

Abstract
Background
Associations between schizophrenia (SCZ) and polymorphisms at the regulator of G-protein signaling 4 (RGS4) gene have been reported (single nucleotide polymorphisms [SNPs] 1, 4, 7, and 18). Yet, similar to other SCZ candidate genes, studies have been inconsistent with respect to the associated alleles.
Methods
In an effort to resolve the role for RGS4 in SCZ susceptibility, we undertook a genotype-based meta-analysis using both published and unpublished family-based and case-control samples (total n = 13,807).
Results
The family-based dataset consisted of 10 samples (2160 families). Significant associations with individual SNPs/haplotypes were not observed. In contrast, global analysis revealed significant transmission distortion (p = .0009). Specifically, analyses suggested overtransmission of two common haplotypes that account for the vast majority of all haplotypes. Separate analyses of 3486 cases and 3755 control samples (eight samples) detected a significant association with SNP 4 (p = .01). Individual haplotype analyses were not significant, but evaluation of test statistics from individual samples suggested significant associations.
Conclusions
Our collaborative meta-analysis represents one of the largest SCZ association studies to date. No individual risk factor arose from our analyses, but interpretation of these results is not straightforward. Our analyses suggest risk due to at least two common haplotypes in the presence of heterogeneity. Similar analysis for other putative susceptibility genes is warranted.
Keywords: RGS4, schizophrenia, meta-analysis, association, polymorphism, linkage
Efforts to identify genetic risk factors for schizophrenia (SCZ; Mendelian Inheritance in Man [MIM] [181500]) using linkage and association studies have yielded several exciting candidate genes, and replicate studies have detected associations at a number of these genes. Yet, the majority of replication studies have been inconsistent with respect to the associated alleles, haplotypes, and conferred risks. For example, the neuregulin 1 locus (NRG1) has recently received considerable attention in replication studies. An initial risk haplotype was identified in an Icelandic population by Stefansson et al (2002) and in a second sample from Scotland (Stefansson et al 2003). At least eight successive studies have attempted to replicate these initial results in European (Williams et al 2003; Corvin et al 2004; Petryshen et al 2005), Chinese (Yang et al 2003; Tang et al 2004; Zhao et al 2004; Li et al 2004), and Japanese (Iwata et al 2004) populations. Only some of these replicate studies have detected associations, and few have detected associations with SCZ from identical alleles, single nucleotide polymorphisms (SNPs), and haplotypes as the original findings. Similarly, complex patterns of associations with reference to replicate studies have been observed following the initial associations with SCZ described by Straub et al (2002) for the dystrobrevin binding protein 1 gene (DTNBP1) (Schwab et al 2003; Morris et al 2003; Van Den Bogaert et al 2003; Tang et al 2003; van den Oord et al 2003b; Williams et al 2004a). Thus, even for these two widely investigated genes, interpretation of replicate studies is difficult.
Several factors, including the overestimation of risk in the initial study, disparate power amongst replicate studies, phenotypic heterogeneity, and population heterogeneity need to be considered (Ioannidis et al 2001). Furthermore, in large genes such as DTNBP1 and NRG1, adequate coverage of all variants across the gene is often challenging.
Meta-analysis might be a means of resolving disparate results. For example, pooling samples from several studies could produce greater power than individual studies or might amplify trends for association in small individual studies. Meta-analyses have been reported for a number of candidate genes for schizophrenia, including COMT, DRD2, DRD3, NOTCH4, and SLC6A3, to name a few (Glatt et al 2003a, 2003b, 2005; Dubertret et al 1998; Jonsson et al 2003, 2004; Gamma et al 2005). In addition to the limitations outlined above, these analyses typically assessed only one variant, used only published data, and predominantly involved case-control designs, not family-based designs. Arguably, it would be better to analyze the original data than published summary statistics from individual studies. Therefore, we conducted meta-analysis involving individual genotype data across all available samples at the regulator of G-protein signaling 4 locus (RGS4).
The regulator of G-protein signaling 4 locus is a member of guanine triphosphate (GTP)ase-activating proteins that regulate the timing and duration of G-protein-mediated receptor signaling through neurotransmitter receptors that have been implicated in the pathophysiology or treatment of SCZ (De Vries et al 2000). Expression of RGS4 transcript, but not other RGS family members, was reduced in the cortex of postmortem brain samples from patients with SCZ (Mirnics et al 2001). A recent study in normal individuals found a dense distribution of RGS4 messenger RNA (mRNA) in most cortical layers examined (Erdely et al 2004). The regulator of G-protein signaling 4 locus is localized to 1q23.3 at 160.2 Mb.
Chowdari et al (2002) first conducted association and linkage analyses of RGS4 using family-based and case-control samples. A panel of 13 SNPs was evaluated in independently ascertained samples from Pittsburgh, the National Institutes of Mental Health (NIMH) Collaborative Genetics Initiative, and New Delhi. In both US samples, transmission distortion of individual alleles and haplotypes was observed at four SNPs, denoted SNPs 1, 4, 7, and 18. However, the associated alleles and haplotypes differed. The overtransmitted haplotypes were G-G-G-G in the Pittsburgh sample and A-T-A-A in the NIMH and Indian samples. Curiously, these risk haplotypes were the two most common in the population, with estimated frequencies of .44 and .39, respectively. Case-control comparisons in the US sample did not reveal significant associations (Chowdari et al 2002).
