

Abstract
The PhoP protein from Mycobacterium tuberculosis is a response regulator of the OmpR/PhoB subfamily, whose structure consists of an N-terminal receiver domain and a C-terminal DNA-binding domain. How the DNA-binding activities are regulated by phosphorylation of the receiver domain remains unclear due to a lack of structural information on the full-length proteins. Here we report the crystal structure of the full-length PhoP of M. tuberculosis. Unlike other known structures of full-length proteins of the same subfamily, PhoP forms a dimer through its receiver domain with the dimer interface involving α4-β5-α5, a common interface for activated receiver domain dimers. However, the switch residues, Thr99 and Tyr118, are in a conformation resembling those of non-activated receiver domains. The Tyr118 side chain is involved in the dimer interface interactions. The receiver domain is tethered to the DNA-binding domain through a flexible linker and does not impose structural constraints on the DNA-binding domain. This structure suggests that phosphorylation likely facilitates/stabilizes receiver domain dimerization, bringing the DNA-binding domains to close proximity, thereby increasing their binding affinity for direct repeat DNA sequences.
The emergence of multiple-drug resistant (MDR) and extremely-drug resistant (XDR) strains of Mycobacterium tuberculosis (MTB) marks a new threat of this old ailment to human health (1). One way to combat the drug resistance problem is to develop new drugs that attack novel targets of the pathogen. Two-component signal transduction systems (TCSs) are attractive novel drug targets because they control major signaling processes involved in bacterial adaptation to changing environments and, importantly, are absent from mammals (2). Most TCSs are composed of two proteins, a sensor histidine kinase (HK) and a response regulator (RR). The HK senses environmental signals and autophosphorylates on a conserved histidine residue. The phosphate group is then transferred to the cognate RR on a conserved aspartate residue to activate this RR for downstream signal propagation (3).
In the MTB genome, there are 30 genes coding for TCS proteins (4, 5). Many of the TCSs are important for MTB virulence. MTB strains with mutations in RR genes, such as prrA (6), devR (7), mprA (8), and phoP (9, 10), are defective for growth in macrophages and mice. PhoP is a major transcription regulator that up regulates 44 genes and down regulates 70 genes (9). An MTB phoP mutant has defects in its cell envelope. It lacks complex lipids such as sulfatides and treheloses, and also shows greatly reduced expression of structural genes for their biosynthesis (9). These results indicate that PhoP is involved in the regulation of complex lipid biosynthesis. However, mutant MTB strains with knockouts of individual genes involved in complex lipid biosynthesis do not have significant defects in virulence (11, 12). Because PhoP regulates more than 110 genes, it is likely that some other genes or a combination of several genes are responsible for the effect of the phoP mutation on virulence (13).
Several recent studies of the avirulent MTB strain, H37Ra, point to the importance of PhoP in virulence of the pathogen (11, 14, 15). A point mutation in phoP, which changes a serine residue, Ser219 on the DNA sequence recognition helix (16), into a leucine, plays an important role in the loss of virulence in H37Ra. The mutation causes the loss of secretion of the major T-cell antigens ESAT-6 and CFP-10 (14). The wild-type H37Rv phoP partially restores the strain’s ability to grow in macrophages and mice. These results suggest that the PhoP-PhoR TCS is a potential target for developing new antibiotics to combat drug-resistant strains of MTB.
Most RRs contain two distinct domains, an N-terminal receiver domain and a C-terminal effector domain (3). The receiver domain contains an absolutely conserved aspartate residue, which receives a phosphate group from the cognate HK. Phosphorylation of the receiver domain causes structural changes to the protein, and thus, in some way, activates the effector domain. In the case of those RRs that function as transcriptional regulators, the effector domain is a DNA-binding domain. The RRs are divided into subfamilies based on structure and/or function of their effector domains. PhoP belongs to the OmpR/PhoB subfamily of RRs, named from their representative members OmpR and PhoB (17, 18). This subfamily is the largest among RRs. The C-terminal domain of the OmpR/PhoB subfamily RRs binds DNA and has a typical structural fold called the winged helix-turn-helix motif. Despite decades of intensive research and accumulation of a large amount of structural and functional data, it is still unclear how phosphorylation regulates the DNA-binding activities of the OmpR/PhoB subfamily RRs.
The N-terminal domain of RRs has a well-conserved structure, composed of a central 5-stranded β-sheet, sandwiched by helices on both sides. Several structures of the isolated receiver domains in complex with beryllium fluoride to mimic the phosphorylation-activated structure have been reported (19–22). These activated receiver domains form symmetric dimers with a common interface through the α4-β5-α5 face. Some receiver domains also form dimers through the same interface in the absence of beryllium fluoride (19, 21), suggesting that the active-form dimer can exist in the absence of phosphorylation. Phosphorylation likely stabilizes the dimer and thus shifts the equilibrium toward the dimeric form. In contrast to the symmetric dimer structure of the activated receiver domains, most of the known DNA binding sequences for the OmpR/PhoB subfamily RRs have been found to be direct tandem repeats, suggesting that these proteins bind DNA as tandem dimers. The structure of the effector domain of PhoB from E. coli in complex with its pho box DNA has been reported (23). The isolated PhoB effector domain binds to DNA as a tandem dimer. Full-length response regulators of the OmpR/PhoB family have been difficult to crystallize, and to date there are only five structures reported, RegX3 (24), MtrA (25) and PrrA (26) from MTB, and DrrB (27) and DrrD (28) from Thermotoga Maritima. All structures show monomers in the crystal and have some intramolecular domain interactions, except RegX3, which forms a domain-swapped dimer through the receiver domain. DrrD shows the least interactions, while MtrA has the strongest domain interface that buries the sequence recognition helix of the DNA-binding domain and the α4-β5-α5 face of the receiver domain, suggesting obstruction of the DNA binding elements by the non-activated receiver domain as a possible regulatory mechanism for DNA-binding activities by phosphorylation. To a lesser extent, the PrrA structure also shows some obstruction of the DNA-binding elements by the α4-β5-α5 face of the receiver domain. However, in solution, unphosphorylated PrrA can exist in a conformation with the DNA-binding elements exposed for binding DNA (26). Both DrrB and DrrD structures have the DNA-binding elements fully exposed, suggesting that they are able to bind DNA without activation by phosphorylation.
