets among CD11b+CD11c+ cells.
The Ly-6C− monocytes patrol the inside of blood vessels in mice 66. Their interaction with the vascular endothelium requires LFA-1 (αLβ2 integrin) and the chemokine receptor CX3CR1 66. CX3CR1 has been shown to be critical for the survival of the Ly-6C− subset 40. Human CD14dim monocytes also show this patrolling behavior when infused into mice 46. Although the patrolling behavior is well described to cover long distances and large areas of the endothelial surface of blood vessels, the kinetics of recruitment, migration into the extravascular space and survival of Ly-6C− cells in the tissue have not been investigated yet. The Ly-6C− monocytes can differentiate into macrophages that produce chemokines like CXCL9 and CXCL10, pro-angiogenic factors like VEGF, participate in tissue remodeling and phagocytosis 1, 64. The relation between the monocyte subsets and M1, M2 and other macrophages (see below) remains unclear.
Monocyte recruitment
Monocytes are thought to reach the arterial wall by transmigrating through the luminal endothelium. However, the evidence for this is circumstantial and monocyte transmigration into atherosclerotic lesions has not been observed directly. Morphologic studies show that monocytes can be found adherent to the luminal endothelium 10. Ex vivo perfusion studies show that monocytes can roll and adhere to the luminal endothelium of atherosclerotic, but not normal arteries 67, 68, but in these experiments the wall shear stress was lower than in vivo, and did not follow the normal cardiac cycle. In vivo observations of leukocyte adhesion in mouse abdominal aorta have been reported 69, but the nature of the leukocytes (monocytes or granulocytes) was not identified. In a recent intravital microscopic study, the chemokine receptor requirement for neutrophil, but not monocyte recruitment into the carotid artery was reported 70.
The traditional leukocyte adhesion cascade involves capture, selectin-dependent rolling, activation by an endothelial surface-bound chemokine, integrin-mediated adhesion, and transendothelial migration 71, 72. The main selectin responsible for monocyte rolling in atherosclerotic mouse arteries appears to be P-selectin 67. L-selectin appears to be involved in lymphocyte homing to mouse aorta 12, but not in monocyte recruitment. Most monocyte firm adhesion is dependent on the α4β1 integrin, also known as VLA-4 68, 73, which can bind to certain isoforms of fibronectin and to VCAM-1, an immunoglobulin family molecule highly expressed in endothelial cells near atherosclerotic lesions 74. Although monocytes express other integrins including αLβ2 (LFA-1) and αMβ2 (Mac-1), their role in recruitment to large arteries has not been documented. A list of adhesion molecules relevant to monocyte recruitment can be found in 75.
The immobilized chemokines responsible for monocyte arrest probably include CCL5, also known as RANTES, CXCL1, also known as KC in the mouse or GRO-α in humans, and IL-8 in humans 76. Whether CCL2, also known as MCP-1, can trigger monocyte adhesion is controversial 73, 76, although CCL2 binding to endothelial cell glycosaminoglycans may provide a reservoir for retaining high local concentrations of CCL2 77.
Blocking experiments have suggested that the Ly-6C+ and Ly-6C− monocyte subsets may use different chemokines to access atherosclerotic lesions 78. Ly-6C− monocyte recruitment was reduced by 40% when CCR5 was blocked, whereas Ly-6C+ monocyte recruitment to sites of atherosclerosis was reduced by 40-50% when CCR5, CCR2 or CX3CR1 were targeted 78. However, in these experiments, only the net effect of blocking these chemokines or their receptors was recorded, but the monocyte recruitment was not observed directly. Consistent with intravital microscopic and flow chamber experiments, CCL5 might be expected to be involved in arrest, and CCL2 perhaps in transendothelial migration. The importance of chemokines in recruiting both Ly-6C+ and Ly-6C− monocytes is emphasized by the finding that combined inhibition of CX3CR1, CCL2and CCR5 almost abolishes atherosclerosis in the Apoe−/− mouse model of atherosclerosis 79.
Among the monocyte subsets, Ly-6Chi monocytes express significantly more functional PSGL-1 than Ly-6C− cells 80. Consistent with this observation, flow chamber assays showed that Ly-6C+ monocytes adhere more avidly to P-selectin and E-selectin 80. This would suggest that Ly-6C+ monocytes may preferentially enter into atherosclerotic lesions, perhaps through a platelet-dependent mechanism. In vivo, Ly-6C+ monocytes were indeed shown to bind to activated endothelium and infiltrate atherosclerotic lesions better than Ly-6C− monocytes 81. Moreover, Ly-6C+ monocytes accumulate in the blood of mice fed a high-fat high-cholesterol “western” diet 81. Ex vivo imaging showed that Ly-6C+ monocytes preferentially localized to lesion-prone sites like the lesser curvature of the aortic arch and arterial branch points 82. Since Ly-6C+ monocytes are known to give rise to CD11b+CD11c+ inflammatory macrophages and Tip-DCs in other models 60, 61, it is tempting to speculate that many of the “macrophages” accumulating in atherosclerotic lesions may actually be Tip-DCs. Interestingly, most CD11b+CD11c+ cells in the atherosclerotic mouse aorta co-express high levels of MHC-II, which is responsible for peptide antigen presentation to CD4 T cells, and F4/80, a classical macrophage marker (Koltsova and Ley, unpublished results, 2011). Similar cells have recently been called resident intimal dendritic cells 56. However, unlike dendritic cells in lymphoid organs, these cells are probably monocyte-derived.
Like other blood cells, monocytes must transmigrate through the endothelium to reach their destination. In vitro systems in the absence of flow show a prominent role of the endothelial cell surface immunoglobulin-like adhesion molecule CD31 in monocyte transendothelial migration 83. Genetic absence of CD31 reduces monocyte transmigration in most mouse strains 84, but not in C57BL/6 mice, which are most commonly used in atherosclerosis studies. Other molecules that have been implicated in monocyte transendothelial migration include ICAM-1, VCAM-1, JAM-A, JAM-C, ESAM, ICAM-2 and CD99 72. However, these molecules are also used by other leukocytes, and no direct in vivo evidence exists that these mechanisms specifically regulate monocyte recruitment to atherosclerotic lesions in vivo. With the exception of VCAM-1, which is also a key adhesion molecule for monocytes 85, the differential effect of blocking or knocking out these molecules on the recruitment of monocyte subsets has not been studied.
Role of platelets
Platelets have long been known to participate in atherogenesis 86, and atherosclerotic lesions express detectable levels of platelet antigens like CD41 and P-selectin. Although it is unlikely that intact platelets survive in lesions, platelets greatly facilitate and accelerate monocyte accumulation 87, 88. Platelets form bridges between monocytes and endothelial cells 88-91. Consistent with this role, infusing activated platelets accelerates atherosclerotic lesion formation 89. Platelet-monocyte adhesion is often dependent on platelet P-selectin and monocyte P-selectin Glycoprotein Ligand-1 (PSGL-1). Indeed, selectively eliminating P-selectin expression from platelets only (and not from endothelial cells) strongly protects mice from atherosclerosis 92. Also, removing Pf4, the gene encoding the platelet chemokine CXCL4, from the mouse genome results in reduced atherosclerosis 93. PF4 is only expressed in platelets and their precursor cells. Heterodimers between CXCL4 (PF4) and CCL5 (RANTES) have been implicated in atherogenesis, because they can promote monocyte arrest 94.
Blocking the main platelet adhesion molecule for von Willebrand factor, GP1bα, also delays atherosclerosis 95. This is probably due to interrupting platelet binding to endothelial vWF, thus preventing immobilized platelets from nucleating monocyte adhesion (figure 1). Platelets bind to circulating monocytes in patients 90 and mice with atherosclerosis 89. They can even promote the formation of heteroaggregates. However, platelets bind neutrophils 96 as avidly as monocytes. It is not known why monocytes, but not neutrophils seem to depend on platelets for recruitment to atherosclerotic lesions.