Following these initial results, four independent studies have been reported (Figure 1), all of which analyzed these same four SNPs. Using a large case-control sample from Cardiff, United Kingdom, Williams et al (2004b) detected significant associations with the T and A alleles at SNPs 4 and 18 but not the 4 SNP haplotype. A case-control study from Dublin, Ireland revealed significant associations at SNPs 1 and 7, as well as multiple haplotypes (Morris et al 2004). Associations were found with the same alleles (G) reported in the Pittsburgh sample when their sample was restricted to a narrow diagnosis of SCZ. The first family-based replicate study utilized multiply affected pedigrees from Ireland and revealed an association with the G allele at SNP 18 (Chen et al 2004). There was also significant overtransmission of the G-G-G haplotype at SNPs 1, 4, and 18 to probands, similar to the Pittsburgh families. Analysis of a sample from Brazil did not yield significant case-control differences, although modest overtransmission of the G allele at SNP 18, as well as the G-G haplotype at SNPs 7 and 18 was reported (Cordeiro et al 2005). A linkage study provided evidence for linkage with SCZ near RGS4 at 1q21-22 (Brzustowicz et al 2000), but recently presented follow-up investigations have not suggested associations with RGS4 in this same sample (Brzustowicz et al 2004).
Figure 1.
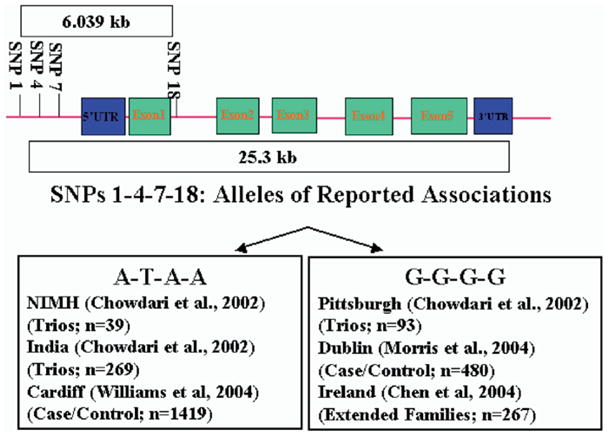
RGS4: Genomic Organization and Reported Associations. Genomic organization of RGS4 and the four SNPs investigated are shown. Previously reported studies that detected significant associations with allele comprising each haplotype are listed.
The published data thus suggest an association with SCZ at RGS4. Similar to other susceptibility candidates, however, the results suggest complex associations that are difficult to interpret. Thus, we sought a comprehensive evaluation of all studies of this gene. We reasoned that analyses of multiple samples might lend clarity, for example, by identifying false-positive results or through highlighting modest effects by the amassing of large samples. We analyzed SNPs 1, 4, 7, and 18 (Chowdari et al 2002). These SNPs have been reported in all previous SCZ studies at this gene, and recent analyses indicate they are substantially correlated (r2 > .8) with 75% of all common polymorphisms spanning the gene (Chowdari et al, unpublished data, 2005). In addition to studying cases and unrelated control samples, we ascertained family-based samples to mitigate against confounding due to population stratification. To reduce the effects of publication bias, namely a bias toward publishing significant associations, we sought genotype data from all published reports as well as any known ongoing investigations. The use of individual genotypes allowed a wide variety of SNP and haplotype analyses.
Methods and Materials
Sample Collection
We organized data from independent investigators worldwide who had either published peer-reviewed manuscripts or abstracts at scientific meetings regarding RGS4 associations with schizophrenia or schizoaffective disorder (SCZA) (Chowdari et al 2002; Williams et al 2004b; Morris et al 2004; Chen et al 2004; Cordeiro et al 2005). We also contacted investigators known to be conducting association studies on SCZ susceptibility genes to identify any additional datasets.
Genotype Assays
Assays were conducted independently at each site, and assay methods varied (Table 1) (Chen et al 1999). Four SNPs were assayed, namely SNPs 1 (rs10917670), 4 (rs951436), 7 (rs951439), and 18 (rs2661319) of RGS4. Single nucleotide polymorphisms 1, 4, and 7 span 849 bases in the 5′ upstream region of the gene. Single nucleotide polymorphism 7 is 5.46 kilobase (kb) from the transcription start site for exon 1. Single nucleotide polymorphism 18 is in the first intron. All four SNPs span 6.935 kb (Figure 1).
Table 1.
Meta-Analysis Site and Sample Information
| Sample | Investigators/ Sample Ethnicity |
Published?