The MTB PhoP protein is able to bind its own promoter in the unphosphorylated state, and the DNA binding site is proposed to contain direct repeats (29, 30). Here, we report the crystal structure of full-length PhoP from MTB. The structure shows that PhoP forms a dimer in the crystal through the α4-β5-α5 face of the receiver domain, a dimer interface similar to that of the activated receiver domain structures. The linker that tethers the receiver and effector domains is disordered. The two effector domains that are tethered to the receiver domain dimer do not interact with each other or with the receiver domains. Our structural results are consistent with observations that the protein is able to bind DNA in the absence of phosphorylation activation. Phosphorylation is likely to enhance the receiver domain dimer association, but unlikely to change the freedom of the effector domains. Therefore, the mechanism of activation by phosphorylation of the MTB PhoP is likely through higher affinity of the receiver domain dimer to bring the effector domains in close proximity, thus increasing their affinity for specific DNA sequences with two direct repeats.
EXPERIMENTAL PROCEDURES
Cloning, expression, and purification
The MTB phoP gene (Rv0757) was amplified from the genomic DNA of MTB strain H37Rv by polymerase chain reactions (PCR) with the KOD Hot Start DNA polymerase (Novagen). The following primers were used for PCR: 5′-primer CCAGCATATGCGGAAAGGGGTTGATCTCG and 3′-primer CCAGAAGCTTTCATCGAGGCTCCCGCAGTACG, which generate NdeI and HindIII restriction sites (underlined). Amplified DNA was cleaved by restriction enzymes NdeI and HindIII, purified by agarose gel extraction, and ligated into a modified pET28a plasmid (Novagen) linearized with the same enzymes. The pET28a vector was modified by inserting a DNA sequence encoding a tobacco etch virus (TEV) main protease cleavage site before the NdeI restriction site as described previously (16). The resulting plasmid pET28-phoP encodes a protein with an N-terminus 6XHis tag, which can be cleaved by the TEV protease. The plasmid was transformed into BL21(DE3) competent cells (Novagen) for protein overexpression.
Cells containing the pET28-phoP plasmid were grown and induced for protein over-expression, and cells were harvested and lysed, as described previously (16). The cell lysate was centrifuged at 37,000×g for 30 min, and the supernatant was applied to a column containing a 1–3 ml bed volume of Ni-NTA agarose (Qiagen). The column was washed with 10 column volumes of wash buffer (50 mM sodium phosphate, pH 7.4, 500 mM NaCl, and 20 mM imidazole) and PhoP was eluted in 50 mM sodium phosphate, pH 7.4, 500 mM NaCl, and 250 mM imidazole. Fractions containing the PhoP protein were pooled, mixed with ~150 μg of His-TEV(S219V) protease (31), and dialyzed against 1 L of buffer containing 50 mM sodium phosphate, pH 7.4, 200 mM NaCl, and 5 mM MgCl2 overnight at 4 ºC. The dialysis mixture was loaded onto a Ni-NTA agarose column to remove the His-TEV protease, the cleaved His-tag, and the undigested protein. Fractions containing PhoP lacking the His tag were pooled, concentrated and the protein was further purified on a Superdex 200 column (GE Healthcare) equilibrated with 20 mM Hepes, pH 7.5, 200 mM NaCl and 10 mM MgCl2. The purified protein was analyzed by SDS-PAGE and MALDI mass spectrometry for purity. Protein concentrations were determined by Bradford assay (32) using BSA as a standard.
For production of selenomethionine-labeled protein (SeMet-PhoP) similar procedures were followed, except that M9 minimal media (33) containing 50 μg/ml kanamycin were used in place of LB. Once an OD600 of 0.6 was reached, amino acids lysine (100mg), phenylalanine (100mg), threonine (100mg), isoleucine (50mg), leucine (50mg) and valine (50mg) along with selenomethionine (60mg) per liter were added directly to the culture as solids. The temperature was lowered to 18 ºC and the cells were induced with 50 μM IPTG for 16–18 hours. Subsequent expression and purification of SeMet-labeled PhoP was identical to that described above for the native protein. The yield of SeMet-labeled protein was ~5–7 mg per gram of cell pellet.
Crystallization and data collection
Purified protein was concentrated to ~3 mg/ml for crystallization. The best crystals were obtained from drops set up with well solutions containing 0.1 M Hepes, pH 7.5, 1.8 M ammonium sulfate, 1.5% PEG 400 and 12% 1,2-propanediol. Before data collection, crystals were soaked for 2–5 min in a cryogenic solution, similar to well solutions but with 1.2 M ammonium sulfate and 20% 1,2 propanediol. Crystals were then flash-cooled in a cryostream of N2 gas at 100 K on the goniometer. Diffraction data were collected at beamline X6A of the National Synchrotron Light Source, Brookhaven National Laboratory, with a Quantum-270 CCD detector. Data reduction and scaling were done with HKL-2000 (34). Data collection and processing statistics are listed in Table 1. The Rpim values were calculated with SCALA (35) using unmerged scaled data from HKL2000 as input. The crystals belong to the space group P21212, with 2 molecules per asymmetric unit and a VM (Matthews volume) of 2.88 Å3/Da. The data were converted to CCP4 format and structure factor amplitudes were calculated by TRUNCATE and other programs in the CCP4 Suite (36).
Table 1.
Data collection statistics for MTB PhoPa.
| Se-Edge | Se-Peak | Se-Remote | |
|---|---|---|---|
| Wavelength (Å) | 0.9804 | 0.9800 | 0.9537 |
| Space group | P21212 | P21212 | P21212 |
| Cell dimensions | |||
| a, b, c (Å) | 79.27, 138.33, 58.93 | 79.18, 138.09, 58.88 | 79.23, 138.24, 58.91 |
| Resolution (Å)b | 30.0-2.5 (2.54-2.50) | 30.0-2.5 (2.54-2.50) | 30.0-2.5 (2.54-2.50) |
| Rmergeb | 0.139 (0.858) | 0.134 (0.669) | 0.138 (0.765) |
| Rpimb | 0.058 (0.261) | 0.057 (0.231) | 0.057 (0.248) |
| I/σ(I)b | 16.44 (2.39) | 17.47 (2.51) | 16.64 (2.77) |
| Completeness (%)b | 99.2 (98.3) | 99.0 (96.5) | 99.3 (98.3) |
| Redundancyb | 8.6 (8.7) | 8.1 (7.4) | 8.5 (8.6) |
One crystal was used for collection of all data sets at three wavelengths; data were collected at 100 K.