Figure 1. Monocyte recruitment to atherosclerotic lesions.
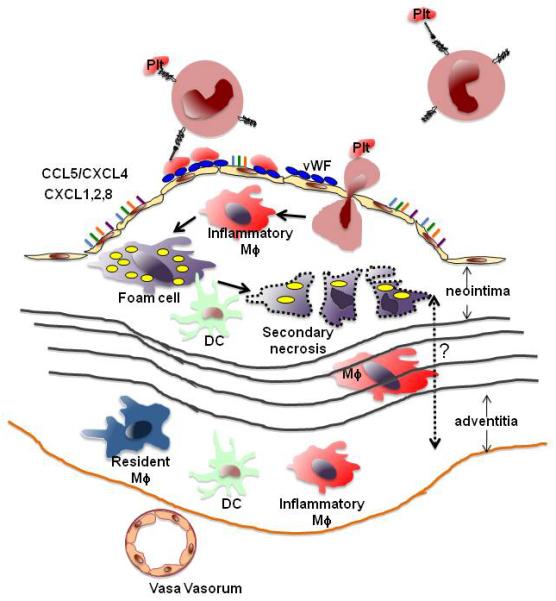
Blood monocytes may be decorated with platelets through P-selectin-PSGL-1 interactions. They can also roll and adhere to platelets bound to von Willebrand Factor (vWF, blue multimers) secreted from endothelial cells. Monocyte arrest can be triggered by chemokines (colored bars) including CCL5/CXCL4 heterodimers, CXCL1, CXCL2 and (in humans) CXCL8. Arrested monocytes are thought to transmigrate into the lesion, where they differentiate to inflammatory macrophages (Mφ) and foam cells, which can undergo secondary necrosis to form the necrotic core in the neointima. Inflammatory macrophages may also migrate through the media (black lines represent laminae elasticae) to the adventitia. Monocytes may also be recruited to the adventitia through vasa vasorum. Resident macrophages (blue) and dendritic cells (green) are constitutively found in the adventitia and may be able to migrate between these two compartments (dashed arrow).
In addition to facilitating monocyte recruitment, platelets also secrete factors that influence the monocyte-macrophage phenotype. An important modifier of macrophage phenotype is platelet factor 4 (PF4 or CXCL4), which induces a unique transcriptome in human blood monocyte-derived macrophages 97 devoid of the hemoglobin-haptgoglobin receptor CD163 98. Consistent with a pro-atherosclerotic role of PF4, Pf4−/− mice show reduced atherosclerotic lesion sizes 93.
Atherosclerotic lesion macrophages
Macrophages are a major cell type of early atherosclerotic lesions and play important roles at all stages of lesion progression. As we noted earlier, macrophage phenotypes in atherosclerotic lesions are likely the result of both lineage commitment and phenotypic changes in response to their environment 58, 99. Because current technology for interrogating macrophage lineages and functions in atherosclerotic lesions has serious limitations, most studies addressed mechanisms of macrophage differentiation in vitro.
In vitro, human monocytes can differentiate into various macrophage subsets. The most common growth factor used to grow macrophages in vitro is M-CSF, generating unpolarized (M0) macrophages. If these macrophages are treated with IFN-γ followed by LPS, they polarize to an M1 phenotype with characteristic expression of TNF-α and IL-12 64. M2 macrophages were originally described as human blood monocyte-derived macrophages differentiated in the presence of M-CSF and IL-4 or IL-13 (M2a), immune complexes and IL-1β or LPS (M2b) or in the presence of IL-10, TGF-β or glucocorticoids (M2c) 100. M2c cells produce the pro-angiogenic growth factor VEGF 100. Monocyte-derived macrophages can also be grown with PF4 without the requirement of M-CSF, resulting in M4 macrophages 97. M4 macrophages have a unique transcriptome that is closer to M2 than M1. In the presence of the oxidized phospholipid oxPAPC, mouse bone marrow-derived macrophages express a small and unique transcriptome (119 characteristic genes) that includes the Hox1 gene encoding the anti-inflammatory enzyme heme oxygenase-1 101. This macrophage phenotype has been called Mox 101 and shares poor phagocytic capacity with M4 macrophages. However, Mox cells are clearly different from M4 cells in many other aspects. The sphingolipid sphingosine-1-phosphate has also been shown to generate a macrophage phenotype closer to a M2 than M1, with increased expression of arginase-I and reduced expression of TNFα 102. Taken together, it is likely that modified LDL and inflammatory mediators like IFN-γ, TNF-α, TGF-β and IL-10 can trigger phenotypic modulation of macrophages in the artery wall during atherogenesis.
Macrophages can be isolated from the peritoneal cavity of mice or grown from bone marrow precursor cells. It is not known whether resident peritoneal macrophages are monocyte-derived. Among other features, resident peritoneal macrophages express high levels of 12/15-lipoxygenase and are extremely efficient efferocytes that take up apoptotic cells 103. By contrast, bone marrow harbors monocyte precursors including MDPs and CDPs, which can give rise to various lineages. Mouse bone marrow-derived macrophages proliferate in vitro and can be differentiated to M1, M2 and a regulatory “Mreg” phenotype 104, which is phenotypically similar to the human M2c phenotype.
Atherosclerotic lesions contain macrophages with M1, M2, M4 and Mox markers 6, 98, 101, but it is unlikely that the in vitro phenotypes in their pure form exist in diseased arteries in vivo. Flow cytometric analysis of aortas from Ldlr−/− mice fed an atherogenic diet for 30 weeks revealed that 39% of the aortic macrophages expressed the M1 marker CD86, 21% the M2 marker CD206 (mannose receptor), 45% the Mox marker heme oxygenase-1, where 10% co-expressed CD86 101. M4 macrophages were not investigated in this mouse study, but in human atherosclerotic coronary arteries, many macrophages are CD163−, a hallmark of the M4 phenotype 98.
The phenotype of lesional macrophages is incompletely understood. Laser capture microdissection has yielded limited numbers of cells and small amounts of mRNA, which has been interrogated for a few gene products 105. Genome-wide analysis of lesional macrophages has not been reported, although the transcriptome of the mixed cell populations contained in atherosclerotic human coronary artery segments has been analyzed 106. Many lesional cells express the macrophage marker F4/80 and the integrins αMβ2 (CD11b CD18) and αxβ2 (CD11c CD18). Because CD11c is also expressed on many dendritic cells, lesional macrophages, likely derived from recently immigrated monocytes, have been called DCs 56,105. Under atherogenic conditions, these inflammatory macrophages accumulate in the aorta and show reduced emigration from lesions 107. During atherosclerosis progression, these cells appear not only in the neointima, but also in the adventitia 12, where they may participate in antigen presentation 9 and cytokine production.
Foam cell formation
An initiating event in the formation of atherosclerotic plaques is excessive lipid accumulation in vascular wall macrophages. Macrophages in atherosclerotic arteries eventually become lipid-laden foam cells 108 through a process regulated by the balance between the uptake of modified LDL and efflux of cholesterol and other lipids 109. In 1913, Nikolai Anitchkow described these cells as ‘Cholesterinesterphagozyten’ observed in the aorta of cholesterol-fed rabbits 110. In the early 1980s, Ross Gerrity was the first to document the early entry of monocytes into the susceptible areas of the vessel wall in cholesterol-fed animals 111. Gerrity described a “monocyte clearance system” in which large numbers of circulating monocytes invade the intima of lesion-prone areas in arteries, become phagocytic, and accumulate lipid 10. It is indeed likely that most of the foam cells differentiate from newly recruited monocytes, but the details of this process are not known. Alternatively or in addition, resident intimal DCs may accumulate lipid and become foam cells 56. Foam cells can migrate back into the circulation by crossing the aortic endothelium in reverse direction 11. It is not known whether this is a major clearance pathway for foam cells.