|
Family-Based Samples
|
Case-Control Samples
|
Sample Collection and Genotyping
|
||||
|---|---|---|---|---|---|---|---|---|---|
| Associationa | Families | n | Trios | Cases | Control Subjects |
Diagnostic Criteria |
Genotyping Method |
||
| 1 | Nimgaonkar/ Chowdari |
Yh | 151 | 463 | 151 | 151 | 127 | DSM-IV | SNaPshot |
| Caucasianc | G-G-G-G | ||||||||
| 2 | Nimgaonkar/ Deshpande/Thelma |
Yh | 269 | 912 | 269 | 0 | 0 | DSM-IV | FP |
| Indian | A-T-A-A | ||||||||
| 3 | NIMHd | Y | 39 | 146 | 39 | 0 | 0 | DSM-IIIR | SNaPshot |
| Caucasian | A-T-A-A | ||||||||
| 4 | Williams/ O’Donovan/Owen |
Yi | 0 | 0 | 0 | 711 | 708 | DSM-IV | FP/RFLPg |
| Caucasian | x-T-x-A | ||||||||
| 5 | Vallada/Cordeiro | Yl | 49 | 147 | 49 | 271 | 576 | DSM-IV | FP |
| Braziliane | x-G-x-G | ||||||||
| 6 | Kendler/Chen | Yk | 267 | 1337 | 46 | 0 | 0 | DSM-IV | FP-TDI |
| Caucasian | G-G-G-G | ||||||||
| 7 | Brzustowicz/ Bassett |
N | 25 | 75 | 25 | 0 | 0 | DSM-IIIR | Pyrosequencing |
| Caucasian | |||||||||
| 8 | Collier/Li | N | 609 | 2,036 | 609 | 609 | 157 | DSM-IIIR | CASP |
| Chinese | |||||||||
| 9 | Kirov/ O’Donovan/Owen |
N | 293 | 905 | 293 | 0 | 0 | DSM-IV | FP |
| Caucasian | |||||||||
| 10 | Sobell/Heston | N | 182 | 609 | 82 | 612 | 704 | DSM-IIIR | Taqman |
| Caucasianf | |||||||||
| 11 | Gill/Morris/Corvin | Yj | 0 | 0 | 0 | 299 | 645 | DSM-IIIR | SNaPshot |
| Caucasianc | G-G-G-Gb | ||||||||
| 12 | Weinberger/ Egan/Straub |
N | 276 | 1180 | 153 | 255 | 242 | DSM-IV | Taqman |
| Caucasian | |||||||||
| 13 | Collier/ Feng/St.Clair |
N | 0 | 0 | 0 | 580 | 596 | DSM-IIIR | CASP |
| Caucasian | |||||||||
| Total | 2,160 | 7,810 | 1,716 | 3,488 | 3,755 | ||||
RFLP, restriction fragment length polymorphisms; SNP, single nucleotide polymorphism; RGS4, regulator of G-protein signaling 4; SCZ, schizophrenia; NIMH, National Institute of Mental Health; FP, fluorescence polarization; FP-TDI, fluorescence polarization with template directed dye terminator incorporation; CASP, competitive allele specific polymerase chain reaction.
Reported associations and associated alleles for SNPs 1– 4 –7–18, respectively, at RGS4.
Reported in narrow SCZ subtype.
Samples 1 and 11 have been expanded from initial populations for the current analyses.
National Institute of Mental Health Collaborative Genetics Initiative.
Due to known ethnic admixture within population, ethnicity for some individuals is unclear. All individuals recoded as “Brazilian.”
The majority of the population is Caucasian, however individuals of other ethnicities exist and have been identified for analyses.
FP assay used to type SNPs 4, 7, 18. RFLP method used for SNP1.
Described in Chowdari et al 2002.
Described in Williams et al 2004b.
Described in Morris et al 2004.
Described in Chen et al 2004.
Described in Cordeiro et al 2005.
Quality Control for Genotype Assays
Quality control measures varied across sites (Table 1). Sites 1, 2, 3, 5, and 10 used known homozygous and heterozygous positive control samples generated by sequencing individuals from the Pittsburgh sample. At site 4, SNP 1 and SNP 7 were genotyped in duplicate for all individuals. Site 6 used a semiautomatic procedure and all genotypes were scored automatically using a script as described (van den Oord et al 2003a). For sites 8, 12, and 13, interplate and intraplate duplicate testing of known DNA samples was performed. At site 10, all genotypes were performed twice and read, blind to affected status, primarily using an automated genotype option. For site 7, initial genotype calls were conducted automatically by pyrosequencing software, and duplicate genotypes were generated for 6% of the sample for quality control.
Comparison of Genotype Assay Methods
Error estimates for five genotyping methods were conducted independently by investigators at Pittsburgh and Dublin, including 1) resequencing (Pittsburgh); 2) SNaPshot assay (Pittsburgh, Dublin); 3) single strand conformational polymorphism method (SSCP) (Pittsburgh); 4) restriction fragment length polymorphisms method (RFLP) (Pittsburgh); and 5) Taqman assay (Dublin).
Statistical Analysis
Family-based and case-control association analyses were conducted separately, using individual SNPs as well as haplotypes. For both sets of analyses, samples from individual sites were analyzed first, followed by the pooled datasets. We tested for Hardy-Weinberg equilibrium among cases, control samples, and parents using the GENEPOP software, version 1.31 (see Appendix 1 for web addresses to all software packages). Mendelian inconsistencies were evaluated using PEDCHECK software (O’Connell and Weeks 1998).
Family-Based Associations
We tested individual SNPs and haplotypes for linkage and association using the transmission disequilibrium test (TDT) (GENEHUNTER software) (Kruglyak et al 1996), followed by analysis of extended pedigrees with a generalization of the TDT, the family-based association test (FBAT). Transmission distortion of all haplotypes was assessed using global tests of association available in TRANSMIT (version 2.5.2) (Clayton and Jones 1999) and FBAT software (Laird et al 2000). TRANSMIT was implemented to conduct bootstrap testing using 10,000 bootstrap samples.
Evaluation of Individual Samples: The pooled data can be influenced by sites with larger samples, and pooled data may obscure associations if risk is conferred by different alleles or haplotypes in individual samples, as has been previously reported for RGS4. Hence, significance tests were performed on the data from each site and on data amalgamated over sites. To test whether the distribution of site-specific p-values deviated from the uniform distribution expected under the null hypothesis, we performed a simple test first described by R.A. Fisher. Under the null hypothesis, two times the negative log of a p-value is distributed as a χ2; for the sum of N independent tests, the sum is distributed as a χ22N statistic.