Values in parentheses are for highest-resolution shell.
Structural determination, model Building, and refinement
The structure of PhoP was determined by multi-wavelength anomalous diffraction using PHENIX AutoSol and AutoBuild (37). The structural model was refined against the diffraction data using REFMAC (38). After each cycle of refinement, the model was manually adjusted with weighted 2Fo - Fc and Fo - Fc electron density maps with COOT (39). The model was refined for a few cycles with TLS refinement (40) before convergence. Final refinement statistics for the refined coordinates are reported in Table 2. Structure validation by MolProbity (41) suggests that over 96% of protein residues fall in the most favored region of the Ramachandran plot.
Table 2.
Refinement statistics for the MTB PhoP structurea.
| Resolution (Å)b | 30.0-2.5 (2.56-2.50) |
| No. reflections | 21622 |
| Rwork/Rfreeb | 21.2 (25.2)/24.5 (28.1) |
| No. atoms | |
| Protein | 3514 |
| Water | 78 |
| B-factors | |
| Protein | 35.0 |
| Water | 33.5 |
| No. other molecules/ions | 5 SO4 (85.5) |
| (Ave. B-factors)c | 1 1,2-propanediol (79.2) |
| rmsd bond lengths (Å) | 0.017 |
| rmsd bond angles (°) | 1.61 |
The data set collected at the Se-remote wavelength was used for structural refinement.
Values in parentheses are for highest-resolution shell.
Values in parentheses are the average B-factors.
Sedimentation velocity analytical ultracentrifugation
Sedimentation velocity scans were obtained using a 4-place An-60 Ti analytical rotor in the Beckman Coulter ProteomeLab XL-A ultracentrifuge. The PhoP protein was in a buffer containing 20 mM Hepes pH 7.5, 200 mM NaCl, 10 mM MgCl2, and 10% glycerol, at two concentrations, 0.9 and 2.3mg/ml. Experiments were carried out using 400 μl of sample in a double-sector 12 mm-thick charcoal filled Epon centerpiece. For each sample, 400 absorbance scans at wavelengths 250 nm and 280 nm were acquired overnight at 20 °C with rotor speeds of 45,000 rpm. SEDFIT (version 12.1b) was used to analyze the sedimentation velocity scans.
Limited trypsin digestion of PhoP
Limited proteolysis of PhoP was carried out at room temperature for 1 hour in 50μl total reaction volume containing 1.2 μg of PhoP and 20 ng of trypsin in 20 mM Hepes (pH 7.5), 50 mM NaCl, 5% glycerol, and 1mM MgCl2. Proteolysis was stopped by addition of 2X Lamelli dye. The trypsin-digested fragments were resolved on a Novex 18% tris-glycine gel and transferred to a polyvinylidene difluoride membrane (Pierce) and visualized by staining with the SimplyBlue safe stain (Invitrogen). Each fragment was then excised and sent to the Protein Core Facility at Columbia University, NY for N-terminal sequencing.
RESULTS
Protein purification and structure determination
The full-length PhoP protein of MTB was expressed in the E. coli strain BL21(DE3) and purified by two passes through a Ni2+ column followed by gel filtration, as described in the Methods section. The His tag was removed by TEV protease digestion before the second pass of the Ni2+ column. The resulting protein contains three extra residues, GluThrHis, at the N-terminus, which originated from the plasmid vector. SeMet-labeled protein was produced and purified in a similar way (see Methods for details). Mass spectroscopy analysis confirmed 100% incorporation of selenium atoms.
The purified PhoP protein has a solubility of ~3 mg/ml in most of the common buffers with pH from 6 to 8. Gel filtration studies gave a single peak with an apparent molecular weight between monomer and dimer, suggesting that the protein exists in solution in rapid equilibrium between monomer and dimer. Analytical ultracentrifugation confirms the existence of dimers in solution. The dimeric form increases from 2% to 16% of the total protein as the protein concentration increases from 0.9 to 2.3 mg/ml.
The best protein crystals were obtained from drops containing 0.1 M Hepes, pH 7.5, 1.8 M ammonium sulfate, 1.5% PEG 400, and 12% 1,2-propanediol. The crystals are thin plates ~1 mm long, but less than 0.02 mm thick. A set of MAD data was collected from one crystal at three wavelengths. The data was processed in space group P21212 at 2.5 Å with 2 molecules in the asymmetric unit (Table 1). The structure was determined from this set of MAD data with PHENIX (37) AutoSol wizard. The resulting initial map was traceable, and the PHENIX AutoBuild wizard built nearly 50% of the model with the rest rebuilt manually. Refinement statistics are reported in Table 2.
The overall structure of PhoP
The refined structural model contains two polypeptide chains. There are 224 residues for chain A and 221 residues for chain B. Each molecule contains two distinct structural domains. At the linker between the two domains, 6 residues in chain A and 8 residues in chain B were not modeled due to lack of electron density, and these residues are presumably disordered. The last residue at the C-terminus for both chains, 19 residues at amino terminus for chain A, and 20 for chain B, including the 3 residues originating from the plasmid vector, also lacked electron density, and therefore were not modeled. Sequence alignments with other homologous RRs suggest that MTB PhoP has an extension of 18 residues at the N-terminus that are absent from other homologs (Figure 1). Most of these residues are disordered in the crystal structure and would extend away from the surface of the globular domain (Figure 2), suggesting that these extra residues are not involved in the N-terminal domain structure but could play a role in interacting with other proteins.
Figure 1.

Sequence alignment of response regulators of the OmpR/PhoB subfamily. Sequences were aligned with the program CLUSTALW (49), and manually adjusted based on structural alignments. The secondary structural elements shown are of the PhoP structure. Identical residues are highlighted in black boxes, while residues with high similarity as defined by CLUSTALW are highlighted in gray boxes. The essential aspartate and other acidic residues at the phosphorylation site, conserved lysine (Lys121 in PhoP), and the switch residues (Thr99 and Tyr118) are labeled with asterisks. A vertical arrow marks the boundary between the N- and C-terminal domains. The linker between domains has variable length as indicated by the gaps in the alignment. This figure was generated with the program ALSCRIPT (50).