Upon differentiation, macrophages display high levels of surface expression of scavenger receptors (SR-A, LOX-1, CXCL16 and CD36), which have the ability to take up modified lipoproteins, such as copper-oxidized, acetylated and malondialdehyde (MDA)-modified LDL 112-119 (figure 2). Atherosclerosis studies with CD36 (and SR-A) knockout mice performed in different laboratories have been contradictory. The Febbraio/Silverstein group demonstrated that CD36 deficiency reduced atherosclerosis 116, 120, but the Moore/Freeman group reported that knocking out CD36 had no effect on atherosclerotic lesion size or even increased aortic root lesions 121. It is possible that scavenger receptors have a differential effect early and late in atherosclerosis, which could reconcile these different findings 116, 117, 121, 122.
Figure 2. Factors that determine macrophage phenotype(s) in atherosclerotic lesions.
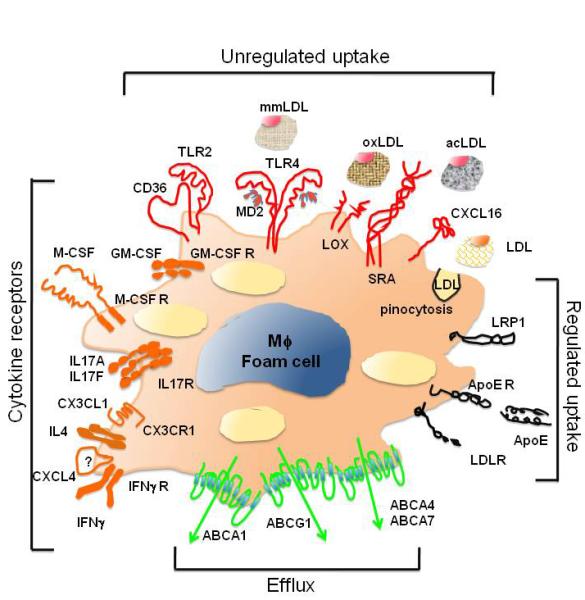
Macrophages in atherosclerotic lesions display markers characteristic of several in vitro-differentiated macrophage phenotypes, which suggest possible factors and receptors that determine their differentiation from monocytes and their function in vivo. The growth factor M-CSF is almost universally required for the monocyte to macrophage differentiation (except M4 macrophages), whereas GM-CSF appears to promote a highly pro-inflammatory phenotype. IFN-γ and LPS polarize macrophages into an M1 phenotype, whereas IL4 (and IL13) and a number of other factors discussed in this review lead to M2 polarization. CXCL4 acts through an unidentified chondroitin sulfate proteoglycan receptor, while IL-17 promotes proliferation of mouse macrophages in vitro and macrophage accumulation in vivo 190. CX3CL1 binding to its receptor CX3CR1 can promote macrophage survival. Macrophages in atherosclerotic lesions accumulate excessive amounts of lipid, and numerous intracellular lipid droplets make the neointimal macrophages look like “foam cells.” Regulated uptake of native LDL via the family of LDL receptors (LDLR, ApoER and LRP1) plays a limited role in foam cell formation. The major mechanism of excessive lipid accumulation is via unregulated uptake of oxidized (oxLDL), minimally modified (mmLDL), acetylated (acLDL) or otherwise modified LDL. This uptake is mediated by CD36 (alone or as heterodimer with TLR2), SRA, LOX-1, CXCL16, TLR4/MD-2 and a number of other receptors. Unmodified LDL also enters macrophages by micro- and macro-pinocytosis. The ABC transporters ABCA1, ABCG1 and ABCA4/7 mediate reverse transport (efflux) of cholesterol, oxysterols and phospholipids, but the presence of foam cells in the lesions indicates that the efflux mechanisms become eventually overwhelmed by unregulated LDL uptake. Lipid accumulation has profound effects on the macrophage gene expression, adhesion, apoptosis, efferocytosis and other characteristics and functions.
Fluid-phase uptake of native and modified LDL is another endocytic pathway that generates macrophage foam cells during atherogenesis 123. This macropinocytosis occurs constitutively in human monocyte-derived macrophages differentiated in vitro with M-CSF. Further studies from the same group recognized that both macropinocytosis and micropinocytosis of native LDL lead to foam cell formation 124. In mouse macrophages, minimally oxidized LDL is recognized by the TLR4 MD-2 complex and induces Syk-dependent membrane ruffling and robust macropinocytosis, resulting in uptake of native and modified LDL and foam cell formation 125, 126. Fluid-phase pinocytosis of fluorescent nanoparticles has been demonstrated in macrophages of mouse atherosclerotic lesions 127, and lipid accumulation and foam cell formation in early lesions of Tlr4−/− apoE−/− mice was reduced by 70-80% compared to apoE−/− controls 128. Under certain dietary conditions, whole body and macrophage TLR4-deficient mice have less atherosclerosis than their Tlr4+/+ counterparts 129, 130. Carotid atherosclerotic plaques dissected from symptomatic patients have higher levels of TLR4 expression compared to the lesions from asymptomatic patients 131, and individuals with inactivating SNPs in the Tlr4 gene have lower risk of atherosclerosis and cardiovascular events, although not all studies agree (reviewed in 132).
Excess cholesterol accumulated in macrophages via scavenger receptor-mediated and/or fluid-phase uptake is removed from macrophages by ATP binding cassette (ABC) transporters. ABCA1 and ABCG1 are upregulated during macrophage differentiation, and these transporters function to regulate cholesterol efflux and reverse cholesterol transport 133-137. PPARγ and LXR agonists function in part to regulate macrophage foam cell formation in atherogenesis 138-142. PPARγ agonists can inhibit foam-cell formation in vivo through ABCA1-dependent 141 and ABCA1-independent pathways 143, 144. Activation of PPARγ reduces cholesterol esterification and induces expression of ABCG1 143. LXR activation in macrophages reduces foam cell formation via induction of both ABCA1 145 and ABCG1 146, 147. Fisher and colleagues recently found that both LXR isoforms were important in atherosclerosis regression. These investigators transplanted aortic arches from atherosclerotic Apoe−/− mice with or without LXRα or LXRβ deficiency into WT recipients. Plaques from both LXRα and LXRβ-deficient Apoe−/− mice exhibited impaired regression and reduced emigration of macrophages from plaques 148. Thus, both LXR and PPARγ signaling inhibit macrophage foam cell formation in atherosclerotic plaques.
During atherogenesis, several eicosanoid-generating enzymatic pathways are induced in macrophages, including 5-lipoxygenase and 12/15-lipoxygenase (5-LO, 12/15-LO) 149-154. Both enzymes have been linked to atherogenesis 151, 155-157. Oxidized fatty acids produced in macrophages contribute to formation of minimally-modified and oxidized LDLs. IL-13, an important cytokine for the alternative activation of macrophages, induces 12/15-LO and CD36 in macrophages 158-160. Engagement of the αMβ2 integrin significantly inhibited IL-13-mediated foam cell formation 161. There is some controversy as to whether the 12/15-LO pathway is pro-atherogenic. Although 12/15LO can clearly generate oxidized lipids in LDL and is pro-atherogenic in mice 156, 157, 162, human 15-LO has been reported to be atheroprotective in rabbits 163-165. This may relate to the concept that the 12/15-LO enzyme can also generate lipoxins and resolvins 166-168, which are important for the resolution of inflammatory responses. Thus, depending on the artery microenvironment, the 12/15-LO enzyme may confer either an atheroprotective or pro-atherogenic role.
