

Abstract
Inhalation of environmental particulate matter (PM) is correlated with adverse health effects in humans, but gene products that couple detection with cellular responses, and the specific properties of PM that target different pathways, have not been fully elucidated. TRPA1 and V1 are two cation channels expressed by sensory neurons and non-neuronal cells of the respiratory tract that have been implicated as possible mediators of PM toxicity. The goals of this research were to determine if environmental PM preferentially activated TRPA1 and to elucidate the criteria responsible for selectivity. Quantification of TRPA1 activation by 4 model PM revealed that diesel exhaust PM (DEP) and coal fly ash PM (CFA1) were TRPA1 agonists at concentrations >0.077 mg/ml. DEP was more potent and approximately 97% of the activity of DEP was recovered by serial extraction of the solid DEP with ethanol and hexane:n-butyl chloride. Modification of the electrophile/agonist binding sites on TRPA1 (C621, C641, C665 and K710) to non-nucleophilic residues reduced TRPA1 activation by DEP and abolished activation by DEP extracts as well as multiple individual electrophilic chemical components of DEP. However, responses to CFA1 and DEP solids were not affected by these mutations. Activity-guided fractionation of DEP and high resolution mass spectroscopy identified several new DEP-derived TRPA1 agonists and activation of mouse dorsal root ganglion neurons demonstrated TRPA1 is a primary target for DEP in a heterogeneous population of primary sensory nerves. It is concluded that TRPA1 is a specific target for electrophilic chemical components of DEP and proposed that activation of TRPA1 in the respiratory tract is likely to be an important mechanism for DEP pneumotoxicity.
Keywords: TRPA1, Diesel, Coal fly ash, Lung
Introduction
Particulate air pollution is associated with numerous acute and chronic adverse health effects in humans, but the biochemical mechanisms linking particle inhalation to specific toxicological processes are poorly understood. One of the major constituents of particulate matter (PM) in urban areas is diesel exhaust particles (DEP) (1). DEP has been studied extensively in vitro and in animal models and has been linked to respiratory and cardiovascular diseases including cardiac arrhythmias, acute lung inflammation, airway irritation, and cancer (2, 3).
A number of studies have correlated responses to urban PM, including DEP, with activation of airway sensory neurons, particularly C- and Aδ- fibers that express Transient Receptor Potential Ankyrin-1 (TRPA1), TRP Vanilloid-1 (TRPV1), and substance P (3–7). TRPA1 and V1 are members of the TRP superfamily of ion channels, and numerous TRP receptors including TRPA1, V1–4, and M8 are highly expressed in the respiratory tract where they can function as environmental sensors. TRPA1, M8, and V1–V4 all respond to specific chemical irritants (8–10) and activation of either TRPA1 or V1 in pulmonary sensory nerves causes neurogenic inflammation via the release of substance P (SP) and neurokinin A (NKA) (11). TRPV1, V3, V4 and M8, but seemingly not TRPA1 or V2, are also expressed by airway epithelial cells, and activation of TRPV1 (7, 12–17) and TRPM8 (18) in these cells by prototype agonists and select forms of environmental PM is coupled with pro-inflammatory cytokine/chemokine production and apoptosis.
Recent studies have demonstrated an essential role of TRPA1 as a mediator of acute respiratory responses to electrophilic and oxidizing environmental pollutants including acrolein and crotonaldehyde, two common constituents of combustion-derived materials (19, 20), as well as to H2O2 and HOCl (21). Additionally, TRPA1 has been implicated in cardiovascular dysfunction associated with pulmonary DEP exposure in rats (3). The evolving role for TRPA1 as a target for both combustion-derived electrophiles and oxidants and the ability of TRPA1 to mediate pulmonary reflex responses and neurogenic inflammation make TRPA1 a prime candidate for a molecular target of DEP and additional combustion-derived PM, as well as a probable mediator of acute respiratory toxicity. In the present study, four unique and environmentally relevant PM were used to explore the scope and selectivity of TRPA1 activation as a possible mechanism of respiratory toxicity for different PM.
Materials and Methods
Chemicals
All chemicals used in this study were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. AITC, nonivamide and the chemicals listed in Tables 2 and 3 have the potential to activate TRPA1 and V1 and therefore are potent irritants and should be handled carefully.
Particles
Diesel exhaust particulate (DEP) was collected from scraping the tailpipe of an in-service 2004 Ford F350 “black smoker” truck. DEP represents a high soot-content PM from a poorly functioning, but environmentally relevant diesel engine. This DEP sample consists of loose agglomerates of submicron primary particles (Figure 1A). DEP was also purchased from the National Institute of Standards and Technology (SRM #2975) and details on this PM are provided on the NIST website. Coal fly ash (CFA1) PM was collected at local power plant and represents a real-world emission, but is enriched in the coarse fraction and depleted in the submicron fraction compared to typical stack emissions. CFA1 is <30µm and consists of mainly spherical 1–10 µm particles, with smaller particles adhering to the surface of larger particles, and some irregular particles (Figure 1B). Residual elemental carbon for CFA1 is 3% and additional details on the composition of this PM can be found in Smith et al. (22). CFA2 (20% elemental carbon) PM, was collected from a laboratory-scale research furnace by combustion of coal under low excess air. The mineral ash component of CFA2 is similar to CFA1 in shape and size, but CFA2 also contains agglomerates of submicron soot particles and irregular particles of unburned coal char (Figure 1C). Crystalline silica (MUS) is Min-U-Sil 5 (5 µm) (US Silica, Mill Creek, OK). MUS represents silica PM produced by mechanical grinding of mineral material and a redox active PM. MUS PM is shown in Figure 1D.
Figure 1.

Scanning electron micrographs (8000X) for (A) diesel exhaust PM (DEP), (B) coal fly ash 1 PM (CFA1), (C) coal fly ash 2 PM (CFA2), and (D) crystalline silica/Min-U-Sil 5 (MUS).
PM suspensions for treating cells were prepared in LHC-9 medium (Invitrogen) at 2.3 mg/ml which yields a particle treatment of 0.77 mg/mL in the well or 180 µg/cm2 after all particles have settled out of solution when 25µL is added to 50 µl of media in a single well of a 96-well plate containing 30–40,000 cells. For dose-response studies, particles were suspended at 2.3, 0.73 and 0.23 mg/mL to yield in-well concentrations of 0.77, 0.24, and 0.077 mg/mL. A concentration of 0.77 mg/mL well concentration was selected for the screening and subsequent mechanistic studies of receptor activation because it produced a maximum response for TRPA1 activation and allowed for the detection of minor activation events involving other TRP channels such that conclusive yes/no data regarding activation of other TRP channels could be obtained. DEP and CFA1 extracts were prepared by suspending PM in either LHC-9, ethanol, or 1:1 hexane:n-butyl chloride at 2.3 mg/mL and vortex-mixed for 1h. The insoluble components were recovered by centrifugation at 20,000×g for 30 minutes and the supernatants filtered through a 0.22 µm filter. Extracts, except LHC-9, were dried and re-suspended in DMSO (2% final concentration) and diluted in LHC-9 to produce a 3X treatment solution that was equivalent to 2.3 mg/mL PM suspension.