Figure 2.

Ribbon diagram of the full-length PhoP dimer. PhoP dimerizes through α4-β5-α5 of the receiver domain with a two-fold symmetry. The N- and C-termini for both subunits are marked with N and C, respectively. Secondary structural elements for one of the subunits are labeled. The two ends of the disordered linker are connected with a dotted line. This figure and all other color figures were generated with PYMOL (www.pymol.org).
The N-terminal domain has a (βα)5 fold, analogous to other receiver domains from the OmpR/PhoB RR subfamily (42). The central core of the structure is made up of a five-stranded parallel β-sheet (β1–β5) surrounded by helices α1 and α5 on one face and helices α2, α3 and α4 on the other side (Figure 2). The β strands are mainly composed of residues valine, leucine, and isoleucine, forming a hydrophobic core. The α helices are amphipathic, with the side facing the β sheet composed of residues with hydrophobic side chains while those exposed to solvent are mostly hydrophilic.
The C-terminal domain has a winged helix-turn-helix fold typical for the OmpR/PhoB family of RRs (Figure 2). The domain starts with a 4-stranded anti-parallel β-sheet, followed by 3 α helices and then a C-terminal β-hairpin. A short strand between helices α6 and α7 and the β hairpin together form a 3-stranded anti-parallel β-sheet. The two β sheets sandwich the helices. The C-terminal domain appears to be more flexible than the N-terminal domain. The overall average B factor for the main chain atoms of the N-terminal domain is 31.1 Å2, while that for the C-terminal domain is 37.6 Å2. The average B factor for all atoms is essentially identical for both protein chains in the asymmetric unit at ~35 Å2.
Acidic pocket at the phosphorylation site
The regulatory domain exhibits an acidic pocket at the C-terminal ends of strands β1 and β3, consisting of side chains from residues Asp27, Asp28, Glu29, and Asp71 (Figure 3) (residues are numbered as encoded in the genome with the initial Met as residue number 1). These residues are well conserved in the OmpR/PhoB subfamily of RRs. The side chain of Asp71 is the site for phosphorylation (30) and is absolutely conserved, while other acidic side chains are either aspartate or glutamate (Figure 1). Side chains of Asp27, Asp28, and Asp71, as well as the carbonyl oxygen of Met73 are involved in coordinating a divalent cation in some structures of RRs crystallized in the presence of divalent cations (43, 44). The PhoP crystal was grown in the absence of divalent cations, and no divalent cation was found in the acidic pocket. However, the side chains are oriented ready to coordinate a divalent cation, except that of Asp71, which would need a slight rotation to orient one of the oxygen atoms toward the bound metal ion. We obtained another PhoP crystal grown in the presence of Ca(OAc)2, and the calcium ion is coordinated by side chains of Asp27, Asp28, Asp71, and the carbonyl of Met73 (data not shown, structure not reported due to disorder of one entire C-terminal domain out of the 2 molecules in the asymmetric unit).
Figure 3.

The phosphorylation site and functional residues. The phosphorylation site is located in a shallow pocket at the C-terminal ends of strands β1 and β3. The side chains of acidic residues, a well-conserved Lys121, and switch residues are shown as sticks. Also shown is the carbonyl group of Met73. Thr99 is located at the end of strand β4, and Tyr118 is in the middle of β5. Hydrogen bonds are shown as dashed lines.
Lys121 is also very well conserved, and its amine group forms a hydrogen bond to the side chain of Asp71 (Figure 3). The Lys121 side chain is likely to assist in the dephosphorylation/phosphorylation reactions or stabilize the structure by reducing the local negative charges around the phosphorylation site. The corresponding side chain in other RR structures shifts slightly to avoid steric clashes with beryllium fluoride when comparing activated with non-activated crystal structures (19, 20). Thr99 and Tyr118 are also well conserved and are proposed to function as switch residues to relay phosphorylation state to structural changes, which are essential for signal transduction on phosphorylation activation. The side chain of Thr99 is oriented away from the acidic pocket, and the side chain of Tyr118 extends outward toward the dimer interface (Figures 2 and 3) and is involved in π-electron stacking with other side chains as part of the dimer interactions.
Receiver domain dimer interface
In the crystal, two N-terminal domains form a dimer with a two-fold symmetry through the α4-β5-α5 face (Figure 4). Helices α4 and α5, strand β5, the loops between them, and the long loop between β4 and α4 together form a relatively flat surface. The two N-terminal domains interact through this surface to bury a total surface area of ~890 Å2 per molecule, calculated with AREAIMOL of the CCP4 Suite (36). The total solvent-accessible area of the N-terminal domain dimer is ~12813 Å2. At the center of the dimer interface, side chains of Tyr118 (one of the switch residues mentioned above), Asp117, Glu127, and Arg131 have π-electron stacking interactions and charge-charge interactions. The side chain of Arg131 of one molecule forms salt bridges to the side chains of Asp117 and Glu127 of the other molecule. There are also hydrogen bonds between these residues, as well as water-mediated interactions (Figure 4). Two small clusters of hydrophobic side chains flank this π-electron stacking core, involving side chains of Ala130 and Val134 of one molecule with those of Leu104 and Ile108 of the other molecule. The dimer interface is mostly hydrophilic, suggesting that the interactions are dynamic in solution. This is consistent with gel filtration and analytical ultracentrifugation results, as described above, which show that in solution there is a monomer-dimer equilibrium that is concentration dependent.
Figure 4.