Fatty acids and eicosanoids regulate expression of both the ABCA1 and ABCG1 transporters 169-171. Arachidonic acid and 12-S-hydroxyeicosatetraenoic acid (12-S-HETE), produced by 12/15-LO, cause reduced ABCA1 and ABCG1 protein expression in macrophages. Both mice 172 and humans 169 with type 2 diabetes have reduced expression and functional activity of ABCG1 in macrophages. As it is known that subjects with type 2 diabetes have increased 12-S-HETE production through an induction in 12/15LO activity 173, it is plausible to speculate that these elevated levels of eicosanoids in type 2 diabetic subjects contribute to loss of ABCG1.
Recently, important links have been made between ABC transporters and vascular inflammation. The interaction of apoA-I with ABCA1 activates signaling molecules, such as Janus kinase 2 (JAK2) 174. ABCA1-mediated activation of JAK2 activates STAT3 independently of the lipid transport function of ABCA1. ABCA1-expressing macrophages suppressed the induction of inflammatory genes in macrophages in response to LPS. LPS-treated macrophages from macrophage-specific ABCA1-deficient mice exhibited enhanced expression of pro-inflammatory cytokines and increased activation of NFκB, which could be inhibited by silencing MyD88 175. This was normalized when excess free cholesterol was removed from macrophages with cyclodextrin, which suggests that increased inflammatory TLR signaling through lipid rafts occurs when ABCA1 is absent. Similar findings have been reported for ABCG1, where macrophages deficient in ABCG1 showed increased cholesterol accumulation and enhanced TLR signaling in response to LPS 176. ABC transporters may also play a role in myeloid proliferation. Tall and colleagues recently reported that proliferation of hematopoietic stem cell precursors is regulated by cholesterol efflux mechanisms involving HDL, ABCG1 and ABCA1. Mice deficient in both ABCA1 and ABCG1 displayed leukocytosis and an expansion of the Lin−Sca-1+Kit+ hematopoietic progenitor cell population in the bone marrow 177. Thus, new evidence is emerging that strongly links cholesterol efflux mechanisms with inflammatory processes in macrophages.
Apoptosis and efferocytosis
As atherosclerosis has been deemed an inflammatory disease 178, inflammatory factors are expected to be involved in both the progression and resolution of atherosclerosis. One important function of macrophages is the clearance of apoptotic cells by phagocytes (a process called ‘efferocytosis’) 179. Efferocytosis is one function of alternatively activated (M2) macrophages (discussed above) 180. As macrophages engulf oxidized lipids and other cellular debris in the arterial wall during early stages of atherogenesis, many of these macrophages undergo apoptosis. In early atherogenesis, macrophage apoptosis is associated with reduced atherosclerosis progression 181, 182. This is most likely due to effective efferocytosis by neighboring phagocytes, which, in turn, reduces pro-inflammatory mediators present in the artery wall 183. Efficient efferocytosis has been shown to induce anti-inflammatory mediators, such as IL-10 and TGFβ 184. However, uptake of excessive apoptotic cells by macrophages induces endoplasmic reticulum (ER) stress and the unfolded protein response. Oxidized phospholipids, free cholesterol and oxysterols can also trigger ER stress 185. Oxidized phospholipids and oxidized lipoproteins trigger apoptosis in ER-stressed macrophages through a mechanism requiring both CD36 and TLR2 186. As atherosclerosis progresses, efferocytosis is thought to become impaired 187. A failure of efferocytosis leads to ‘secondary necrosis’, in which macrophages die and release their cellular contents, including debris, oxidized lipids and pro-inflammatory mediators. Secondary necrosis amplifies the inflammatory response and leads to the development of a necrotic core in the plaque. The possible effects of defective efferocytosis are detailed in a recent review 179. One possibility, among many, may be a change in macrophage phenotype that occurs in the artery wall during atherogenesis, leading to an accumulation of poorly phagocytic macrophages in the artery wall 188. Minimally oxidized LDL prolongs the survival of foam cells loaded with free cholesterol or extensively oxidized LDL, but also inhibits efferocytosis 125, 189, and thereby may exacerbate vascular inflammation. Thus, the balance between apoptosis, efferocytosis and secondary necrosis determines atherosclerosis progression and severity.
In summary, monocytes play important roles in the initiation, progression and complications of atherosclerosis. Their recruitment to the artery wall, their differentiation to macrophages, and their phenotypes can be modulated by factors present within the microenvironment of the artery wall, including oxidized lipids, TLR ligands, hematopoietic growth factors, cytokines, and chemokines. Within atherosclerotic plaques, the dynamic modulation of macrophage phenotypes impacts atherosclerosis progression by modulating ongoing inflammatory responses within the vessel wall, by regulating apoptotic cell clearance within the developing plaque, and by egress mechanisms. Thus, the dynamic roles that macrophages play in early and advanced atherosclerotic plaques make macrophage phenotype modulation an attractive therapeutic targets for the prevention and treatment of cardiovascular disease.
Acknowledgement
The authors thank Dr. E. Koltsova for generating the figures.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- (1).Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Swirski FK, Weissleder R, Pittet MJ. Heterogeneous in vivo behavior of monocyte subsets in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1424–32. doi: 10.1161/ATVBAHA.108.180521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cybulsky MI, Won D, Haidari M. Leukocyte recruitment to atherosclerotic lesions. Can J Cardiol. 2004;20(Suppl B):24B–8B. [PubMed] [Google Scholar]
- (4).Soehnlein O, Weber C. Myeloid cells in atherosclerosis: initiators and decision shapers. Semin Immunopathol. 2009;31:35–47. doi: 10.1007/s00281-009-0141-z. [DOI] [PubMed] [Google Scholar]
- (5).Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–15. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- (6).Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–23. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- (7).Koltsova EK, Ley K. Dendritic cells in atherosclerosis. Trends Immunol. 2011 doi: 10.1016/j.it.2011.07.001. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005;26:1700–4. doi: 10.1093/eurheartj/ehi282. [DOI] [PubMed] [Google Scholar]
- (9).Niessner A, Weyand CM. Dendritic cells in atherosclerotic disease. Clin Immunol. 2009 doi: 10.1016/j.clim.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gerrity RG. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am J Pathol. 1981;103:181–90. [PMC free article] [PubMed] [Google Scholar]
- (11).Gerrity RG. The role of the monocyte in atherogenesis: II. Migration of foam cells from atherosclerotic lesions. Am J Pathol. 1981;103:191–200. [PMC free article] [PubMed] [Google Scholar]
- (12).Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;203:1273–82. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Van Furth R, Diesselhoff-den Dulk MM. Dual origin of mouse spleen macrophages. J Exp Med. 1984;160:1273–83. doi: 10.1084/jem.160.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–92. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- (15).Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–6. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–43. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- (17).Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113:3418–27. doi: 10.1182/blood-2008-12-180646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, Cumano A, Geissmann F. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science. 2006;311:83–7. doi: 10.1126/science.1117729. [DOI] [PubMed] [Google Scholar]
- (19).Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol. 2007;8:1207–16. doi: 10.1038/ni1518. [DOI] [PubMed] [Google Scholar]