Cloning and Site-Directed Mutagenesis
Human TRPA1, V2, V3 and V4 cDNA was obtained from Open Biosystems (Huntsville, AL). Human TRPM8 cDNA was obtained from Origene (Rockville, MD). Human TRPV1 cDNA was amplified from fetal brain mRNA (Stratagene; La Jolla, CA). All genes were sub-cloned into the pcDNA 3.1D-V5/His6 mammalian expression vector (Invitrogen, Carlsbad, CA), as previously described for TRPV1 (23). The TRPA1–3CK mutant was constructed from the pcDNA 3.1-hTRPA1–V5/His6 construct using the QuickChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) and the following primers: C621A (+) 5' – GAT TTT CAG TCA TAA TTC TCC AGG CAA TAA AGC TCC AAT TAC AGA AAT GAT AGA AT - 3' (-) 5' – ATT CTA TCA TTT CTG TAA TTG GAG CTT TAT TGC CTG GAG AAT TAT GAC TGA AAA TC - 3'; C641A (+) 5' – CAT GAA GGT ACT TTT AGA TTT CGC CAT GTT GCA TTC CAC AGA AGA C - 3' (-) 5' – GTC TTC TGT GGA ATG CAA CAT GGC GAA ATC TAA AAG TAC CTT CAT G - 3'; C665A (+) 5' – CGA GAC TAT TAT ATC GAG TAT AAT TTC AAA TAT CTT CAA GCT CCA TTA GAA TTC ACC AAA A -3' (-) 5'- TTT TGG TGA ATT CTA ATG GAG CTT GAA GAT ATT TGA AAT TAT ACT CGA TAT AAT AGT CTC G -3' K710R (+) 5'- GTG TAA AGA ATA TTT ACT CAT GAG ATG GTT GGC TTA TGG ATT TAG AG -3' (-) 5'- CTC TAA ATC CAT AAG CCA ACC ATC TCA TGA GTA AAT ATT CTT TAC AC -3'.
Cell Culture and TRP Channel Overexpression
HEK-293 cells (ATCC; Rockville, MD) were used as a model for studying TRP channel activation by PM because they exhibit low basal response to the TRP channel agonists and the PM used in this study and are reproducibly and uniformly manipulated by transfection which is necessary for mechanistic studies of the responses. HEK-293 cells were grown in DMEM:F12 (Invitrogen; Carlsbad, CA) containing 5% FBS and 1X penicillin/streptomycin (Invitrogen; Carlsbad, CA). Cells were sub-cultured using trypsin and plated into 1% gelatin-coated 96-well plates for calcium imaging experiments. Human TRPA1, V2, V3, V4, and M8 were stably over-expressed in HEK-293 cells, created by transfecting and selecting for transformed cells using Geneticin (400 µg/ml) (Invitrogen) and isolating homogenous colonies by dilution and expansion of the most sensitive populations determined by calcium flux elicited by the respective prototype agonist. The following agonists were used: TRPA1 – 150 µM AITC (allyl isothiocyanate); TRPM8 – 20 µM icilin; TRPV2 – 100 µM Δ9-tetrahydrocannabinol, TRPV3 – 300 µM carvacrol; TRPV4 – 12.5 nM GSK 1016790A. These concentrations yielded the maximum calcium response relative to wild-type HEK-293 cells. Over-expressing cells were maintained in DMEM:F12 supplemented with 5% FBS and 300 µg/ml Geneticin. TRPV1, TRPA1 and TRPA1–3CK required transient transfection into HEK-293 cells to evaluate channel activation. Purified plasmid (175ng/well in a 96-well plate) was transfected into HEK-293 cells using Lipofectamine 2000 (Invitrogen) and a lipid:DNA ratio of 2:1 in a single well of a 96-well plate. Over 95% of cells were transfected using pMaxGFP as a reference and cells were assayed 48h post transfection. Nonivamide (20 µM) was used as the TRPV1 agonist.
Calcium Imaging
Cells were grown to 80–90% confluence in 1% gelatin-coated 96-well cell culture plates and loaded with Fluo-4 AM, a fluorogenic calcium indicator using the Fluo-4 Direct Kit (Invitrogen) and LHC-9 as the diluent, for 60 min at 37°C in the dark. Cells were washed for 30 min at 37°C in the dark with LHC-9 containing 1 mM probenecid and 0.75 mM trypan red (ATT Bioquest) to stabilize the concentration of active Fluo-4 within the cells. Changes in cellular fluorescence in response to treatments were determined microscopically using an Olympus 1×50 inverted microscope, essentially as described (24). Images were collected immediately prior to and every 30 s for 3 min after treatment, followed by the addition of ionomycin (10 µM final concentration) to establish the maximum attainable fluorescence value for normalization of responses. All agonist and antagonist solutions were prepared in LHC-9 at 3X concentration and 25 µl was added to the cells at room temperature, with the exception of Δ9-tetrahydrocannabinol which was prepared as a 2X stock in LHC-9 containing 5% v/v DMSO. Addition of the solid particles was done manually and the particles were vortex-mixed immediately prior to addition to the cells to ensure the particles were fully suspended before treatment. Images were analyzed for the change in fluorescence of all cells in the field over time. Data are represented as percent maximum response elicited by ionomycin with the vehicle control value subtracted. For screening of HPLC fractions, responses were measured using a NOVOstar fluorescent plate reader (BMG LABTECH, Offenberg, Germany).
Characterization of TRPA1 Agonists from DEP
An ethanol extract of DEP was prepared, concentrated, and fractionated by reversed-phase HPLC using a prep-scale (100×10.0 mm i.d. 5µm) C18 column. Fractionation was achieved by collecting 1 min (2 mL) fractions over a 60 min linear gradient of 20%–100% acetonitrile (ACN). Adjacent active fractions identified by calcium flux were pooled, concentrated, and re-fractionated by HPLC using a 60 min gradient with minimal change in ACN (determined from the original elution conditions) and collecting individual peaks. Active fractions were again identified using calcium flux, concentrated, and submitted to the University of Utah Metabolomics Core for identification of the unknowns. Samples were assayed as MSTFA-derivatized and underivatized samples using accurate mass gas chromatography-electron impact mass spectrometry (GC-EIMS) using a Waters GCT Premier time-of-flight mass spectrometer mated to an Agilent 6890 gas chromatograph equipped with a Gerstel MPS auto sampler and cooled inlet system. GC-EIMS spectral library matching used both NIST and Wiley libraries. Each sample yielded several potential compounds, ranked by probability score, representative of the quality of the spectral match between the unknown and the reference compound. The top 5 chemicals predicted for each peak in each sample were considered and standards of these chemicals (or highly similar analogues) were purchased and tested for TRPA1 agonist activity.