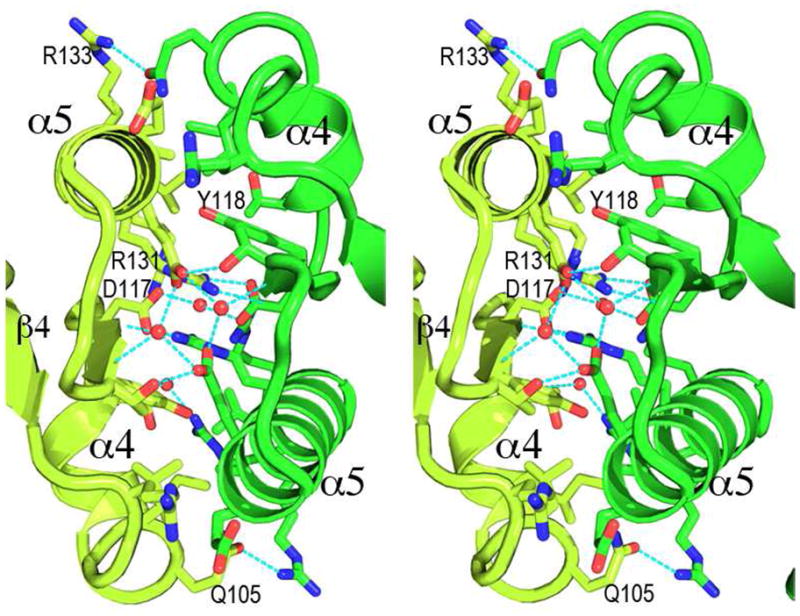
A stereo view of the receiver domain dimer interface. Side chains and water molecules not involved in dimer interactions are not shown for clarity. Hydrogen bonds are depicted as dashed lines. A cluster of side chains with flat groups in the center of the interface interact with π-electron stacking, salt bridges, and hydrogen bonds. Most pronounced are side chains of Tyr118 and Arg131, with 2 rings and 2 guanidinium groups stack in parallel with a spacing of ~3.5 Å. There are also two small patches of hydrophobic interactions flanking the central polar interactions.
Although well conserved in amino acid sequence, helix α5 has variable length among known structures of PhoB/OmpR subfamily RRs. In PhoP, α5 has 15 residues from Leu125 to Ala139. In some other RR structures, such as MtrA from MTB, helix α5 ends at a Leu residue corresponding to Leu136 of PhoP, 3 residues shorter. In the crystal structure of PrrA from M. tuberculosis, there are two molecules in the asymmetric unit, and the length of helix α5 is different between them. In one subunit there are 15 residues in α5 similar to that of PhoP, but in the other subunit the helix extends two more residues at its C-terminal end. Helix α4 and the loops flanking it also have variable structures among RRs and adopt different structures between activated and non-activated structures of PhoB from E. coli (20). The flexibility of α4, α5, and loops allows the dimer interface to be modulated by phosphorylation and adds to the flexibility of the domain liker.
Lack of interactions between the receiver and effector domain
As shown in Figure 2, the two domains of the same polypeptide chain do not have any direct interactions with each other. The two C-terminal domains of the dimer also do not have any direct interactions. In addition, the loop connecting the N- and C-terminal domains is disordered and thus has no role in structural interactions between domains other than tethering them together. In the crystal packing, the N-terminal domain of a neighboring molecule interacts with both domains of the same polypeptide chain, thus keeping their relative orientation.
The lack of interactions between the N- and C-terminal domains and between the two C-terminal domains of the dimer suggests that in solution the C-terminal domains are free to move relative to the N-terminal domains. In the sequence alignments (Figure 1) PhoP of MTB has a longer linker than most of other homologous RRs. This long linker also contributes to the freedom of relative movements of the two domains in solution.
Limited protease digestion
In order to assess the flexibility of the linker in solution, we carried out limited protease digestion of PhoP by trypsin. Two major fragments were visible on SDS-PAGE after trypsin digestion (Figure 5). N-terminal sequencing of the tryptic fragments indicated that the top band starts with residues K3GVDLV and the lower band begins with N148VRLTF (single letter codes of amino acid with the residue number of the first one labeled). The N-terminal sequence of the top band indicates a trypsin cleavage site between Arg2 and Lys3, removing 5 residues from N-terminus including those that are vector-generated. The sequence of the lower band indicates another trypsin cleavage site after Arg147, which is within the linker. The results are consistent with the crystal structure, which indicates that residues of the N-terminal extension, as well as the linker residues, are disordered.
Figure 5.
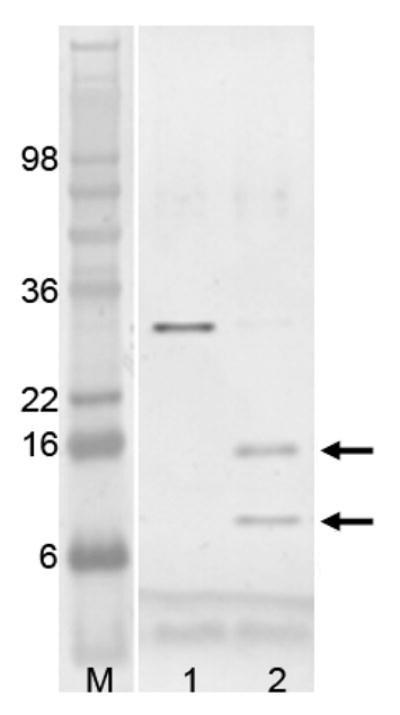
SDS polyacrylamide gel electrophoresis of limited trypsin digestion of PhoP. The lane labeled “M” is the molecular weight marker. Lane 1 is PhoP before trypsin digestion, and lane 2 is after digestion. Calculated molecular weight for PhoP is 27.8 kDa. Protein samples were resolved by SDS-PAGE, and visualized by staining with SimplyBlue. Two bands marked with arrows are sequenced by Edman degradation to identify trypsin cleavage sites.
The flexibility and relatively long length of the inter-domain linker would allow the N-terminal domain to adopt a symmetric dimer while the C-terminal domain could form a tandem dimer when bound to DNA direct repeats. PhoP has been proposed to bind tandem direct repeat DNA as a dimer (30). Most of the known DNA sequences recognized by the OmpR/PhoB subfamily RRs contain direct repeats. The E. coli PhoB has been crystallized in complex with its pho box DNA, and the structure shows PhoB binding to DNA as a tandem dimer. The N-terminal domain of PhoB is expected to form a typical symmetric dimer through the α4-β5-α5 face when activated, as suggested by the crystal structure in the presence of beryllium fluoride (20). These observations suggest that it is possible for the PhoP effector domain to bind DNA direct repeats as a tandem dimer while the receiver domain forms a symmetric dimer.