- (20).Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, von Andrian UH. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99:111–20. doi: 10.1182/blood.v99.1.111. [DOI] [PubMed] [Google Scholar]
- (22).Wiktor-Jedrzejczak W, Gordon S. Cytokine regulation of the macrophage (M phi) system studied using the colony stimulating factor-1-deficient op/op mouse. Physiol Rev. 1996;76:927–47. doi: 10.1152/physrev.1996.76.4.927. [DOI] [PubMed] [Google Scholar]
- (23).Hromas R, Orazi A, Neiman RS, Maki R, Van BC, Moore J, Klemsz M. Hematopoietic lineage- and stage-restricted expression of the ETS oncogene family member PU.1. Blood. 1993;82:2998–3004. [PubMed] [Google Scholar]
- (24).Chen HM, Zhang P, Voso MT, Hohaus S, Gonzalez DA, Glass CK, Zhang DE, Tenen DG. Neutrophils and monocytes express high levels of PU.1 (Spi-1) but not Spi-B. Blood. 1995;85:2918–28. [PubMed] [Google Scholar]
- (25).McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–58. [PMC free article] [PubMed] [Google Scholar]
- (26).Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–7. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- (27).Nguyen HQ, Hoffman-Liebermann B, Liebermann DA. The zinc finger transcription factor Egr-1 is essential for and restricts differentiation along the macrophage lineage. Cell. 1993;72:197–209. doi: 10.1016/0092-8674(93)90660-i. [DOI] [PubMed] [Google Scholar]
- (28).Krishnaraju K, Hoffman B, Liebermann DA. Early growth response gene 1 stimulates development of hematopoietic progenitor cells along the macrophage lineage at the expense of the granulocyte and erythroid lineages. Blood. 2001;97:1298–305. doi: 10.1182/blood.v97.5.1298. [DOI] [PubMed] [Google Scholar]
- (29).Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. 2000;13:155–65. doi: 10.1016/s1074-7613(00)00016-9. [DOI] [PubMed] [Google Scholar]
- (30).Nagamura-Inoue T, Tamura T, Ozato K. Transcription factors that regulate growth and differentiation of myeloid cells. Int Rev Immunol. 2001;20:83–105. doi: 10.3109/08830180109056724. [DOI] [PubMed] [Google Scholar]
- (31).Lord KA, Abdollahi A, Hoffman-Liebermann B, Liebermann DA. Proto-oncogenes of the fos/jun family of transcription factors are positive regulators of myeloid differentiation. Mol Cell Biol. 1993;13:841–51. doi: 10.1128/mcb.13.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mollinedo F, Vaquerizo MJ, Naranjo JR. Expression of c-jun, jun B and jun D proto-oncogenes in human peripheral-blood granulocytes. Biochem J. 1991;273:477–9. doi: 10.1042/bj2730477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Passegue E, Wagner EF. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000;19:2969–79. doi: 10.1093/emboj/19.12.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000;19:2056–68. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Passegue E, Jochum W, Schorpp-Kistner M, Mohle-Steinlein U, Wagner EF. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell. 2001;104:21–32. doi: 10.1016/s0092-8674(01)00188-x. [DOI] [PubMed] [Google Scholar]
- (36).Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119:431–43. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- (37).Mullican SE, Zhang S, Konopleva M, Ruvolo V, Andreeff M, Milbrandt J, Conneely OM. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat Med. 2007;13:730–5. doi: 10.1038/nm1579. [DOI] [PubMed] [Google Scholar]
- (38).Feinberg MW, Wara AK, Cao Z, Lebedeva MA, Rosenbauer F, Iwasaki H, Hirai H, Katz JP, Haspel RL, Gray S, Akashi K, Segre J, Kaestner KH, Tenen DG, Jain MK. The Kruppel-like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J. 2007;26:4138–48. doi: 10.1038/sj.emboj.7601824. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hegde SP, Zhao J, Ashmun RA, Shapiro LH. c-Maf induces monocytic differentiation and apoptosis in bipotent myeloid progenitors. Blood. 1999;94:1578–89. [PubMed] [Google Scholar]
- (40).Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E, Lira SA, Weissman IL, Weber C, Jung S. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113:963–72. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- (41).Goto Y, Hogg JC, Suwa T, Quinlan KB, Van Eeden SF. A novel method to quantify the turnover and release of monocytes from the bone marrow using the thymidine analog 5′-bromo-2′-deoxyuridine. Am J Physiol Cell Physiol. 2003;285:C253–C259. doi: 10.1152/ajpcell.00035.2003. [DOI] [PubMed] [Google Scholar]
- (42).Van Furth R. Origin and turnover of monocytes and macrophages. Curr Top Pathol. 1989;79:125–50. [PubMed] [Google Scholar]
- (43).Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- (44).Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, Habenicht AJ, Ziegler-Heitbrock L, Randolph GJ. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;115:e10–e19. doi: 10.1182/blood-2009-07-235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. N. [PubMed] [Google Scholar]
- (46).Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, Puel A, Biswas SK, Moshous D, Picard C, Jais JP, D’Cruz D, Casanova JL, Trouillet C, Geissmann F. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–86. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Varol C, Landsman L, Fogg DK, Greenshtein L, Gildor B, Margalit R, Kalchenko V, Geissmann F, Jung S. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J Exp Med. 2006;204:171–80. doi: 10.1084/jem.20061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Methe H, Kim JO, Kofler S, Weis M, Nabauer M, Koglin J. Expansion of circulating Toll-like receptor 4-positive monocytes in patients with acute coronary syndrome. Circulation. 2005;111:2654–61. doi: 10.1161/CIRCULATIONAHA.104.498865. [DOI] [PubMed] [Google Scholar]
- (49).Geng HL, Lu HQ, Zhang LZ, Zhang H, Zhou L, Wang H, Zhong RQ. Increased expression of Toll like receptor 4 on peripheral-blood mononuclear cells in patients with coronary arteriosclerosis disease. Clin Exp Immunol. 2006;143:269–73. doi: 10.1111/j.1365-2249.2005.02982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Xie P, Cao YS, Su P, Li YH, Gao ZL, Borst MM. Expression of toll-like receptor 4, tumor necrosis factor- alpha, matrix metalloproteinase-9 and effects of benazepril in patients with acute coronary syndromes. Clin Med Insights Cardiol. 2010;4:89–93. doi: 10.4137/CMC.S5659. 89-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Coen PM, Flynn MG, Markofski MM, Pence BD, Hannemann RE. Adding exercise to rosuvastatin treatment: influence on C-reactive protein, monocyte toll-like receptor 4 expression, and inflammatory monocyte (CD14+CD16+) population. Metabolism. 2010;59:1775–83. doi: 10.1016/j.metabol.2010.05.002. [DOI] [PubMed] [Google Scholar]
- (52).Geissmann F, Auffray C, Palframan R, Wirrig C, Ciocca A, Campisi L, Narni-Mancinelli E, Lauvau G. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol Cell Biol. 2008;86:398–408. doi: 10.1038/icb.2008.19. [DOI] [PubMed] [Google Scholar]
- (53).Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211:609–18. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- (54).Varol C, Yona S, Jung S. Origins and tissue-context-dependent fates of blood monocytes. Immunol Cell Biol. 2009;87:30–8. doi: 10.1038/icb.2008.90. [DOI] [PubMed] [Google Scholar]
- (55).Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–83. doi: 10.1084/jem.20060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–90. doi: 10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- (57).Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annual Review of Immunology. 2009;27:165–97. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Taylor PR, Gordon S. Monocyte heterogeneity and innate immunity. Immunity. 2003;19:2–4. doi: 10.1016/s1074-7613(03)00178-x. [DOI] [PubMed] [Google Scholar]
- (59).Varol C, Landsman L, Jung S. Probing in vivo origins of mononuclear phagocytes by conditional ablation and reconstitution. Methods Mol Biol. 2009;531:71–87. 71–87. doi: 10.1007/978-1-59745-396-7_6. [DOI] [PubMed] [Google Scholar]