Cladistic Analysis: We performed cladistic analyses using EHAP software (Seltman et al 2003). Conceptually, this approach uses the evolutionary relationship among sampled haplotypes to structure tests of association in case-control designs or differential transmission in family-based designs (Seltman et al 2001, 2003). This methodology takes into account the uncertainty in phase determination rather than using only the most likely phase. To assure no recombination between generations, EHAP evaluates possible recombination events and generates an alert if recombination is detected.
When multilocus genotypes are compatible with multiple haplotype pairs (configurations), EHAP computes statistics on the basis of the joint probability of phenotype and haplotype configurations. For our analyses, we eliminated multilocus genotypes as uninformative if they were consistent with too many haplotype configurations (>10) or if haplotypes were rare relative to sample size. Also, we connected nodes (haplotypes) only if they were separated by a single mutational step or two steps if the node was not connected to the rest of the cladogram (or network). Permutation with score testing was employed for tests of association as incorporated in EHAP.
Case-Control Associations
Genotype comparisons for individual samples were evaluated using the Armitage trends test (SAS software, SAS Institute Inc., Cary, North Carolina). Haplotype frequencies were estimated using PHASE software, version 2.0.2 (Stephens et al 2001; Stephens and Donnelly 2003), and case-control differences were evaluated with an estimation maximization algorithm using an omnibus likelihood ratio test with SNPEM software (Fallin et al 2001).
For cladistic analyses, a haplotype relative risk model implemented as a general linear model was used. All case-control studies were analyzed using both measured haplotype analysis (MHA) permutation and permutation of the overall (full cladogram vs. single collapsed node) test. Each of these was performed with both likelihood ratio and score testing. Good agreement of p-values was found for all four analyses, and only likelihood ratio, overall permutation method results are reported here.
Regression analyses were performed successively for each SNP. The dependent variable was case/control status, and the independent variables were SNP genotype and site of ascertainment. As a measure of heterogeneity between samples ascertained across sites, we examined the interaction between site of ascertainment and SNP genotype on case status. This model is likely to account for heterogeneity between varying ethnic groups (Caucasian, Indian, Chinese, and “Brazilian”), as well as heterogeneity introduced between sites ascertaining similar ethnic groups (e.g., by genotyping variation or unknown admixture). These analyses were conducted in the entire sample, the Caucasian only sample (six samples), and the Caucasians ascertained in European countries (three samples). Based on these results, haplotype associations were assessed by pooling the Caucasian samples (six samples, 5596 individuals). Similar to the family sample, we also evaluated statistical distributions from individual studies as described above, followed by analysis of pooled samples.
We examined population dispersion amongst the Caucasian samples (see Weir and Hill 2002 for review) by the standardized measure of variation among subpopulations first put forth by Wright (1950). As an estimator of θ, or the degree of allele sharing identical by descent between populations, we used FSTAT software (Goudet 1994, 1995) to obtain the unbiased estimate of Fst as described by Weir and Cockerham (1984).
Publication Bias
We analyzed published as well as unpublished data. At the time of our analyses, the majority of the unpublished data were ongoing studies intended for peer-reviewed publication on completion, so formal analyses of publication bias would not be fruitful. Nevertheless, it is possible that we were more likely to become aware of an unpublished dataset due to some common feature, e.g., if they were positive. It is also possible that datasets not showing a significant association were not submitted for publication as quickly as those with significant associations. Thus, we evaluated our results using the published and unpublished datasets separately.
Results
Sample Description
Thirteen groups submitted individual genotype data, with two declinations. Six provided unpublished genotype data (samples 7, 8, 9, 10, 12, 13) (Table 1). Two of the previously reported samples were enlarged (Chowdari et al 2002; Morris et al 2004) and are reported here. In sum, genotypes for 13,807 individuals were obtained (Table 1). Most probands were diagnosed with schizophrenia or schizoaffective disorder (DSM III or DSM IV criteria). Nine cases (<.002% of the total sample) had other diagnoses: psychosis NOS (n = 6), schizoid personality disorder (n = 1), and schizotypal personality disorders (n = 2).
Family-Based Datasets
Parents of probands and available family members were ascertained at 10 sites. The dataset included probands with both available parents (case-parent trios, “trios,” n = 1716 families), as well as probands with one available parent (n = 444 families). The latter group also included families with multiple affected and/or unaffected siblings and relatives. Thus, the entire family dataset incorporated 7810 individuals from 2160 families (“extended family sample”).
Case-Control Datasets
Eight sites ascertained unrelated control individuals (Table 1). The recruitment of control individuals varied and included both screened and unscreened individuals with respect to psychiatric illness (Chowdari et al 2002; Cordeiro et al 2005; Egan et al 2000; Morris et al 2004; Williams et al 2004b). A total of 3755 control individuals were available. They were compared with 3486 cases, including 2242 persons without available relatives and 1244 probands from the family-based samples (one proband per family was randomly selected when multiple affected individuals were available from the same pedigree). In this sample, 77.28% (n = 5596) reported Caucasian ancestry and were recruited from six sites (samples 1, 4, 10, 11, 12, and 13) (Table 1).