Mouse Dorsal Root Ganglion (DRG) Assays
Primary mouse DRG neurons were used to model responses of pulmonary sensory neurons to DEP components. DRG were isolated from 3-week C57Bl/6 mice, as described (25). Sixteen to 23h after plating, cultures were loaded with Fura-2 AM (Molecular Probes) for ~50 min, then washed with pH 7.4 oxygenated observation medium containing 145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1 mM citrate, 10 mM glucose, 10 mM MES, 10 mM HEPES. Cells were imaged using the Meta Imaging Series Metafluor program (Universal Imaging). Neurons were selected from a phase image based on a round shape and a clear, single nucleus and monitored throughout the assay. Agonists and antagonists were sequentially evaluated by exchanging the solution using two pipettes, one to remove and one to add the solutions, with a wash between treatments. Following DEP treatments, AITC 50 µM was applied, followed by a wash, and then 50 mM KCl to identify TRPA1-expressing (AITC responsive) and viable (AITC or KCl responsive) neurons and to establish the maximum calcium response for each neuron (KCl). A response to either AITC or KCl indicated viability and only viable neurons were analyzed. An average of 110 ± 10 cells were tracked in real time using the 340/380nm fluorescence ratio. Values were normalized to a maximum of 1.0 and minimum of 0.0 and were loaded into an Excel spreadsheet for further analysis. Positive responses were defined as an increase in 340/380 >2SD above baseline obtained from 30–60 s of imaging prior to addition of DEP-EtOH.
Statistical Analysis
Values are expressed as the mean ± SEM. Statistical significance was determined using Student’s t-test or ANOVA with Bonferroni correction using a 95% confidence interval, as indicated in the figure legends.
Results
Treatment of TRPA1 over-expressing HEK-293 cells with PM (0.77 mg/mL) revealed that TRPA1 was robustly activated by DEP and to a lesser extent by CFA1, but not by CFA2 or MUS (Figures 2 and 3). Activation of TRPA1 by DEP was substantially greater than that observed for all other TRP channels tested with relative changes in fluorescence of ~45% maximum response (80% AITC) for TRPA1, versus ~15% maximum response (15% icilin) for TRPM8, 10% maximum response (20% GSK 1016790A) for TRPV4, and ~5% maximum response (15% nonivamide) for TRPV1 (Figure 4). Activation of TRPA1 by “black smoker” DEP and SRM #2975 was qualitatively similar (Table 1 and Figure 3A), with the differences in magnitude reflecting a slower rate of activation by SRM #2975 due to apparent differences in the relative concentrations of electrophilic chemical agonists present on the two DEP samples, as indicated by the differences in the extent and rates of activation by the PM and DEP-EtOH extracts (Figures 3A and B). Data in Table 1 and Figures 3A and B support a mechanism whereby the release of TRPA1 agonists requires cell contact or prior extraction to render the substances soluble.
Figure 2.

Activation of TRPA1 by PM. Human TRPA1 over-expressing HEK-293 cells and untransfected HEK-293 cells were treated with 150 µM AITC or diesel exhaust PM (DEP), coal fly ash PM (CFA1 and CFA2), and crystalline silica (MUS) to a final in-well concentration of 0.77 mg/ml. (A) Data are the percent maximum response elicited by ionomycin (10 µM) with the vehicle control subtracted. *Indicates p<0.05 using Student’s t-test (n=3). (B) Fluorescence micrographs of TRPA1 over-expressing HEK-293 cells treated with the various particles.
Figure 3.

Dose- and time-dependent activation of TRPA1 by CFA1, DEP, and ethanol extracts of DEP. (A) TRPA1-overexpressing cells were treated with CFA1, DEP (black smoker) and DEP-EtOH (black smoker) with in-well concentrations of 0.077, 0.24, and 0.77 mg/ml or equivalent ethanol extract concentrations. Data from the 2 min time point are represented as the mean and SEM of at least 3 separate treatment wells. (B) TRPA1-overexpressing cells were treated with DEP (black smoker), DEP (SRM 2975), and DEP-EtOH (black smoker) using an in-well concentration of 0.77 mg/ml for up to 5 min with measurements taken at 30 s intervals. Data are represented as the mean and SEM of at least 3 separate treatment wells.
Figure 4.

Activation of other human TRP channels by DEP. Over-expressing (TRPA1, M8, V2, V3, and V4), transiently transfected (TRPV1), and untransfected HEK-293 cells were treated with DEP (0.77 mg/ml in-well concentration) or a solution of a prototype TRP channel agonist at a concentration that yielded the maximum response relative to control HEK-293 cells. Agonists were TRPA1 – 150 µM AITC; TRPM8 – 20 µM icilin; TRPV1 – 20 µM nonivamide; TRPV2 – 100 µM Δ9-tetrahydrocannabinol TRPV3 – 300 µM carvacrol; TRPV4 – 12.5 nM GSK 1016790A. Data are the percent maximum response elicited by ionomycin (10 µM) with the vehicle control subtracted. N.D. = no response detected. *Indicates p<0.05 using ANOVA with Bonferroni correction (n=3).
Table 1.
Activation of TRPA1 by DEP and CFA1 particles (0.77 mg/) and equivalent particle extracts.
| Extract | |||||
|---|---|---|---|---|---|
| PM | Aqueous | EtOH | Hex:nBuCl | Residual material |
|
| DEP (black smoker) | 49 ± 10* | N.D. | 84 ± 6* | 48 ± 15* | 3 ± 2 |
| DEP (SRM 2975) | 7 ± 3* | N.D. | 19 ± 7* | 18 ± 5* | N.D. |
| CFA1 | 15 ± 7* | 2 ± 2 | N.D. | N.D. | 4 ± 2 |
Data are % maximum response at 2 min.
Indicates significant induction of calcium flux compared to non-transfected controls using Student’s t-test, p<0.05 (n=3).
The mechanism of TRPA1 activation by DEP and CFA1 was investigated using two strategies: First, particle extraction using LHC-9 media (aqueous), ethanol, or n-butyl chloride:hexane was used to determine the physical/chemical nature of the agonist(s), and second, by site-directed mutagenesis of the TRPA1 electrophile binding sites. Both ethanol and n-butyl chloride:hexane extracts of DEP activated TRPA1, with the ethanol extract being substantially more potent than DEP (Table 1 and Figure 3). Only ~3% of the original activity of DEP remained after serial extraction and none of the CFA1 extracts activated TRPA1 (Table 1). Prototype agonists of TRPA1, including AITC, activate TRPA1 by covalent modification of C621, C641, C665, and K710 (26, 27). Calcium flux elicited by DEP in TRPA1–3CK mutant transfected cells was ~40% of wild-type TRPA1, and responses to DEP-EtOH were completely abolished (Figure 5). In contrast, the response of TRPA1 to CFA1 was unaffected by the 3CK mutation (Figure 5) indicating that activation of TRPA1 by DEP and CFA1 occurred via two different mechanisms, the first involving electrophilic modification by DEP-associated electrophiles, and the second potentially involving mechanical activation of TRPA1 (28–30) at the cell surface by insoluble components of DEP and CFA1.
Figure 5.

Mutation of C621, C641, and C665 to A and lysine K710 to A of TRPA1 (the TRPA1–3CK mutant) inhibits DEP-induced calcium flux. HEK-293 cells were transiently transfected with human TRPA1 or the TRPA1–3CK mutant and treated with 150 µM AITC, suspensions of DEP or CFA1 to a final in-well concentration of 0.77 mg/mL, or an equivalent concentration of DEP-EtOH extract. Data are the percent maximum response elicited by ionomycin (10 µM) with the vehicle control subtracted. N.D. = no response detected. *Indicates significant (p<0.05) calcium influx compared to HEK-293 controls, and # indicates a significant reduction in TRPA1–3CK mutant transfected cells compared to wild-type TRPA1 using ANOVA with Bonferroni correction.