- (60).Varol C, Vallon-Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, Fehling HJ, Hardt WD, Shakhar G, Jung S. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31:502–12. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- (61).Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- (62).Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–52. 421–52. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–6. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- (64).Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. J Immunol. 2006;177:7303–11. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- (65).Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. 1995;25:1101–4. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
- (66).Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–70. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- (67).Ramos CL, Huo Y, Jung U, Ghosh S, Manka DR, Sarembock IJ, Ley K. Direct demonstration of P-selectin and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ Res. 1999;84:1237–44. doi: 10.1161/01.res.84.11.1237. [DOI] [PubMed] [Google Scholar]
- (68).Huo Y, Hafezi-Moghadam A, Ley K. Role of Vascular Adhesion Molecule-1 (VCAM-1) and fibronectin connecting segment-1 (CS-1) in monocyte adherence on early atherosclerotic lesions. Circ Res. 2000;87:153–9. doi: 10.1161/01.res.87.2.153. [DOI] [PubMed] [Google Scholar]
- (69).Eriksson EE, Werr J, Guo Y, Thoren P, Lindbom L. Direct observations in vivo on the role of endothelial selectins and alpha4 integrin in cytokine-induced leukocyte-endothelium interactions in the mouse aorta. Circ Res. 2000;86:526–33. doi: 10.1161/01.res.86.5.526. [DOI] [PubMed] [Google Scholar]
- (70).Drechsler M, Megens RT, van ZM, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714. [DOI] [PubMed] [Google Scholar]
- (71).Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- (72).Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–89. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- (73).Huo Y, Weber C, Forlow SB, Sperandio M, Thatte J, Mack M, Jung S, Littman DR, Ley K. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest. 2001;108:1307–14. doi: 10.1172/JCI12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Richardson M, Hadcock SJ, DeReske M, Cybulsky MI. Increased expression in vivo of VCAM-1 and E-selectin by the aortic endothelium of normolipemic and hyperlipemic diabetic rabbits. Arteriosclerosis & Thrombosis. 1994;14:760–9. doi: 10.1161/01.atv.14.5.760. [DOI] [PubMed] [Google Scholar]
- (75).Galkina E, Ley K. Leukocyte influx in atherosclerosis. Current Drug Targets. 2007;8:1239–48. doi: 10.2174/138945007783220650. [DOI] [PubMed] [Google Scholar]
- (76).Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA, Luster AD, Luscinskas FW, Rosenzweig A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718–23. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- (77).Lau EK, Paavola CD, Johnson Z, Gaudry JP, Geretti E, Borlat F, Kungl AJ, Proudfoot AE, Handel TM. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J Biol Chem. 2004;279:22294–305. doi: 10.1074/jbc.M311224200. [DOI] [PubMed] [Google Scholar]
- (78).Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–57. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- (80).An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, Huo Y, Xia L. P-Selectin Glycoprotein Ligand-1 Is Highly Expressed on Ly-6Chi Monocytes and a Major Determinant for Ly-6Chi Monocyte Recruitment to Sites of Atherosclerosis in Mice. Circulation. 2008;24:3227–37. doi: 10.1161/CIRCULATIONAHA.108.771048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103:10340–5. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Muller WA. The role of PECAM-1 (CD31) in leukocyte emigration: Studies in vitro and in vivo. J Leukocyte Biol. 1995;57:523–8. doi: 10.1002/jlb.57.4.523. [DOI] [PubMed] [Google Scholar]
- (84).Schenkel AR, Chew TW, Muller WA. Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J Immunol. 2004;173:6403–8. doi: 10.4049/jimmunol.173.10.6403. [DOI] [PubMed] [Google Scholar]
- (85).Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–62. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).HORLICK L. Platelet adhesiveness in normal persons and subjects with atherosclerosis. Effect of high fat meals and anticoagulants on the adhesive index. Am J Cardiol. 1961;8:459–70. doi: 10.1016/0002-9149(61)90119-9. [DOI] [PubMed] [Google Scholar]
- (87).Bradfield PF, Scheiermann C, Nourshargh S, Ody C, Luscinskas FW, Rainger GE, Nash GB, Miljkovic-Licina M, Aurrand-Lions M, Imhof BA. JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood. 2007;110:2545–55. doi: 10.1182/blood-2007-03-078733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Kuckleburg CJ, Yates C, Kalia N, Zhao Y, Nash GB, Watson SP, Rainger GE. Endothelial cell borne platelet bridges selectively recruit monocytes in human and mouse models of vascular inflammation. Cardiovasc Res. 2011 doi: 10.1093/cvr/cvr040. [DOI] [PubMed] [Google Scholar]
- (89).Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nature Med. 2003;9:61–7. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- (90).Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, Hechtman HB, Michelson AD. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. Journal of the American College of Cardiology. 1998;31:352–8. doi: 10.1016/s0735-1097(97)00510-x. [DOI] [PubMed] [Google Scholar]
- (91).Tull SP, Anderson SI, Hughan SC, Watson SP, Nash GB, Rainger GE. Cellular pathology of atherosclerosis: smooth muscle cells promote adhesion of platelets to cocultured endothelial cells. Circ Res. 2006;98:98–104. doi: 10.1161/01.RES.0000198386.69355.87. [DOI] [PubMed] [Google Scholar]
- (92).Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101:2661–6. doi: 10.1182/blood-2002-07-2209. [DOI] [PubMed] [Google Scholar]
- (93).Sachais BS, Turrentine T, McKenna JM Dawicki, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE−/− mice. Thromb Haemost. 2007;98:1108–13. [PubMed] [Google Scholar]
- (94).Koenen RR, von HP, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15:97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- (95).Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, Bergmeier W, Richter T, Lorenz M, Konrad I, Nieswandt B, Gawaz M. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–96. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2006;21:99–111. doi: 10.1016/j.blre.2006.06.001. [DOI] [PubMed] [Google Scholar]
- (97).Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. 2010;184:4810–8. doi: 10.4049/jimmunol.0901368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Gleissner CA, Shaked I, Erbel C, Bockler D, Katus HA, Ley K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ Res. 2010;106:203–11. doi: 10.1161/CIRCRESAHA.109.199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages and dendritic cells. Science. 2010;327:656–61. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. 453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- (101).Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, Kensler T, Ravichandran KS, Isakson BE, Wamhoff BR, Leitinger N. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–46. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res. 2008;102:950–8. doi: 10.1161/CIRCRESAHA.107.170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Miller YI, Chang MK, Funk CD, Feramisco JR, Witztum JL. 12/15-lipoxygenase translocation enhances site-specific actin polymerization in macrophages phagocytosing apoptotic cells. J Biol Chem. 2001;276:19431–9. doi: 10.1074/jbc.M011276200. [DOI] [PubMed] [Google Scholar]
- (104).Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–6. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).King JY, Ferrara R, Tabibiazar R, Spin JM, Chen MM, Kuchinsky A, Vailaya A, Kincaid R, Tsalenko A, Deng DX, Connolly A, Zhang P, Yang E, Watt C, Yakhini Z, Ben-Dor A, Adler A, Bruhn L, Tsao P, Quertermous T, Ashley EA. Pathway analysis of coronary atherosclerosis. Physiol Genomics. 2005;23:103–18. doi: 10.1152/physiolgenomics.00101.2005. [DOI] [PubMed] [Google Scholar]
- (107).Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779–84. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Current Pharmaceutical Design. 2005;11:3061–72. doi: 10.2174/1381612054865064. [DOI] [PubMed] [Google Scholar]