Missing Data
Overall, 5.4% of genotypes from the case-control sample were unavailable (10.9%, 4.1%, 3.2%, 3.3% for SNPs 1, 4, 7, 18, respectively). Genotypes were unavailable in the family dataset for 10.9%, 10.2%, 10.3%, and 8.4% of samples for SNPs 1, 4, 7, and 18, respectively, excluding sample 9. Sample 9 typed a subset of samples for all SNPs and subsequently genotyped all samples based on the LD patterns, resulting in missing data rates of 23.1%, 28.8%, 90.7%, and 0% at SNPs 1, 4, 7, and 18, respectively.
Comparison of Genotype Accuracy Rates
At Pittsburgh, no discrepancies were detected in genotypes of 72 individuals between the SNaPshot assay and the sequencing method at all four SNPs. Hence, the SNaPshot method was used as a reference. The SNaPshot and RFLP methods were compared for SNPs 1 and 7, and unacceptably high discrepancy rates were noted (SNP 1: 7.51%, n = 493 samples; SNP 7: 3.95%, n = 506 samples). SNaPshot and SSCP methods were compared at SNPs 4 and 18, where lower discrepancy rates were observed (.59%, n = 507, and .79%, n = 509, respectively). Due to the low concordance rates between assays, all data submitted for meta-analysis from samples 1, 3, and 5 were genotyped using the SNaPshot assay. At Dublin, 48 individuals were compared using the SNaPshot assay and Taqman assay for all SNPs. No discrepancies were detected between these two methods.
Linkage Disequilibrium
We estimated LD for all locus pairs for each sample using EMLD software. Analyses were performed on control samples or parents of probands from trio samples. Table 2 provides LD information for all samples and all SNP combinations.
Table 2.
Linkage Disequilibrium Across All Samples
| Sample | Ethnicity | Locus Pairs (D′/r
2)
|
|||||
|---|---|---|---|---|---|---|---|
| 1&4 | 1&7 | 1&18 | 4&7 | 4&18 | 7&18 | ||
| 1 | Caucasian | 1.00/.75 | 1.00/.98 | .86/.64 | 1.00/.72 | .98/.84 | .85/.59 |
| 3 | Caucasian | 1.00/.60 | 1.00/.82 | .91/.56 | 1.00/.49 | 1.00/.88 | .89/.44 |
| 4 | Caucasian | .87/.60 | .93/.80 | .74/.48 | .93/.63 | .96/.84 | .78/.49 |
| 6 | Caucasian | .85/.51 | .94/.77 | .72/.37 | 1.00/.61 | .83/.69 | .92/.52 |
| 7 | Caucasian | .99/.72 | 1.00/.97 | .91/.55 | .97/.72 | .91/.74 | .90/.55 |
| 9 | Caucasian | 1.00/.73 | 1.00/1.00 | .86/.61 | 1.00/.73 | .98/.86 | .86/.61 |
| 10 | Caucasian | .97/.74 | 1.00/.99 | .89/.67 | .97/.74 | .97/.86 | .89/.67 |
| 11 | Caucasian | .97/.74 | 1.00/.99 | .89/.67 | .97/.74 | .97/.86 | .89/.67 |
| 12 | Caucasian | 1.00/.62 | 1.00/.79 | .85/.50 | .90/.67 | .98/.86 | .76/.55 |
| 13 | Caucasian | 1.00/.62 | 1.00/.79 | .85/.50 | .90/.67 | .98/.86 | .76/.55 |
| 2 | Indian | .87/.38 | .89/.74 | .78/.32 | .93/.39 | .80/.61 | .87/.36 |
| 5 | Brazilian | 1.00/.52 | 1.00/.99 | .93/.48 | 1.00/.52 | .98/.91 | .93/.47 |
| 8 | Chinese | .99/.62 | 1.00/1.00 | .86/.52 | 1.00/.63 | .96/.82 | .95/.56 |
LD is provided for each locus pair. D= values are given first, followed by the r 2 values.
LD, linkage disequilibrium.
Family-Based Associations
We found seven transmissions inconsistent with Mendelian inheritance among 2160 families across all four SNPs. No sample had more than three non-Mendelising families. Genotypes for these individuals were set to null. To evaluate the distribution of genotypes amongst the parent populations included in the analyses, we assessed Hardy-Weinberg Equilibrium (HWE) in the parents at each sample for each of the four SNPs. We found only the parents of the Indian sample deviated significantly from HWE at SNP1 (p < .01). This rate is about what we would expect by chance. No recombination events were detected by EHAP.
Analysis of Individual Sample Data
Single nucleotide polymorphism and/or haplotype based associations were observed in four samples (p < .055), all of which have been previously reported: samples 2, 3 (Chowdari et al 2002); sample 6 (Chen et al 2004); and sample 5 (Cordeiro et al 2005). Cladistic analyses revealed associations at two of these samples (samples 3 and 6). Global tests of association of all haplotypes using FBAT were significant for samples 2 (p = .02), 3 (p = .002), and 6 (p = .007) (Chowdari et al 2002; Chen et al 2004).
For individual SNPs, the distribution of sample-specific p-values was consistent with a uniform, but the p-values for global haplotype tests showed greater mass toward small p-values (χ2 = 47.4, df = 20, p = .0005). In fact, p-values were less than .5 for all tests. This conclusion was not altered by removing sample 3, the most significantly associated sample (χ2 = 34.95, df = 18, p = .009). Inspection of Table 3 shows deviation from expected haplotype transmissions for one of the two common haplotypes at most samples (p ≤ .2 for 8 of the 10 samples).