The identities of soluble DEP-derived TRPA1 agonists were also pursued. A set of chemicals that comprise DEP was compiled based on the OSHA list of DEP components (31) and Schauer et al. (32) and were tested as TRPA1 agonists (Table 2). Acrolein, a known TRPA1 agonist (19, 20, 33–37), formaldehyde, 2-methyl-1,4-napthoquinone (menadione), 1,3-dihydroxynaphthalene, 1-nitronaphthalene, 1,2-naphthoquinone, hydroquinone, and o-anisaldehyde all activated TRPA1 via electrophilic modification based on a lack of activation of the TRPA1–3CK mutant. None of these chemicals activated TRPM8, but 1,2-naphthoquinone activated TRPV1 to ~5% nonivamide at both 25 and 250 µM. However, responses were not statistically significant for TRPV1 and likely were an artifact based on the lack of dose response.
Table 2.
Activation of TRPA1 by select known chemical components of DEP.
| Compound |
aCitations with DEP |
aCitations with TRPA1 |
Ca++ Flux (% max response) 2.5 µM |
Ca++ Flux (% max response) 25 µM |
Ca++ Flux (% max response) 250 µM |
Ca++ Flux TRPA1–3CK |
|---|---|---|---|---|---|---|
| Coumarin | (31, 53) | None | - | - | N.D. | - |
| 6-Methycoumarin | (31) | None | - | - | N.D. | - |
| Formaldehyde | (31, 43, 54) | (36, 55) | N.D. | N.D. | 12 ± 2* | N.D. |
| Acrolein | (31, 43, 54) | (19, 20, 33–37) | 12 ± 6* | 23 ± 12* | 66 ± 2* | N.D. |
| o-Anisaldehyde | (31, 54) | None | - | N.D. | 43 ± 8* | N.D. |
| Phenanthrene | (31, 56, 57) | None | - | - | N.D | - |
| Naphthalene | (31, 46, 56–58) | (46) | - | - | N.D | - |
| 1,3-Dihydroxynapththalene | None | None | - | N.D. | 22 ± 5* | N.D. |
| 2-Methyl-1,4-naphthoquinone | (31, 59) | None | - | N.D. | 2 ± 1* | N.D. |
| 1,2-Naphthoquinone | (7, 59) | (7) | 5 ± 6 | 85 ± 16* | 91 ± 1* | N.D. |
| 1-Nitronaphthalene | (60, 61) | None | - | N.D. | 13 ± 5* | N.D. |
| Hydroquinone | (62) | None | N.D. | N.D. | 7 ± 1* | N.D. |
| AITC | None | (63) | N.D. | 5 ± 4 | 75 ± 4* | N.D. |
Indicates significant induction of calcium flux compared to non-transfected controls using Student’s t-test, p<0.05 (n=3).
N.D. =Response was not detected. -=Not tested.
Literature search was performed with NLM and other similar database search programs. The search terms were: compound name and diesel, compound name and DEP, compound name and TRPA1, compound name and Transient Receptor Potential.
Subsequently, the ethanol extract from DEP was fractionated by HPLC to purify TRPA1 agonists from this potent preparation. After several rounds of enrichment, five fractions were obtained. GCEIMS analysis and EI spectral library searching provided a number of potential identities for the unknowns. Eleven of these chemicals (or structural analogues of the predicted compounds) were purchased and tested as TRPA1 agonists (Table 3). 4-Phenyl-1,2-dihydronaphthalene, [(3E)-1-phenyl-1,3-pentadienyl]benzene, (1-phenyl-1-pentenyl)benzene, [(2Z)-3-phenyl-2-butenyl]benzene, 3,5-ditert-butylphenol, and 2,4-ditert-butylphenol activated TRPA1 and these chemicals were either conjugated alkenes similar to the TRPA1 agonist 1,3-butadiene (20, 38) or were ditert-butylphenols, which represent a previously unknown class of TRPA1 agonists presumably arising from the addition of stabilizers to lubricants and diesel (39). None of these compounds activated TRPV1, but 2,4-ditert-butylphenol activated TRPM8 to ~9% icilin, which was not statistically significant. Additionally, all of these chemicals were inactive in TRPA1–3CK-transfected cells.
Table 3.
Activation of TRPA1 by DEP-derived chemicals, or structurally similar analogues, isolated from an ethanol extract of DEP by HPLC and identified by GC-EIMS.
| Name | Structure | TRPA1 Activity % AITC |
|---|---|---|
| trans,trans-1,4-Diphenyl-1,3-butadiene | 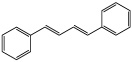 |
N.D. |
| 4-Phenyl-1,2-dihydronaphthalene | 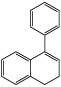 |
14 ± 3* |
| [(3E)-1-Phenyl-1,3-pentadienyl]benzene | 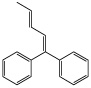 |
73 ± 27* |
| (1-Phenyl-1-pentenyl)benzene | 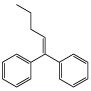 |
26 ± 3* |
| [(2Z)-1-Methyl-3-phenyl-2-butenyl]benzene | 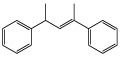 |
6 ± 8 |
| [(2Z)-3-Phenyl-2-butenyl]benzene | 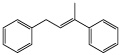 |
5 ± 2* |
| 4-Hydroxybenzyl alcohol | N.D | |
| 2-Hydroxybenzyl alcohol | 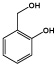 |
N.D |
| 3,5-Ditert-butylphenol | 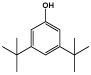 |
87 ± 26* |
| 2,4-Ditert-butylphenol | 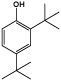 |
89 ± 11* |
| Dibutylphthalate | 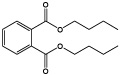 |
2 ± 2 |
All treatments were at 250 µM and are reported as the % response relative to the positive control AITC (150µM).
Indicates significant induction of calcium flux compared to non-transfected controls using Student’s t-test, p<0.05 (n=3).
Finally, activation of TRPA1 by DEP-EtOH was evaluated in cultured mouse DRG neurons to test for selectivity in a highly complex and biologically relevant model where TRPA1, TRPV1-V4, M8 and countless other potential molecular targets of DEP or DEP components are expressed. DEP-EtOH induced calcium influx that overlapped ~90% with responses to AITC (Figure 6), indicating that the overall cellular response to DEP-EtOH was principally attributable to TRPA1-expressing cells. Activation of TRPA1 in DRG neurons by both AITC and DEP-EtOH was equivalently reduced by ~75% by pre/co-treatment with the TRPA1 antagonist, HC-030031, further indicating the critical role of TRPA1 in mediating neuronal responses to electrophilic DEP components.
Figure 6.

Activation of TRPA1 by DEP-EtOH in mouse DRG neurons. DRG were treated with 50 µM AITC or the DEP-EtOH extract prepared from a 2.3 mg/mL suspension in the presence or absence of the TRPA1 antagonist HC-030031 (50 µM). Data are represented as percent maximum response as elicited by KCl (50 mM). Each data point is the mean ± SEM response from DRG cultures of four separate animals. *Indicates significant inhibition by HC-030031 (p<0.05 using Student’s t-test).