- (109).Linton MF, Fazio S. Macrophages, inflammation, and atherosclerosis. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S35–40. doi: 10.1038/sj.ijo.0802498. S35-S40. [DOI] [PubMed] [Google Scholar]
- (110).Anitchkow N. Ueber die Veraenderungen der Kaninchenaorta bei experimenteller Cholesterinsteatose. Beitr Pathol Anat. 1913;56:379–404. [Google Scholar]
- (111).Gerrity RG, Naito HK. Ultrastructural identification of monocyte-derived foam cells in fatty streak lesions. Artery. 1980;8:208–14. [PubMed] [Google Scholar]
- (112).Mehta JL. The role of LOX-1, a novel lectin-like receptor for oxidized low density lipoprotein, in atherosclerosis. Can J Cardiol. 2004;20(Suppl B):32B–6B. [PubMed] [Google Scholar]
- (113).Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277:49982–8. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- (114).Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002;277:38517–23. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- (115).Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem. 2002;277:38503–16. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- (116).Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–56. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203:2613–25. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4:211–21. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Manning-Tobin JJ, Moore KJ, Seimon TA, Bell SA, Sharuk M, Alvarez-Leite JI, de Winther MP, Tabas I, Freeman MW. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2009;29:19–26. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest. 2008:35535. doi: 10.1172/JCI35535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Witztum JL. You are right too! J Clin Invest. 2005;115:2072–5. doi: 10.1172/JCI26130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, Combs CA, Malide D, Zhang WY. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2005;280:2352–60. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- (124).Anzinger JJ, Chang J, Xu Q, Buono C, Li Y, Leyva FJ, Park BC, Greene LE, Kruth HS. Native low-density lipoprotein uptake by macrophage colony-stimulating factor-differentiated human macrophages is mediated by macropinocytosis and micropinocytosis. Arterioscler Thromb Vasc Biol. 2010;30:2022–31. doi: 10.1161/ATVBAHA.110.210849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem. 2003;278:1561–8. doi: 10.1074/jbc.M209634200. [DOI] [PubMed] [Google Scholar]
- (126).Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, Witztum JL, Bae YS, Miller YI. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ Res. 2009;104:1355–63. doi: 10.1161/CIRCRESAHA.108.192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Buono C, Anzinger JJ, Amar M, Kruth HS. Fluorescent pegylated nanoparticles demonstrate fluid-phase pinocytosis by macrophages in mouse atherosclerotic lesions. J Clin Invest. 2009;119:1373–81. doi: 10.1172/JCI35548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Higashimori M, Tatro JB, Moore KJ, Mendelsohn ME, Galper JB, Beasley D. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:50–7. doi: 10.1161/ATVBAHA.110.210971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–84. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Coenen KR, Gruen ML, Lee-Young RS, Puglisi MJ, Wasserman DH, Hasty AH. Impact of macrophage toll-like receptor 4 deficiency on macrophage infiltration into adipose tissue and the artery wall in mice. Diabetologia. 2009;52:318–28. doi: 10.1007/s00125-008-1221-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Katsargyris A, Tsiodras S, Theocharis S, Giaginis K, Vasileiou I, Bakoyiannis C, Georgopoulos S, Bastounis E, Klonaris C. Toll-like receptor 4 immunohistochemical expression is enhanced in macrophages of symptomatic carotid atherosclerotic plaques. Cerebrovasc Dis. 2011;31:29–36. doi: 10.1159/000320259. [DOI] [PubMed] [Google Scholar]
- (132).Miller YI, Choi SH, Fang L, Harkewicz R. Toll-like receptor-4 and lipoprotein accumulation in macrophages. Trends Cardiovasc Med. 2009;19:227–32. doi: 10.1016/j.tcm.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Oram JF, Lawn RM. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. J Lipid Res. 2001;42:1173–9. [PubMed] [Google Scholar]
- (134).Oram JF. Novel approaches to treating cardiovascular disease: lessons from Tangier disease. Expert Opin Investig Drugs. 2001;10:427–38. doi: 10.1517/13543784.10.3.427. [DOI] [PubMed] [Google Scholar]
- (135).Wang N, Silver DL, Thiele C, Tall AR. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J Biol Chem. 2001;276:23742–7. doi: 10.1074/jbc.M102348200. [DOI] [PubMed] [Google Scholar]
- (136).Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–31. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- (138).Castrillo A, Tontonoz P. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu Rev Cell Dev Biol. 2004;20:455–80. doi: 10.1146/annurev.cellbio.20.012103.134432. [DOI] [PubMed] [Google Scholar]
- (139).Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol. 2003;17:985–93. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- (140).Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- (141).Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–71. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- (142).Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. Ppargamma promotes monocyte/macrophage differentiation and uptake of oxidized ldl. Cell. 1998;93:241–52. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- (143).Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, Valledor AF, Davis RA, Willson TM, Witztum JL, Palinski W, Glass CK. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–76. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Li AC, Glass CK. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J Lipid Res. 2004;45:2161–73. doi: 10.1194/jlr.R400010-JLR200. [DOI] [PubMed] [Google Scholar]
- (145).Larrede S, Quinn CM, Jessup W, Frisdal E, Olivier M, Hsieh V, Kim MJ, Van EM, Couvert P, Carrie A, Giral P, Chapman MJ, Guerin M, Le GW. Stimulation of cholesterol efflux by LXR agonists in cholesterol-loaded human macrophages is ABCA1-dependent but ABCG1-independent. Arterioscler Thromb Vasc Biol. 2009;29:1930–6. doi: 10.1161/ATVBAHA.109.194548. [DOI] [PubMed] [Google Scholar]
- (146).Baldan A, Bojanic DD, Edwards PA. The ABCs of sterol transport. J Lipid Res. 2009 April;(50 Suppl):S80–S85. doi: 10.1194/jlr.R800044-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Bultel S, Helin L, Clavey V, Chinetti-Gbaguidi G, Rigamonti E, Colin M, Fruchart JC, Staels B, Lestavel S. Liver X receptor activation induces the uptake of cholesteryl esters from high density lipoproteins in primary human macrophages. Arterioscler Thromb Vasc Biol. 2008;28:2288–95. doi: 10.1161/ATVBAHA.108.175042. [DOI] [PubMed] [Google Scholar]
- (148).Feig JE, Pineda-Torra I, Sanson M, Bradley MN, Vengrenyuk Y, Bogunovic D, Gautier EL, Rubinstein D, Hong C, Liu J, Wu C, van RN, Bhardwaj N, Garabedian M, Tontonoz P, Fisher EA. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J Clin Invest. 2010;120:4415–24. doi: 10.1172/JCI38911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Yla-Herttuala S, Rosenfeld ME, Parthasarathy S, Sigal E, Sarkioja T, Witztum JL, Steinberg D. Gene expression in macrophage-rich human atherosclerotic lesions. 15-lipoxygenase and acetyl low density lipoprotein receptor messenger RNA colocalize with oxidation specific lipid-protein adducts. J Clin Invest. 1991;87:1146–52. doi: 10.1172/JCI115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Yla-Herttuala S, Rosenfeld ME, Parthasarathy S, Glass CK, Sigal E, Witztum JL, Steinberg D. Colocalization of 15-lipoxygenase mRNA and protein with epitopes of oxidized low density lipoprotein in macrophage-rich areas of atherosclerotic lesions. Proc Natl Acad Sci U S A. 1990;87:6959–63. doi: 10.1073/pnas.87.18.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Mehrabian M, Allayee H, Wong J, Shi W, Wang XP, Shaposhnik Z, Funk CD, Lusis AJ, Shih W. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91:120–6. doi: 10.1161/01.res.0000028008.99774.7f. [erratum appears in Circ Res 2002;91(8):e27.][Note:Shih Weibin [corrected to Shi Weibin]] [DOI] [PubMed] [Google Scholar]