Structural comparison between the subunits of the dimer
The two subunits in the dimer are related by a two-fold non-crystallographic symmetry. The rmsd for the two subunits over all matched Cα atoms (residues 18–141 and 150–246) is 1.0 Å. The two N-terminal domains alone superpose very well, with an rmsd of 0.085 Å for Cα atoms of residues 18 to 140. The two C-terminal domains have an rmsd of 0.24 Å for Cα atoms of residues 150 to 245. The last residues at the C-terminus are partially disordered and have a large deviation. The first β-strand of the C-terminal domain, β6, also shows measurable shifts. The rmsd between the two subunits in the dimer is mainly from a shift of relative domain positions between the subunits. When the two N-terminal domains are aligned, the two C-terminal domains shift ~4 Å. This shift is expected because the only restrictions on their relative positions are from the crystal packing, and it is consistent with conclusions above that in solution the two domains are likely to be able to move freely relatively to each other.
DISCUSSION
The effector domain of MTB PhoP shows a large degree of structural flexibility, consistent with its proposed function of interacting with DNA and other proteins to regulate gene expression. The average B factor of the effector domain is consistently higher than that of the receiver domain in both protein molecules of the asymmetric unit. Structural alignment between the isolated effector domain and that of the full-length PhoP gives an rmsd of 1.66 Å for the Cα atoms of residues 150 to 246. Most of the deviations are in the transactivation loop, i.e. residues 204 to 212. The corresponding loop of PhoB from E. coli is proposed to interact with the σ70 subunit of the RNA polymerase (23). This loop is likely to be disordered in solution and adopts a fixed structure upon binding to the RNA polymerase. There is a slight rotation of the N-terminal β-sheet, similar to what observed among the protomers in the crystal structure of the isolated C-terminal domain of PhoP (16). The sequence recognition helix, α8, aligns very well for the main chain atoms. Several residues in this helix are well conserved (Figure 1). These residues have hydrophobic side chains and are involved in packing with the hydrophobic core of the domain to maintain the same domain folding of this group of proteins, suggesting that the proteins bind DNA in a similar fashion. One exception is Arg222, which is well conserved but not involved in the domain packing. This residue might be important for binding DNA by interacting with the phosphate backbone or stacking with DNA bases. Most of other residues with side chains exposed to the surface are not conserved, consistent with their proposed roles in recognizing different DNA sequences for different RRs. A recent paper (45) reports that mutation of these residues, including Arg222, into alanine abolishes DNA binding, confirming our earlier prediction based on the structure of the DNA-binding domain (16).
There are currently six full-length structures of the OmpR/PhoB subfamily RRs, DrrB (27) and DrrD (28) from Thermotoga maritima, RegX3 (24), PrrA (26) and MtrA (25) from MTB, and the current structure of PhoP. All these structures have different organization of the domain interactions. The PhoP structure is unique in that its receiver domain forms a dimer with the dimer interface involving the α4-β5-α5 face, which is a common interface among isolated receiver domain dimers that are activated by binding beryllium fluoride, mimicking the phosphorylation state. PhoP also has a completely disordered linker, and its N- and C-terminal domains do not interact with each other or with the domain linker. The closest structure in this respect is the DrrD structure, which has a very small inter-domain interface. DrrD has a shorter linker (Figure 1), which is ordered in the crystal structure but has higher temperature factors, suggesting that the linker is flexible. There is minimal interaction between helix α5 and the N-terminal β-sheet of the C-terminal domain, allowing the sequence recognition helix to be fully exposed for insertion into the major groove of DNA (23). DrrB has the shortest linker of 3 residues, which is involved in the domain interface and well ordered in the crystal structure. In addition to the linker, the domain interface also involves the interactions between the helix α4 and the N-terminal β-sheet of the effector domain. The DNA-binding elements are also fully exposed in the DrrB crystal structure. Crystal structures of PrrA and MtrA show that the DNA-binding elements are obstructed by the domain interface. Both proteins involve the α4-β5-α5 face of the N-terminal domain to pack against the recognition helix and some other elements of the C-terminal domain, although the domain interfaces are different between the two proteins. In solution, PrrA can exist in an open conformations, allowing the DNA recognition helix and the β-hairpin exposed for binding DNA (26). The MtrA crystal structure has the most significant inter-domain interface that buries ~1600 Å2 surface area. However, most of the interface interactions are polar or water-mediated, suggesting that in solution the open conformation is likely present. The recombinant protein purified from expression in E. coli binds promoter DNA of genes regulated by MtrA without phosphorylation (46).
The receiver domain dimer with the interface involving the α4-β5-α5 face has been proposed to be the active conformation. Several isolated receiver domains have been crystallized in complex with beryllium fluoride, which occupies the phosphorylation site and mimics the phospho-aspartate (19–21, 47). All activated receiver domains form a symmetric dimer with the same interface involving α4-β5-α5. Some isolated receiver domains, such as that of PhoP from E. coli (19), form the same activated dimer interface in the absence of phosphorylation or phosphorylation mimics. However, there are some consistent differences between these unphosphorylated active dimers and the phosphorylation-activated dimers. The switch residues, a Ser/Thr residue and a Tyr residue (Thr99 and Tyr118 in PhoP of MTB, see sequence alignments in Figure 1), are in the same inward conformation facing toward the phosphorylation site in the beryllium fluoride activated dimers. In the unphosphorylated active dimers, the switch residues are in outward conformations. This is also the case for PhoP of MTB; both Thr99 and Tyr118 side chains are pointing away from the phosphorylation site (Figure 2). Although the PhoP dimer involves the α4-β5-α5 face, there is a slight shift of the dimer interface when compared to those of the activated dimers (Figure 6). Phosphorylation of the essential aspartate will create favorable hydrogen-bond interactions that shift the side chain of Thr99 toward the phosphate group, causing a shift of the loop β4-α4 and helix α4, which is the part of the structure that is the most flexible and shows the largest variation among known receiver domain structures. These shifts make the inward conformation of the Tyr118 side chain more energetically favorable. Flipping of the Tyr118 side chain inward should result in a rearrangement of the dimer interface because Tyr118 is one of the interface residues (see above for details). Several studies have shown that phosphorylation stabilizes dimer formation of full-length RRs and isolated receiver domains (19, 25). It is possible that upon phosphorylation, Thr99 and Tyr118 of the MTB PhoP protein adopt the inward conformation and the dimer interface rearranges to have similar interactions to the activated receiver dimers, which has a more compact dimer interface (Figure 6). However, the shift should not have much influence on the effector domain structure or its interactions with the receiver domain. This is in sharp contrast to the structure of MtrA, which would release the effector domain for binding DNA if phosphorylation results in the favorable formation of activated receiver domain dimer through the α4-β5-α5 face. It is likely that for MTB PhoP, rearrangement of the dimer interface upon phosphorylation strengthens dimer interactions. Therefore, a larger fraction of PhoP forms dimers through the dimerization of the receiver domain. This, in turn, results in higher DNA-binding affinity by bringing two copies of the DNA-binding domain in close proximity. Current knowledge in the field of the OmpR/PhoB subfamily RRs suggests that these transcription regulators bind DNA as a dimer on direct repeats of DNA sequence. A recent study (48) found that PhoP binds to the promoter DNA of msl3, a gene strongly regulated by PhoP (9), at a much higher affinity upon phosphorylation. This is consistent with the proposed mechanism that phosphorylation promotes dimerization of the receiver domain to bring two DNA-binding domains in close proximity for binding direct-repeat DNA sequences.