Discussion
Mechanisms underlying the morbidity and mortality associated with PM are not fully understood. DEP is a pneumotoxicant that causes pulmonary inflammation and tissue damage, in part, via the activation of pro-inflammatory signaling in the lung through both neurogenic and non-neurogenic pathways (7, 40). However, gene products that detect DEP and initiate specific cellular responses are not well defined. This study corroborates recent studies suggesting a role for TRPA1 in acute cardiovascular responses elicited by DEP (3) and provides evidence that TRPA1 is a probable molecular target for DEP and DEP-associated electrophilic combustion by-products in the respiratory tract. Accordingly, activation of TRPA1-expressing chemosensory nerves by DEP may contribute to the acute adverse respiratory responses elicited by DEP and possibly other similar combustion-derived PM, including airway irritation, inflammation and cough.
Figures 2–4 summarize results from a quantitative assessment of human TRPA1, V1–V4, and M8 activation by 4 different model environmental PM. At concentrations of PM in the range used in many in vitro particle toxicology studies (i.e., 0.077–0.77 mg/mL or 18–180 µg/cm2), which are believed to be relevant to human exposures, DEP activated TRPA1. To this end, it is emphasized that the applied dose of PM does not necessarily equal the cellular exposure due to the length of time required for the various PM to settle onto the cells and activate TRPA1. Taking into account the time the particles are exposed to the cells (<5 min in all cases), the particle sizes, lipophilicity, and/or ability to become hydrated, and the predicted settling velocities, it is estimated that the actual cellular dose for DEP, but not CFA1, CFA2, or MUS, may be much lower, perhaps only 10% of the applied dose versus 50% or more for CFA1, CFA2, and MUS, making the doses used even more relevant to human exposures. This concept is reviewed by Teeguarden et al. (41), where it is shown that particles <10 µm may require hours to fully settle out of solution. Regardless, data in Figures 2–4 support the conclusion of Hazari et al. (3) that TRPA1 is a key mediator of acute DEP-induced cardiovascular effects.
TRPA1 is also activated by a number of specific environmental pollutants including cigarette smoke extracts containing acrolein, crotonaldehyde and many other toxic substances (3, 20, 21, 33, 34, 42). Acrolein, crotonaldehyde, and other aldehydes are also common components of diesel exhaust PM (43) and other combustion-derived PM that commonly occur in urban air pollution including wood smoke PM (44, 45). While the activation of TRPA1 by DEP in rats has been shown previously by Hazari et al., (3) this study only tested whole DEP in vivo and suggested that soluble components of DEP may activate TRPA1. Here we show activation of TRPA1 by DEP in a simplified genetically modified cell model and confirm that the DEP components 1,2-naphthoquinone, 1-nitronaphthalene, 1,3-dihydroxynaphthalene, and others (Tables 2 and 3) activate TRPA1 through the known electrophile/oxidant recognition sites. The lack of significant activation of several other TRP channels by DEP and these specific chemicals, and the results of Hazari et al. combined, suggest that TRPA1 is likely to be the primary neuronal sensor for DEP in the respiratory tract.
TRP channels are often highly expressed by sensory neurons of DRG and trigeminal (TG) and vagal fibers that innervate the respiratory tract. TRPA1 is highly expressed by neurons that express TRPV1, substance P, and neurokinin A (6), but has not yet been found in non-neuronal cells of the lung. Mouse DRG neurons were used as a surrogate for vagal and trigeminal airway sensory nerves to assess the selectivity of DEP for TRPA1 expressing pulmonary chemosensitive neurons in vivo. DRG neurons were robustly activated by DEP-EtOH and the responses exhibited near complete (~90%) overlap with AITC and were inhibited ~75% by HC-030031 pre/co-treatment, similar to AITC (Figure 6). These data indicate that TRPA1 is the principal determinant of neuronal responses to DEP-EtOH and supports the conjecture that TRPA1 is likely a significant mediator of DEP toxicity in the lung. Data from Teles et al. (7) also support this conclusion based on results that neonatal capsaicin treatment to obliterate TRPV1-positive sensory nerves ameliorated DEP and 1,2-naphthoquinone toxicity. Specifically, because TRPV1 and TRPA1 are highly co-expressed in airway sensory neurons (4–6), neonatal capsaicin exposure would eliminate TRPV1- and TRPA1-expressing nerves that largely control neurogenic inflammation, and thus would also inhibit toxicities attributable to TRPA1-regulated pathways.
DEP is a complex material that can potentially activate TRPA1 in many ways, and like other TRP channels TRPA1 appears to be polymodal. To identify how DEP activated TRPA1, extracts of DEP, CFA1, and the PM solids were evaluated for differences in potency using TRPA1 (Table 1) and the TRPA1–3CK mutant (Figure 6). Approximately 97% of the activity of DEP was extractable (Table 1). All of the DEP components assayed in Tables 2 and 3 are ethanol soluble, and as shown in Table 2, activate TRPA1 by modification of amino acid residues C621, C641, C665, and K710 located on the intracellular N-terminal tail (26, 27). Figure 5 and Table 2 show that the TRPA1–3CK mutant was not activated by the DEP extracts or reactive chemical components of DEP. However, the TRPA1–3CK mutant was equally activated by DEP, fully extracted DEP, CFA1, and fully extracted CFA1 (Figure 5 and Table 1), implying a second mechanism of activation, presumably via mechanical gating involving the ankyrin domains, as suggested by other studies of TRPA1 activation mechanisms (28–30). However, while mechanistically intriguing, the toxicological relevance of mechanical activation by DEP in normal airways with an intact epithelium is unclear.
Fractionation of the ethanol extract of DEP to identify specific agonists de novo also yielded several new TRPA1 agonists including the ditert-butylphenols, and several conjugated alkenes (Table 3). From Tables 2 and 3 it is clear that the number and diversity of direct TRPA1 agonists associated with DEP is immense. However, it is also likely that additional agonists can be generated in the lung from polycyclic aromatic hydrocarbons (PAHs) as shown for naphthalene and styrene by Lanosa et al. (46, 47) and confirmed for the P450-generated naphthalene metabolites 1,2-naphthoquinone and 1,3-naphthalenediol in Table 2. In fact 1,2-naphthoquinone was one of the most potent TRPA1 agonist studied. These collective results further emphasize TRPA1 as a sensor for DEP, as well as the importance of pulmonary cytochrome P450 enzymes in regulating TRPA1 activation by common PAH components of DEP that also occur on other environmental combustion-derived PM such as CFA1 and wood smoke PM.
While TRPA1 was the only channel for which statistically significant responses to DEP were observed (Figure 4), this does not exclude other TRP channels, including TRPM8, TRPV1, and TRPV4 which showed a small response to DEP, or even other gene products, as additional targets for DEP in the respiratory tract. For example, Li et al. (48) demonstrated the activation of TRPV4 in bronchial epithelial cells by DEP through proteinase-activated receptor 2 (PAR-2). Although significant activation of TRPV4 was not observed in this study, measurable, but highly variable responses occurred. Since TRPA1 is not expressed by airway epithelial cells, these data suggest the possibility for complimentary roles for TRPA1 and V4 in DEP pneumotoxicity where activation of TRPA1 in sensory nerves mediates select responses to DEP (e.g., cough, dyspnea, neurogenic inflammation), as shown for other TRPA1 agonists (33, 34, 42, 49), while activation of PAR2/TRPV4 promotes matrix degradation and remodeling through matrix metalloproteinases, and possibly cytokine release (IL-6, -8, others) and cytotoxicity as observed for TRPV1 and M8 agonists and select forms of PM in these same cells (23, 24, 50–52). Thus, collateral activation of TRPA1, V1, PAR2/V4, and M8 by DEP in multiple lung cell types provides a fascinating working model (Figure 7) that may explain how different TRP channels and different cell types cooperatively contribute to DEP pneumotoxicity.