- (152).Benz DJ, Mol M, Ezaki M, Mori-Ito N, Zelan I, Miyanohara A, Friedmann T, Parthasarathy S, Steinberg D, Witztum JL. Enhanced levels of lipoperoxides in low density lipoprotein incubated with murine fibroblast expressing high levels of human 15-lipoxygenase. J Biol Chem. 1995;270:5191–7. doi: 10.1074/jbc.270.10.5191. [DOI] [PubMed] [Google Scholar]
- (153).Miller YI, Worrall DS, Funk CD, Feramisco JR, Witztum JL. Actin polymerization in macrophages in response to oxidized LDL and apoptotic cells: role of 12/15-lipoxygenase and phosphoinositide 3-kinase. Mol Biol Cell. 2003;14:4196–206. doi: 10.1091/mbc.E03-02-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Sigari F, Lee C, Witztum JL, Reaven PD. Fibroblasts that overexpress 15-lipoxygenase generate bioactive and minimally modified LDL. Arterioscler Thromb Vasc Biol. 1997;17:3639–45. doi: 10.1161/01.atv.17.12.3639. [DOI] [PubMed] [Google Scholar]
- (155).Reilly KB, Srinivasan S, Hatley ME, Patricia MK, Lannigan J, Bolick DT, Vandenhoff G, Pei H, Natarajan R, Nadler JL, Hedrick CC. 12/15-Lipoxygenase activity mediates inflammatory monocyte/endothelial interactions and atherosclerosis in vivo. J Biol Chem. 2004;279:9440–50. doi: 10.1074/jbc.M303857200. [DOI] [PubMed] [Google Scholar]
- (156).Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apoE-deficient mice. J Clin Invest. 1999;103:1597–604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Cyrus T, Pratico D, Zhao L, Witztum JL, Rader DJ, Rokach J, Fitzgerald GA, Funk CD. Absence of 12/15-lipoxygenase expression decreases lipid peroxidation and atherogenesis in apolipoprotein e-deficient mice. Circulation. 2001;103:2277–82. doi: 10.1161/01.cir.103.18.2277. [DOI] [PubMed] [Google Scholar]
- (158).Roy B, Bhattacharjee A, Xu B, Ford D, Maizel AL, Cathcart MK. IL-13 signal transduction in human monocytes: phosphorylation of receptor components, association with Jaks, and phosphorylation/activation of Stats. J Leukoc Biol. 2002;72:580–9. [PubMed] [Google Scholar]
- (159).Roy B, Cathcart MK. Induction of 15-lipoxygenase expression by IL-13 requires tyrosine phosphorylation of Jak2 and Tyk2 in human monocytes. J Biol Chem. 1998;273:32023–9. doi: 10.1074/jbc.273.48.32023. [DOI] [PubMed] [Google Scholar]
- (160).Xu B, Bhattacharjee A, Roy B, Xu HM, Anthony D, Frank DA, Feldman GM, Cathcart MK. Interleukin-13 induction of 15-lipoxygenase gene expression requires p38 mitogen-activated protein kinase-mediated serine 727 phosphorylation of Stat1 and Stat3. Mol Cell Biol. 2003;23:3918–28. doi: 10.1128/MCB.23.11.3918-3928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Yakubenko VP, Bhattacharjee A, Pluskota E, Cathcart MK. {alpha}M{beta}2 Integrin Activation Prevents Alternative Activation of Human and Murine Macrophages and Impedes Foam Cell Formation. Circ Res. 2011;108:544–54. doi: 10.1161/CIRCRESAHA.110.231803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, Forlow SB, Stark MA, Smith DF, Clarke S, Srinivasan S, Hedrick CC, Pratico D, Witztum JL, Nadler JL, Funk CD, Ley K. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–31. doi: 10.1161/01.CIR.0000143628.37680.F6. [DOI] [PubMed] [Google Scholar]
- (163).Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, Colgan SP, Stahl GL, Merched A, Petasis NA, Chan L, Van Dyke TE. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. 2003;171:6856–65. doi: 10.4049/jimmunol.171.12.6856. [DOI] [PubMed] [Google Scholar]
- (164).Shen J, Kuhn H, Petho-Schramm A, Chan L. Transgenic rabbits with the integrated human 15-lipoxygenase gene driven by a lysozyme promoter: macrophage-specific expression and variable positional specificity of the transgenic enzyme. FASEB J. 1995;9:1623–31. doi: 10.1096/fasebj.9.15.8529842. [DOI] [PubMed] [Google Scholar]
- (165).Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kuhn H, Guevara NV, Chan L. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J Clin Invest. 1996;98:2201–8. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008;22:3595–606. doi: 10.1096/fj.08-112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Norling LV, Serhan CN. Profiling in resolving inflammatory exudates identifies novel anti-inflammatory and pro-resolving mediators and signals for termination. J Intern Med. 2010;268:15–24. doi: 10.1111/j.1365-2796.2010.02235.x. [DOI] [PubMed] [Google Scholar]
- (168).Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. 2010;177:1576–91. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Mauldin JP, Nagelin MH, Wojcik AJ, Srinivasan S, Skaflen MD, Ayers CR, McNamara CA, Hedrick CC. Reduced expression of ATP-binding cassette transporter G1 increases cholesterol accumulation in macrophages of patients with type 2 diabetes mellitus. Circulation. 2008;117:2785–92. doi: 10.1161/CIRCULATIONAHA.107.741314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Nagelin MH, Srinivasan S, Nadler JL, Hedrick CC. Murine 12/15-lipoxygenase regulates ATP-binding cassette transporter G1 protein degradation through p38- and JNK2-dependent pathways. J Biol Chem. 2009;284:31303–14. doi: 10.1074/jbc.M109.028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Nagelin MH, Srinivasan S, Lee J, Nadler JL, Hedrick CC. 12/15-Lipoxygenase activity increases the degradation of macrophage ATP-binding cassette transporter G1. Arterioscler Thromb Vasc Biol. 2008;28:1811–9. doi: 10.1161/ATVBAHA.108.167908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Mauldin JP, Srinivasan S, Mulya A, Gebre A, Parks JS, Daugherty A, Hedrick CC. Reduction in ABCG1 in Type 2 diabetic mice increases macrophage foam cell formation. J Biol Chem. 2006;281:21216–24. doi: 10.1074/jbc.M510952200. [DOI] [PubMed] [Google Scholar]
- (173).Antonipillai I, Nadler J, Vu EJ, Bughi S, Natarajan R, Horton R. A 12-lipoxygenase product, 12-hydroxyeicosatetraenoic acid, is increased in diabetics with incipient and early renal disease. J Clin Endocrinol Metab. 1996;81:1940–5. doi: 10.1210/jcem.81.5.8626861. [DOI] [PubMed] [Google Scholar]
- (174).Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–43. doi: 10.1074/jbc.M109.047472. 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, Hiltbold EM, Fessler MB, Parks JS. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–206. doi: 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–47. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–93. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- (179).Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. 451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- (181).Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25:174–9. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–64. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- (183).Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, Boyle JJ, Haskard DO. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol. 2007;170:416–26. doi: 10.2353/ajpath.2007.060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Henson PM, Bratton DL, Fadok VA. Apoptotic cell removal. Curr Biol. 2001;11:R795–R805. doi: 10.1016/s0960-9822(01)00474-2. [DOI] [PubMed] [Google Scholar]
- (185).Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–50. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–82. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Xu W, Roos A, Schlagwein N, Woltman AM, Daha MR, van KC. IL-10-producing macrophages preferentially clear early apoptotic cells. Blood. 2006;107:4930–7. doi: 10.1182/blood-2005-10-4144. [DOI] [PubMed] [Google Scholar]
- (189).Boullier A, Li Y, Quehenberger O, Palinski W, Tabas I, Witztum JL, Miller YI. Minimally oxidized LDL offsets the apoptotic effects of extensively oxidized LDL and free cholesterol in macrophages. Arterioscler Thromb Vasc Biol. 2006;26:1169–76. doi: 10.1161/01.ATV.0000210279.97308.9a. [DOI] [PubMed] [Google Scholar]
- (190).von Vietinghoff S, Komatsu S, Mestas J, Diehl CJ, Witztum JL, Ley K. Mycophenolate mofetil decreases atherosclerotic lesion size by depression of aortic T lymphocyte and IL-17-mediated macrophage accumulation. J Am Coll Cardiol. 2011 doi: 10.1016/j.jacc.2010.12.030. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]