Table 3.
Family-Based Analyses: Haplotype Transmission Results
| Sample | Families | Overtransmitted Haplotype/p-Value |
Global Test p-Valueb |
|---|---|---|---|
| 1 | 151 | G-G-G-G/.20 | .307 |
| 2 | 269 | A-T-A-A/.055 | .021 |
| 3 | 39 | A-T-A-A/.0006 | .002 |
| 5 | 49 | G-G-G-G/.1 | .393 |
| 6 | 267 | G-G-G-G/.01a | .007 |
| 7c | 25 | G-G-G-G/.78 | .469 |
| 8c | 609 | A-T-A-A/.43 | .214 |
| 9c | 293 | A-T-A-A/.2 | .49 |
| 10c | 182 | G-G-G-G/.19 | .193 |
| 12c | 276 | A-T-A-A/.13a | .396 |
Family-based transmission distortion for the two most common haplotypes in individual samples. Results show overtransmitted haplotype and corresponding individual haplotype p-value.
FBAT, family-based association test; TDT, transmission disequilibrium test.
p-values are results of haplotype analyses (FBAT software) with extended pedigrees. All other p-values are single haplotype TDT results.
Global p-values for individual samples are whole marker results generated using the FBAT permutation test (100,000 permutations).
Unpublished sample.
When evaluating individual haplotype transmissions, i.e., TDT analyses, the G-G-G-G haplotype was overtransmitted in four samples (samples 1, 5, 6, 10 only: 110 transmitted haplotypes, 79 not tranmsitted haplotypes) and the A-T-A-A haplotype was overtransmitted in four other samples (samples 2, 3, 8, 12 only: 110 transmitted haplotypes, 79 nontranmsitted haplotypes). Consistent with disparate haplotypes being overtransmitted amongst different samples in Table 3, cladistic analyses across all trio datasets did not detect a significant individual risk haplotype (p = .289).
Analysis of the Pooled Sample
Transmission disequilibrium test analyses were conducted for individual SNP/haplotype transmissions using the case-parent trios (n = 1716), and analyses of the extended pedigree datasets were performed using FBAT (n = 2160 families; 7810 individuals, mean of 3.62 individuals/pedigree). Of note, there were more extended pedigrees than trios from samples 6, 10, and 12 (Chen et al 2004) (Table 1). Significant transmission distortion of individual SNPs was not noted for TDT analyses of the trio sample (numbers of G alleles transmitted/not transmitted, T/NT: SNP 1, 588/584, p = .91; SNP 4, 554/559, p = .88; SNP 7, 499/484, p = .63; SNP 18, 639/616, p = .52). Similar results were obtained from FBAT analyses of the extended pedigree dataset (p > .4 for all SNPs).
Consistent with published frequency estimates, we find two common haplotypes in our population, namely G-G-G-G (42.4%) and A-T-A-A (38.9%). The frequency of the next common haplotype, G-T-G-A, was 8.2%. All other haplotypes had a frequency of 5% or less. Analyses of individual haplotype transmissions using the TDT suggested no significant distortions for any of the common haplotypes when they were analyzed individually (G-G-G-G: 246/243 [p = .89]; A-T-A-A: 273/244 [p = .2]; G-T-G-A: 97/100 [p = .83]). Individual haplotype analyses also did not reveal significant associations in the extended pedigrees (G-G-G-G, p = .18; A-T-A-A, p = .39; G-T-G-A, p = .72).
By contrast, global tests of transmission distortion for all haplotypes revealed significant associations in the trio as well as the extended family samples using TRANSMIT software (trios: χ2 = 33.63, df = 15, p = .003; extended family samples: χ2 = 37.3, df = 15, p = .001). Similar results were detected using the FBAT permutation test (whole marker results: trios, p = .010; entire dataset, p = .0006). Bootstrap testing was carried out using transmit software, and the global results from 10,000 bootstrap samples indicated significant transmission distortion (p = .0019).
We evaluated the discrepancy in significance values between the initial analysis of individual haplotypes and the subsequent global tests. Inspection of the results of global tests suggested overtransmission of the two common haplotypes at the expense of other haplotypes. We examined this result in two ways. 1) We assessed the impact of excluding rare haplotypes using the extended family sample. When the global tests were restricted to haplotypes with a frequency greater than 1% (6 haplotypes) or 5% (3 haplotypes), significant associations persisted (p = .0007 and p = .01, respectively). Bootstrap testing (10,000 samples) was also carried out on these restricted haplotypes, and the results remained unaltered (global significance for haplotypes greater than 1% and 5% = .0006 and .012, respectively). 2) We constructed specific contrasts using EHAP software. When either of the two common haplotypes was individually contrasted against a bin encompassing all other haplotypes, significant transmission distortion was not detected; however, when the two most common haploytpes were combined and contrasted against all other haplotypes combined, significant overtransmission of the common haplotypes was detected (p = .004). Thus, the initial significant p-values for the global tests are due to overtransmission of the G-G-G-G and A-T-A-A haplotypes at the expense of all other haplotypes.
To determine if the global analyses were influenced by ethnic variation, we conducted separate global tests of the Caucasian (n = 1233 families) and non-Caucasian (n = 925) families. In the Caucasian families, the same pattern of association was observed for the global test of transmission distortion as in the entire sample, although it was not quite significant (p = .08). Consistent with our observations in the entire dataset, results were significant when restricting the haplotypes to those greater than 1% (p = .003) or 5% frequency (p = .03) in the Caucasian sample. All global analyses were significant in the non-Caucasian families (all haplotypes, p = .002; haplotypes > 1%, p = .01; haplotypes > 5%, p = .05).