Figure 7.

Hypothetical scheme outlining possible contributions of different TRP channels shown to be activated by DEP and different lung cell types to DEP-induced pneumotoxicity. This scheme integrates relevant data and concepts from Li et al. (48), Hazari et al. (3), Teles et al. (7), Agopyan et al. (12, 13, 52), Veronesi et al. (14–16), Oortgiesen et al. (17), Sabnis et al. (18), and this study.
In conclusion, TRPA1 is proposed to be an important molecular target for DEP and electrophilic chemical components of DEP implying that activation of TRPA1 in airway sensory nerves may be an early and critical determinant of the acute adverse respiratory and cardiovascular effects associated with urban air pollution, including DEP.
Acknowledgements
Funding Support: This work was supported by NIH grant ES017431, the 2011 Colgate-Palmolive Postdoctoral Fellowship (CEDR), and the University of Utah Department of Anesthesiology.
Abbreviations
- AITC
allyl isothiocyanate
- BEAS-2B
human bronchial epithelial cells
- CFA1
power plant coal fly ash
- CFA2
laboratory-generated-coal fly ash
- DEP
diesel exhaust particles
- MUS
Min-U-Sil 5 µM crystalline silica
- DRG
dorsal root ganglion
- HEK-293
human embryonic kidney-293 cells
- LHC-9
Lechner and LaVeck media
- PM
particulate matter
- SRM
Standard Reference Material
- TRPA1
transient receptor potential ankyrin-1
- TRPM8
transient receptor potential melastatin-8
- TRPV1
transient receptor potential vanilloid-1
- TRPV2
transient receptor potential vanilloid-2
- TRPV3
transient receptor potential vanilloid-3
- TRPV4
transient receptor potential vanilloid-4
- HC-030031
1,2,3,6-Tetrahydro-1,3-dimethyl-N-[4-(1-methylethyl)phenyl]-2,6-dioxo-7H-purine-7-acetamide, 2-(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N-(4-isopropylphenyl)acetamide
- MSTFA
N-methy-N-(trimethylsilyl) trifluoroacetamide
Footnotes
Supporting Information:
None
Contributor Information
Cassandra E. Deering-Rice, Email: cassandra.deering@utah.edu.
Erin G. Romero, Email: ering.romero@yahoo.com.
Darien Shapiro, Email: darien.shapiro@utah.edu.
Ronald W. Hughen, Email: shikoyama@gmail.com.
Alan R. Light, Email: Alan.Light@hsc.utah.edu.
Garold S. Yost, Email: gyost@pharm.utah.edu.
John M. Veranth, Email: john.veranth@m.cc.utah.edu.
Christopher A. Reilly, Email: Chris.Reilly@pharm.utah.edu.
References
- 1.Hammond DM, Dvonch JT, Keeler GJ, Parker EA, Kamal AS, Barres JA, Yip FY, Brakefield-Caldwell W. Sources of ambient fine particulate matter at two community sites in Detroit, Michigan. Atmospheric Environment. 2008;42:720–732. [Google Scholar]
- 2.Mauderly JL. Current Status of the Toxicology of Diesel Engine Exhaust - and the ACES Project. ZBL Arbeitsmed. 2010;50:412–417. [Google Scholar]
- 3.Hazari MS, Haykal-Coates N, Winsett DW, Krantz QT, King C, Costa DL, Farraj AK. TRPA1 and Sympathetic Activation Contribute to Increased Risk of Triggered Cardiac Arrhythmias in Hypertensive Rats Exposed to Diesel Exhaust. Environ Health Perspect. 2011 doi: 10.1289/ehp.1003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nassenstein C, Kwong K, Taylor-Clark T, Kollarik M, MacGlashan DM, Braun A, Undem BJ. Expression and function of the ion channel TRPA1 in vagal afferent nerves innervating mouse lungs. The Journal of Physiology. 2008;586:1595–1604. doi: 10.1113/jphysiol.2007.148379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi K, Fukuoka T, Obata K, Yamanaka H, Dai Y, Tokunaga A, Noguchi K. Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with aδ/c-fibers and colocalization with trk receptors. The Journal of Comparative Neurology. 2005;493:596–606. doi: 10.1002/cne.20794. [DOI] [PubMed] [Google Scholar]
- 6.Anand U, Otto WR, Facer P, Zebda N, Selmer I, Gunthorpe MJ, Chessell IP, Sinisi M, Birch R, Anand P. TRPA1 receptor localisation in the human peripheral nervous system and functional studies in cultured human and rat sensory neurons. Neuroscience Letters. 2008;438:221–227. doi: 10.1016/j.neulet.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 7.Teles A, Kumagai Y, Brain S, Teixeira S, Varriano A, Barreto M, de Lima W, Antunes E, Muscará M, Costa S. Involvement of sensory nerves and TRPV1 receptors in the rat airway inflammatory response to two environment pollutants: diesel exhaust particles (DEP) and 1,2-naphthoquinone (1,2-NQ) Archives of Toxicology. 2009;84:109–117. doi: 10.1007/s00204-009-0427-x. [DOI] [PubMed] [Google Scholar]
- 8.Venkatachalam K, Montell C. TRP Channels. Annual Review of Biochemistry. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voets T, Talavera K, Owsianik G, Nilius B. Sensing with TRP channels. Nat Chem Biol. 2005;1:85–92. doi: 10.1038/nchembio0705-85. [DOI] [PubMed] [Google Scholar]
- 10.Vriens J, Appendino G, Nilius B. Pharmacology of Vanilloid Transient Receptor Potential Cation Channels. Molecular Pharmacology. 2009;75:1262–1279. doi: 10.1124/mol.109.055624. [DOI] [PubMed] [Google Scholar]
- 11.Baraldi PG, Preti D, Materazzi S, Geppetti P. Transient Receptor Potential Ankyrin 1 (TRPA1) Channel as Emerging Target for Novel Analgesics and Anti-Inflammatory Agents. Journal of Medicinal Chemistry. 2010;53:5085–5107. doi: 10.1021/jm100062h. [DOI] [PubMed] [Google Scholar]
- 12.Agopyan N, Head J, Yu S, Simon SA. TRPV1 receptors mediate particulate matter-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L563–L572. doi: 10.1152/ajplung.00299.2003. [DOI] [PubMed] [Google Scholar]
- 13.Agopyan N, Li L, Yu S, Simon SA. Negatively charged 2- and 10-µm particles activate vanilloid receptors, increase cAMP, and induce cytokine release. Toxicology and Applied Pharmacology. 2003;186:63–76. doi: 10.1016/s0041-008x(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 14.Veronesi B, Wei G, Zeng J-Q, Oortgiesen M. Electrostatic charge activates inflammatory vanilloid (VR1) receptors. NeuroToxicology. 2003;24:463–473. doi: 10.1016/S0161-813X(03)00022-6. [DOI] [PubMed] [Google Scholar]
- 15.Veronesi B, Oortgiesen M. Neurogenic Inflammation and Particulate Matter (PM) Air Pollutants. NeuroToxicology. 2001;22:795–810. doi: 10.1016/s0161-813x(01)00062-6. [DOI] [PubMed] [Google Scholar]