Figure 6.

Structural alignment of the receiver domain dimer of PhoP with that of activated PhoB from E. coli (PDB code 1ZES). The PhoP structure is colored in green, and PhoB is colored in orange. The structural superposition was carried out on one of the subunits, shown on the left, with 111 matched Cα atoms and an rmsd of 1.5 Å. The other subunit, shown on the right, has a shift between the two proteins. Panel (a) has the two-fold symmetric axis of the PhoP dimer perpendicular to the paper similar to the view in Figure 2, and panel (b) is a 90° rotation from the view in (a). Despite that both involve α4-β5-α5, the interactions at the dimer interface are different in PhoP and PhoB. The activated PhoB dimer has a more compact interface, with helices from subunits packing more closely.
Acknowledgments
We thank Drs. Vivian Stojanoff, Jean Kakoncic, and Kun Qian at the beamline X6A, NSLS, Brookhaven National Laboratory, Upton, NY, USA for assistance in data collection, Dr. Stephen Techtmann for help with analytical ultracentrifugation experiments, and Drs. Issar Smith, Chou-zen Giam, and Earnest Maynard for helpful comments and revisions on the manuscript.
Abbreviations
- HK
histidine kinase
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- MAD
multi-wavelength anomalous diffraction
- MTB
Mycobacterium tuberculosis
- NCS
non-crystallographic-symmetry
- PMSF
phenylmethanesulfonyl fluoride
- PCR
polymerase chain reaction
- RR
response regulator
- SeMet
selenomethionine
- TEV
tobacco etch virus
- rmsd
root mean square deviation
- TCS
two-component system
Footnotes
Atomic coordinates and structure factors have been deposited with the RCSB Protein Data Bank under accession code 3R0J. This work was supported by the National Institute of Health grant R01GM079185 and the Uniformed Services University of the Health Sciences intramural grant R071IR.
References
- 1.Haydel SE. Extensively Drug-Resistant Tuberculosis: A Sign of the Times and an Impetus for Antimicrobial Discovery. Pharmaceuticals (Basel) 2010;3:2268–2290. doi: 10.3390/ph3072268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gotoh Y, Eguchi Y, Watanabe T, Okamoto S, Doi A, Utsumi R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr Opin Microbiol. 2010;13:232–239. doi: 10.1016/j.mib.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Galperin MY. Diversity of structure and function of response regulator output domains. Curr Opin Microbiol. 2010;13:150–159. doi: 10.1016/j.mib.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Barrell BG, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 5.Tucker PA, Nowak E, Morth JP. Two-component systems of Mycobacterium tuberculosis: structure-based approaches. Methods Enzymol. 2007;423:479–501. doi: 10.1016/S0076-6879(07)23023-X. [DOI] [PubMed] [Google Scholar]
- 6.Ewann F, Jackson M, Pethe K, Cooper A, Mielcarek N, Ensergueix D, Gicquel B, Locht C, Supply P. Transient requirement of the PrrA-PrrB two-component system for early intracellular multiplication of Mycobacterium tuberculosis. Infect Immun. 2002;70:2256–2263. doi: 10.1128/IAI.70.5.2256-2263.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malhotra V, Sharma D, Ramanathan VD, Shakila H, Saini DK, Chakravorty S, Das TK, Li Q, Silver RF, Narayanan PR, Tyagi JS. Disruption of response regulator gene, devR, leads to attenuation in virulence of Mycobacterium tuberculosis. FEMS Microbiol Lett. 2004;231:237–245. doi: 10.1016/S0378-1097(04)00002-3. [DOI] [PubMed] [Google Scholar]
- 8.Zahrt TC, Deretic V. Mycobacterium tuberculosis signal transduction system required for persistent infections. Proc Natl Acad Sci U S A. 2001;98:12706–12711. doi: 10.1073/pnas.221272198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol. 2006;60:312–330. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 10.Perez E, Samper S, Bordas Y, Guilhot C, Gicquel B, Martin C. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol. 2001;41:179–187. doi: 10.1046/j.1365-2958.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- 11.Chesne-Seck ML, Barilone N, Boudou F, Gonzalo Asensio J, Kolattukudy PE, Martin C, Cole ST, Gicquel B, Gopaul DN, Jackson M. A point mutation in the two-component regulator PhoP-PhoR accounts for the absence of polyketide-derived acyltrehaloses but not that of phthiocerol dimycocerosates in Mycobacterium tuberculosis H37Ra. J Bacteriol. 2008;190:1329–1334. doi: 10.1128/JB.01465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Domenech P, Reed MB, Dowd CS, Manca C, Kaplan G, Barry CE., 3rd The role of MmpL8 in sulfatide biogenesis and virulence of Mycobacterium tuberculosis. J Biol Chem. 2004;279:21257–21265. doi: 10.1074/jbc.M400324200. [DOI] [PubMed] [Google Scholar]
- 13.Ryndak M, Wang S, Smith I. PhoP, a key player in Mycobacterium tuberculosis virulence. Trends Microbiol. 2008;16:528–534. doi: 10.1016/j.tim.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Frigui W, Bottai D, Majlessi L, Monot M, Josselin E, Brodin P, Garnier T, Gicquel B, Martin C, Leclerc C, Cole ST, Brosch R. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS pathogens. 2008;4:e33. doi: 10.1371/journal.ppat.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JS, Krause R, Schreiber J, Mollenkopf HJ, Kowall J, Stein R, Jeon BY, Kwak JY, Song MK, Patron JP, Jorg S, Roh K, Cho SN, Kaufmann SH. Mutation in the transcriptional regulator PhoP contributes to avirulence of Mycobacterium tuberculosis H37Ra strain. Cell host & microbe. 2008;3:97–103. doi: 10.1016/j.chom.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Wang S, Engohang-Ndong J, Smith I. Structure of the DNA-binding domain of the response regulator PhoP from Mycobacterium tuberculosis. Biochemistry. 2007;46:14751–14761. doi: 10.1021/bi700970a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondo H, Nakagawa A, Nishihira J, Nishimura Y, Mizuno T, Tanaka I. Escherichia coli positive regulator OmpR has a large loop structure at the putative RNA polymerase interaction site. Nat Struct Biol. 1997;4:28–31. doi: 10.1038/nsb0197-28. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Hackert E, Stock AM. Structural relationships in the OmpR family of winged-helix transcription factors. J Mol Biol. 1997;269:301–312. doi: 10.1006/jmbi.1997.1065. [DOI] [PubMed] [Google Scholar]