Case-Control Associations
We assessed HWE in the cases and control samples from individual samples (eight samples, two groups) for all four SNPs (64 tests), and found deviations in two samples (p < .01; control samples in sample 12 at SNP 7 and control samples from sample 8 at SNP 4), roughly equal to what is expected by chance.
A reasonable concern is whether these samples are too heterogenous with respect to ancestry. Naturally, the samples with Asian, African, and Amerindian ancestry would be expected to differ somewhat from the samples of European ancestry; but do the six samples of European ancestry differ substantially? To assess this question, we examined their degree of divergence, as estimated by Fst. Estimated Fst was .001 and .002 overall for cases and control samples, respectively, and individual loci showed similar estimates (cases: .001, .000, .003, .000; control samples: .001, .001, .003, .001 for SNPs 1, 4, 7, and 18, resectively). Fst ranges from 0 to 1, and the estimate for these RGS4 SNPs are consistent with the frequent observation of little heterogeneity among samples of European ancestry (Morton 1992; Devlin and Roeder 1999; Devlin et al 2001; Chakraborty 1993).
Analysis of Individual Sample Data
Significant associations with individual SNPs were noted in three samples (Sample 4: SNPs 4 and 18, Sample 8: SNP4, Sample 13: SNPs 1, 7, and 18; Table 4). All significant associations were observed with alleles constituting the A-T-A-A haplotype. Global tests incorporating all four SNP haplotypes (i.e., SNPEM omnibus likelihood ratio tests) detected associations in three samples (p < .05; samples 8, 12, and 13; Table 5). Cladistic analyses revealed haplotype associations at two of these samples (samples 8, 12) but only a trend at sample 13 (p = .10).
Table 4.
Case-Control Analyses: SNP-Based Results
| Sample | Cases/Control Subjects |
Ethnicity | Case Frequency (G Allele)a |
Control Frequency (G Allele) | p-Valueb | Odds Ratio (Allele)c | 95% CI |
|---|---|---|---|---|---|---|---|
| SNP1 | |||||||
| 1 | 151/127 | Caucasian | .551 | .556 | .92 | ||
| 4 | 711/708 | Caucasian | .579 | .583 | .886 | ||
| 10d | 612/704 | Caucasian | .602 | .573 | .132 | ||
| 11 | 299/645 | Caucasian | .589 | .552 | .12 | ||
| 12d | 255/242 | Caucasian | .541 | .59 | .122 | ||
| 13d | 580/596 | Caucasian | .567 | .611 | .033 | 1.26(A) | 1.644–.965 |
| 5 | 271/576 | Brazilian | .576 | .568 | .747 | ||
| 8d | 609/157 | Chinese | .6 | .544 | .095 | ||
| SNP4 | |||||||
| 1 | 151/127 | Caucasian | .487 | .469 | .676 | ||
| 4 | 711/708 | Caucasian | .486 | .532 | .021 | 1.2(T) | 1.388–1.037 |
| 10d | 612/704 | Caucasian | .508 | .496 | .518 | ||
| 11 | 299/645 | Caucasian | .52 | .487 | .178 | ||
| 12d | 255/242 | Caucasian | .477 | .474 | .938 | ||
| 13d | 580/596 | Caucasian | .491 | .528 | .075 | ||
| 5 | 271/576 | Brazilian | .416 | .408 | .75 | ||
| 8d | 609/157 | Chinese | .526 | .631 | .002 | 1.544(T) | 2.022–1.179 |
| SNP7 | |||||||
| 1 | 151/127 | Caucasian | .557 | .551 | .89 | ||
| 4 | 711/708 | Caucasian | .579 | .606 | .145 | ||
| 10d | 612/704 | Caucasian | .602 | .573 | .132 | ||
| 11 | 299/645 | Caucasian | .594 | .554 | .105 | ||
| 12d | 255/242 | Caucasian | .5 | .53 | .392 | ||
| 13d | 580/596 | Caucasian | .561 | .615 | .006 | 1.254(A) | 1.482–1.061 |
| 5 | 271/576 | Brazilian | .576 | .569 | .777 | ||
| 8d | 609/157 | Chinese | .603 | .553 | .121 | ||
| SNP18 | |||||||
| 1 | 151/127 | Caucasian | .514 | .504 | .819 | ||
| 4 | 711/708 | Caucasian | .51 | .549 | .042 | 1.171(A) | 1.363–1.007 |
| 10d | 612/704 | Caucasian | .534 | .525 | .644 | ||
| 11 | 299/645 | Caucasian | .548 | .509 | .128 | ||
| 12d | 255/242 | Caucasian | .498 | .502 | .891 | ||
| 13d | 580/596 | Caucasian | .516 | .56 | .03 | 1.194(A) | 1.407–1.014 |
| 5 | 271/576 | Brazilian | .445 | .424 | .381 | ||
| 8d | 609/157 | Chinese | .509 | .466 | .19 | ||
Results for the SNP-based analyses from individual samples.
Samples are grouped by ethnicity.
SNP, single nucleotide polymorphism; CI, confidence interval.
Frequencies are given with reference to the G allele at each SNP.
p-values are trends test results from genotype data.
Odds ratio, associated allele, and 95% confidence intervals (CI) for significant associations.