- 16.Veronesi B, Oortgiesen M, Roy J, Carter JD, Simon SA, Gavett SH. Vanilloid (Capsaicin) Receptors Influence Inflammatory Sensitivity in Response to Particulate Matter. Toxicology and Applied Pharmacology. 2000;169:66–76. doi: 10.1006/taap.2000.9040. [DOI] [PubMed] [Google Scholar]
- 17.Oortgiesen M, Veronesi B, Eichenbaum G, Kiser PF, Simon SA. Residual oil fly ash and charged polymers activate epithelial cells and nociceptive sensory neurons. Am J Physiol Lung Cell Mol Physiol. 2000;278:L683–L695. doi: 10.1152/ajplung.2000.278.4.L683. [DOI] [PubMed] [Google Scholar]
- 18.Sabnis AS, Shadid M, Yost GS, Reilly CA. Human Lung Epithelial Cells Express a Functional Cold-Sensing TRPM8 Variant. Am. J. Respir. Cell Mol. Biol. 2008;39:466–474. doi: 10.1165/rcmb.2007-0440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mio T, Romberger DJ, Thompson AB, Robbins RA, Heires A, Rennard SI. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med. 1997;155:1770–1776. doi: 10.1164/ajrccm.155.5.9154890. [DOI] [PubMed] [Google Scholar]
- 20.Simon SA, Liedtke W. How irritating: the role of TRPA1 in sensing cigarette smoke and aerogenic oxidants in the airways. The Journal of Clinical Investigation. 2008;118:2383–2386. doi: 10.1172/JCI36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt S-E. TRPA1 is a major oxidant sensor in murine airway sensory neurons. The Journal of Clinical Investigation. 2008;118:1899–1910. doi: 10.1172/JCI34192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith KR, Veranth JM, Kodavanti UP, Aust AE, Pinkerton KE. Acute pulmonary and systemic effects of inhaled coal fly ash in rats: comparison to ambient environmental particles. Toxicol Sci. 2006;93:390–399. doi: 10.1093/toxsci/kfl062. [DOI] [PubMed] [Google Scholar]
- 23.Reilly CA, Taylor JL, Lanza DL, Carr BA, Crouch DJ, Yost GS. Capsaicinoids Cause Inflammation and Epithelial Cell Death through Activation of Vanilloid Receptors. Toxicol. Sci. 2003;73:170–181. doi: 10.1093/toxsci/kfg044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansen ME, Reilly CA, Yost GS. TRPV1 Antagonists Elevate Cell Surface Populations of Receptor Protein and Exacerbate TRPV1-Mediated Toxicities in Human Lung Epithelial Cells. Toxicol. Sci. 2006;89:278–286. doi: 10.1093/toxsci/kfi292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Light AR, Hughen RW, Zhang J, Rainier J, Liu Z, Lee J. Dorsal Root Ganglion Neurons Innervating Skeletal Muscle Respond to Physiological Combinations of Protons, ATP, and Lactate Mediated by ASIC, P2X, and TRPV1. Journal of Neurophysiology. 2008;100:1184–1201. doi: 10.1152/jn.01344.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinman A, Chuang H-h, Bautista DM, Julius D. TRP channel activation by reversible covalent modification. Proceedings of the National Academy of Sciences. 2006;103:19564–19568. doi: 10.1073/pnas.0609598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- 28.Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin S-Y, Vollrath MA, Amalfitano A, Cheung ELM, Derfler BH, Duggan A, Geleoc GSG, Gray PA, Hoffman MP, Rehm HL, Tamasauskas D, Zhang D-S. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432:723–730. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 29.Lee G, Abdi K, Jiang Y, Michaely P, Bennett V, Marszalek PE. Nanospring behaviour of ankyrin repeats. Nature. 2006;440:246–249. doi: 10.1038/nature04437. [DOI] [PubMed] [Google Scholar]
- 30.Howard J, Bechstedt S. Hypothesis: A helix of ankyrin repeats of the NOMPC-TRP ion channel is the gating spring of mechanoreceptors. Current Biology. 2004;14:R224–R226. doi: 10.1016/j.cub.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 31.OSHA. Partial List of Chemicals Associated with Diesel Exhaust. Washington, DC: OSHA; 2010. [Google Scholar]
- 32.Schauer JJ, Kleeman MJ, Cass GR, Simoneit BRT. Measurement of Emissions from Air Pollution Sources. 2. C1 through C30 Organic Compounds from Medium Duty Diesel Trucks. Environmental Science & Technology. 1999;33:1578–1587. [Google Scholar]
- 33.Andrè E, Gatti R, Trevisani M, Preti D, Baraldi PG, Patacchini R, Geppetti P. Transient receptor potential ankyrin receptor 1 is a novel target for pro-tussive agents. British Journal of Pharmacology. 2009;158:1621–1628. doi: 10.1111/j.1476-5381.2009.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birrell MA, Belvisi MG, Grace M, Sadofsky L, Faruqi S, Hele DJ, Maher SA, Freund-Michel V, Morice AH. TRPA1 Agonists Evoke Coughing in Guinea Pig and Human Volunteers. Am. J. Respir. Crit. Care Med. 2009;180:1042–1047. doi: 10.1164/rccm.200905-0665OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilburn KH, McKenzie WN. Leukocyte recruitment to airways by aldehyde-carbon combinations that mimic cigarette smoke. Lab Invest. 1978;38:134–142. [PubMed] [Google Scholar]
- 36.Kunkler PE, Ballard CJ, Oxford GS, Hurley JH. TRPA1 receptors mediate environmental irritant-induced meningeal vasodilatation. PAIN. 2011;152:38–44. doi: 10.1016/j.pain.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leikauf GD, Leming LM, O'Donnell JR, Doupnik CA. Bronchial responsiveness and inflammation in guinea pigs exposed to acrolein. J Appl Physiol. 1989;66:171–178. doi: 10.1152/jappl.1989.66.1.171. [DOI] [PubMed] [Google Scholar]
- 38.Nilius B, Prenen J, Owsianik G. Irritating Channels: the case of TRPA1. The Journal of Physiology. 2010 doi: 10.1113/jphysiol.2010.200717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizvi SQA. Additives and Additive Chemistry. In: Totten GE, Westbrook SR, Shah RJ, editors. In Fuels and Lubricants Handbook: Technology, Properties, Performance, and Testing. 2003. pp. 199–248. [Google Scholar]
- 40.Boland S, Baeza-Squiban A, Fournier T, Houcine O, Gendron M-C, Chévrier M, Jouvenot G, Coste A, Aubier M, Marano F. Diesel exhaust particles are taken up by human airway epithelial cells in vitro and alter cytokine production. American Journal of Physiology - Lung Cellular and Molecular Physiology. 1999;276:L604–L613. doi: 10.1152/ajplung.1999.276.4.L604. [DOI] [PubMed] [Google Scholar]
- 41.Teeguarden JG, Hinderliter PM, Orr G, Thrall BD, Pounds JG. Particokinetics in vitro: dosimetry considerations for in vitro nanoparticle toxicity assessments. Toxicol Sci. 2007;95:300–312. doi: 10.1093/toxsci/kfl165. [DOI] [PubMed] [Google Scholar]