- 19.Bachhawat P, Stock AM. Crystal structures of the receiver domain of the response regulator PhoP from Escherichia coli in the absence and presence of the phosphoryl analog beryllofluoride. J Bacteriol. 2007;189:5987–5995. doi: 10.1128/JB.00049-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachhawat P, Swapna GV, Montelione GT, Stock AM. Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure. 2005;13:1353–1363. doi: 10.1016/j.str.2005.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toro-Roman A, Mack TR, Stock AM. Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the alpha4-beta5-alpha5 face. J Mol Biol. 2005;349:11–26. doi: 10.1016/j.jmb.2005.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toro-Roman A, Wu T, Stock AM. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 2005;14:3077–3088. doi: 10.1110/ps.051722805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanco AG, Sola M, Gomis-Ruth FX, Coll M. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure. 2002;10:701–713. doi: 10.1016/s0969-2126(02)00761-x. [DOI] [PubMed] [Google Scholar]
- 24.King-Scott J, Nowak E, Mylonas E, Panjikar S, Roessle M, Svergun DI, Tucker PA. The structure of a full-length response regulator from Mycobacterium tuberculosis in a stabilized three-dimensional domain-swapped, activated state. J Biol Chem. 2007;282:37717–37729. doi: 10.1074/jbc.M705081200. [DOI] [PubMed] [Google Scholar]
- 25.Friedland N, Mack TR, Yu M, Hung LW, Terwilliger TC, Waldo GS, Stock AM. Domain orientation in the inactive response regulator Mycobacterium tuberculosis MtrA provides a barrier to activation. Biochemistry. 2007;46:6733–6743. doi: 10.1021/bi602546q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nowak E, Panjikar S, Konarev P, Svergun DI, Tucker PA. The structural basis of signal transduction for the response regulator PrrA from Mycobacterium tuberculosis. J Biol Chem. 2006;281:9659–9666. doi: 10.1074/jbc.M512004200. [DOI] [PubMed] [Google Scholar]
- 27.Robinson VL, Wu T, Stock AM. Structural analysis of the domain interface in DrrB, a response regulator of the OmpR/PhoB subfamily. J Bacteriol. 2003;185:4186–4194. doi: 10.1128/JB.185.14.4186-4194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buckler DR, Zhou Y, Stock AM. Evidence of intradomain and interdomain flexibility in an OmpR/PhoB homolog from Thermotoga maritima. Structure (Camb) 2002;10:153–164. doi: 10.1016/s0969-2126(01)00706-7. [DOI] [PubMed] [Google Scholar]
- 29.Sinha A, Gupta S, Bhutani S, Pathak A, Sarkar D. PhoP-PhoP interaction at adjacent PhoP binding sites is influenced by protein phosphorylation. J Bacteriol. 2008;190:1317–1328. doi: 10.1128/JB.01074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta S, Sinha A, Sarkar D. Transcriptional autoregulation by Mycobacterium tuberculosis PhoP involves recognition of novel direct repeat sequences in the regulatory region of the promoter. FEBS Lett. 2006;580:5328–5338. doi: 10.1016/j.febslet.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 31.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 32.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 33.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1996;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 36.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 41.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bourret RB. Receiver domain structure and function in response regulator proteins. Curr Opin Microbiol. 2010;13:142–149. doi: 10.1016/j.mib.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Birck C, Chen Y, Hulett FM, Samama JP. The crystal structure of the phosphorylation domain in PhoP reveals a functional tandem association mediated by an asymmetric interface. J Bacteriol. 2003;185:254–261. doi: 10.1128/JB.185.1.254-261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davies KM, Lowe ED, Venien-Bryan C, Johnson LN. The HupR receiver domain crystal structure in its nonphospho and inhibitory phospho states. J Mol Biol. 2009;385:51–64. doi: 10.1016/j.jmb.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 45.Das AK, Pathak A, Sinha A, Datt M, Singh B, Karthikeyan S, Sarkar D. A single-amino-acid substitution in the C terminus of PhoP determines DNA-binding specificity of the virulence-associated response regulator from Mycobacterium tuberculosis. J Mol Biol. 2010;398:647–656. doi: 10.1016/j.jmb.2010.03.056. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Zeng J, Zhang H, He ZG. The characterization of conserved binding motifs and potential target genes for M. tuberculosis MtrAB reveals a link between the two-component system and the drug resistance of M. smegmatis. BMC Microbiol. 2010;10:242. doi: 10.1186/1471-2180-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbieri CM, Mack TR, Robinson VL, Miller MT, Stock AM. Regulation of response regulator autophosphorylation through interdomain contacts. J Biol Chem. 2010;285:32325–32335. doi: 10.1074/jbc.M110.157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pathak A, Goyal R, Sinha A, Sarkar D. Domain structure of virulence-associated response regulator PhoP of Mycobacterium tuberculosis: role of the linker region in regulator-promoter interaction(s) J Biol Chem. 2010;285:34309–34318. doi: 10.1074/jbc.M110.135822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barton GJ. ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]