- 42.Andrè E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, Baraldi PG, Poole DP, Bunnett NW, Geppetti P, Patacchini R. Cigarette smoke-induced neurogenic inflammation is mediated by α,β-unsaturated aldehydes and the TRPA1 receptor in rodents. The Journal of Clinical Investigation. 2008;118:2574–2582. doi: 10.1172/JCI34886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song C, Zhao Z, Lv G, Song J, Liu L, Zhao R. Carbonyl compound emissions from a heavy-duty diesel engine fueled with diesel fuel and ethanol-diesel blend. Chemosphere. 2010;79:1033–1039. doi: 10.1016/j.chemosphere.2010.03.061. [DOI] [PubMed] [Google Scholar]
- 44.Schauer JJ, Kleeman MJ, Cass GR, Simoneit BRT. Measurement of Emissions from Air Pollution Sources. 3. C1-C29 Organic Compounds from Fireplace Combustion of Wood. Environmental Science & Technology. 2001;35:1716–1728. doi: 10.1021/es001331e. [DOI] [PubMed] [Google Scholar]
- 45.Jordan TB, Seen AJ. Effect of airflow setting on the organic composition of woodheater emissions. Environ. Sci. Technol. 2005;39:3601–3610. doi: 10.1021/es0487628. [DOI] [PubMed] [Google Scholar]
- 46.Lanosa MJ, Willis DN, Jordt S, Morris JB. Role of Metabolic Activation and the TRPA1 Receptor in the Sensory Irritation Response to Styrene and Naphthalene. Toxicological Sciences. 2010;115:589–595. doi: 10.1093/toxsci/kfq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Totlandsdal A, Cassee F, Schwarze P, Refsnes M, Lag M. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells. Particle and Fibre Toxicology. 2010;7:41. doi: 10.1186/1743-8977-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Kanju P, Patterson M, Chew W-L, Cho S-H, Gilmour I, Oliver T, Yasuda R, Ghio A, Simon SA, Liedtke W. TRPV4-Mediated Calcium-influx into Human Bronchial Epithelia upon Exposure to Diesel Exhaust Particles. Environ Health Perspect. 2011 doi: 10.1289/ehp.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bessac BF, Jordt S-E. Breathtaking TRP Channels: TRPA1 and TRPV1 in Airway Chemosensation and Reflex Control. Physiology. 2008;23:360–370. doi: 10.1152/physiol.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas KC, Sabnis AS, Johansen ME, Lanza DL, Moos PJ, Yost GS, Reilly CA. Transient Receptor Potential Vanilloid 1 Agonists Cause Endoplasmic Reticulum Stress and Cell Death in Human Lung Cells. Journal of Pharmacology and Experimental Therapeutics. 2007;321:830–838. doi: 10.1124/jpet.107.119412. [DOI] [PubMed] [Google Scholar]
- 51.Reilly CA, Johansen ME, Lanza DL, Lee J, Lim J-O, Yost GS. Calcium-dependent and independent mechanisms of capsaicin receptor (TRPV1)-mediated cytokine production and cell death in human bronchial epithelial cells. Journal of Biochemical and Molecular Toxicology. 2005;19:266–275. doi: 10.1002/jbt.20084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agopyan N, Bhatti T, Yu S, Simon SA. Vanilloid receptor activation by 2- and 10-[mu]m particles induces responses leading to apoptosis in human airway epithelial cells. Toxicology and Applied Pharmacology. 2003;192:21–35. doi: 10.1016/s0041-008x(03)00259-x. [DOI] [PubMed] [Google Scholar]
- 53.Kaneko T, Yoshida H, Suzuki S. The determination by gas chromatography with atomic emission detection of total sulfur in fuels used as forensic evidence. Forensic Science International. 2008;177:112–119. doi: 10.1016/j.forsciint.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 54.Ban-Weiss GA, McLaughlin JP, Harley RA, Kean AJ, Grosjean E, Grosjean D. Carbonyl and Nitrogen Dioxide Emissions From Gasoline- and Diesel-Powered Motor Vehicles. Environmental Science & Technology. 2008;42:3944–3950. doi: 10.1021/es8002487. [DOI] [PubMed] [Google Scholar]
- 55.McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proceedings of the National Academy of Sciences. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sobus JR, Waidyanatha S, McClean MD, Herrick RF, Smith TJ, Garshick E, Laden F, Hart JE, Zheng Y, Rappaport SM. Urinary naphthalene and phenanthrene as biomarkers of occupational exposure to polycyclic aromatic hydrocarbons. Occupational and Environmental Medicine. 2009;66:99–104. doi: 10.1136/oem.2008.041418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsien A, Diaz-Sanchez D, Ma J, Saxon A. The Organic Component of Diesel Exhaust Particles and Phenanthrene, a Major Polyaromatic Hydrocarbon Constituent, Enhances IgE Production by IgE-Secreting EBV-Transformed Human B Cellsin Vitro. Toxicology and Applied Pharmacology. 1997;142:256–263. doi: 10.1006/taap.1996.8063. [DOI] [PubMed] [Google Scholar]
- 58.Rhead MM, Pemberton RD. Sources of Naphthalene in Diesel Exhaust Emissions. Energy & Fuels. 1996;10:837–843. [Google Scholar]
- 59.Jakober CA, Riddle SG, Robert MA, Destaillats H, Charles MJ, Green PG, Kleeman MJ. Quinone Emissions from Gasoline and Diesel Motor Vehicles. Environmental Science & Technology. 2007;41:4548–4554. doi: 10.1021/es062967u. [DOI] [PubMed] [Google Scholar]
- 60.Sauer J-M, Eversole RR, Lehmann CL, Johnson DE, Beuving LJ. An ultrastructural evaluation of acute 1-nitronaphthalene induced hepatic and pulmonary toxicity in the rat. Toxicology Letters. 1997;90:19–27. doi: 10.1016/s0378-4274(96)03817-9. [DOI] [PubMed] [Google Scholar]
- 61.Wing CY, Fine DH, Chiu KS, Biemann K. Determination of nitrated polycyclic aromatic hydrocarbons in diesel particulates by gas chromatography with chemiluminescent detection. Analytical Chemistry. 1984;56:1158–1162. doi: 10.1021/ac00271a024. [DOI] [PubMed] [Google Scholar]
- 62.Jakober CA, Riddle SG, Robert MA, Destaillats H, Charles MJ, Green PG, Kleeman MJ. Quinone emissions from gasoline and diesel motor vehicles. Environ Sci Technol. 2007;41:4548–4554. doi: 10.1021/es062967u. [DOI] [PubMed] [Google Scholar]
- 63.Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Högestätt ED, Julius D, Jordt S-E, Zygmunt PM. Pungent products from garlic activate the sensory ion channel TRPA1. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12248–12252. doi: 10.1073/pnas.0505356102. [DOI] [PMC free article] [PubMed] [Google Scholar]