Unpublished samples.
Table 5.
Case-Control Analyses: Four SNP Haplotype Results by Sample
| Sample | Ethnicity | Cases/Control Subjects |
G-G-G-G Frequency (Case) |
G-G-G-G Frequency (Control) |
A-T-A-A Frequency (Case) |
A-T-A-A Frequency (Control) |
Omnibus p-Valuea |
|---|---|---|---|---|---|---|---|
| 1 | Caucasian | 151/127 | .486 | .484 | .416 | .408 | .596 |
| 4 | Caucasian | 711/708 | .459 | .497 | .37 | .343 | .445 |
| 10b | Caucasian | 612/704 | .505 | .491 | .372 | .395 | .566 |
| 11 | Caucasian | 299/645 | .521 | .502 | .368 | .402 | .64 |
| 12b | Caucasian | 255/242 | .478 | .463 | .438 | .378 | .037 |
| 13b | Caucasian | 580/596 | .477 | .525 | .408 | .359 | .032 |
| 5 | Brazilian | 271/576 | .412 | .402 | .399 | .416 | .739 |
| 8b | Chinese | 609/157 | .481 | .284 | .376 | .234 | .0001 |
Estimated haplotype frequencies in cases and control subjects from individual samples.
Samples are grouped by ethnicity.
SNP, single nucleotide polymorphism.
p-values represent EM algorithm estimations of SNP haplotypes omnibus likelihood ratio tests of association for all haplotypes.
Unpublished samples.
The distribution of p-values for sample specific tests deviated significantly from a uniform for two of the SNPs, SNP 4 (χ2 = 31.59, df = 16, p = .01) and SNP 7 (χ2 = 29.2, df = 16, p = .02), while the other two SNPs showed similar but not significant deviations (SNP 1 [p = .07]; SNP 18 [p = .09]). Similar to the family sample, sample-specific global tests of association using the four SNP haplotypes suggested a significant deviation from a uniform distribution for all samples (χ2 = 49.6, df = 16, p = .002), as well samples of Caucasian ancestry only (χ2 = 29.7, df = 12, p = .002) (Table 5). However, in this case, the conclusion was not robust to removing the most significantly associated sample (sample 8 excluded: χ2 = 18.75, df = 14, p = .17). Cladistic analyses suggested a modest trend for association (p = .11) across all samples.
Analyses of All Samples
Regression analyses indicated significant effects of SNP genotype on case status for SNPs 4 (χ2 = 10.75, df = 3, p = .01) and 7 (χ2 = 7.91, df = 3, p = .05) across all samples. No associations were observed when analyses were restricted to Caucasian samples.
The interaction of SNP genotype and site of ascertainment on case status suggested significant heterogeneity for SNPs 1 (χ2 = 34.95, df = 21, p = .001), 4 (χ2 = 41.9, df = 21, p = .004), and 18 (χ2 = 37.7, df = 21, p = .01) across all samples, SNP 1 only in the Caucasian samples (six samples; p = .003), and no heterogeneity in Caucasians ascertained in European countries (three samples). Thus, heterogeneity is present across all samples, but is diminished when restricting analyses to the Caucasian samples.
Analysis of Pooled Data for Haplotypes
Due to observed heterogeneity amongst all samples, haplotype analyses were conducted by pooling the case-control samples of Caucasian ancestry (n = 5596). These analyses did not yield significant associations with any of the individual haplotypes or global tests of association across all haplotypes (4 SNP omnibus likelihood ratio: p = .51). We also designed specific contrasts using EHAP software to determine associations with the two most common haplotypes, similar to analyses in the family sample. When the two common haplotypes were combined and contrasted against all other haplotypes combined, a modest association was detected (frequency of two common haplotypes/frequency of rare haplotypes; cases: .834/.166, control samples: .817/.183, p = .09).
Publication Bias
Six of the seven published datasets detected significant or marginally significant associations at the SNP or haplotype level (samples 1, 2, 3, 4, 6, 11). We ascertained six unpublished case-control datasets. We detected associations in case-control analyses at the SNP (samples 8, 13) or haplotype level (sample 12) in three of the six additional samples. No associations were detected in any of the five unpublished family-based samples included in these analyses.
Discussion
Genetic analyses of complex diseases are fraught with challenges because we often know so little about how to control for the genetic and environmental sources of heterogeneity. For this reason, when studies attempting to replicate initial associations of complex disorders produce different results, it is difficult to interpret their significance. Recently, a number of candidates for schizophrenia susceptibility genes have emerged from the analyses of linkage-defined positional candidates, some of which have been motivated by other biological information such as gene expression. In addition, there have been studies replicating these initial findings, in a sense, but the interpretation of these results is often obscure. In fact, most often neither the associated alleles nor the associated haplotypes are consistent across these studies (Shirts and Nimgaonkar 2004). Here, we investigate the results for one of the genes recently described as a positional and functional SCZ susceptibility candidate, RGS4. Unlike many of the other candidates, RGS4 is a relatively small gene in which patterns of LD have been investigated and associations reported for a limited number of SNPs. Yet, like the other candidates, studies of the association between SCZ and RGS4 alleles and haplotypes have been plagued by inconsistency. In this report, we performed meta-analysis in an attempt to understand these inconsistencies.
Our goal was to elicit greater evidence for, or against, RGS4 as a gene containing variations affecting susceptibility to SCZ. Moreover, if the evidence was positive, then we hoped the analyses would elucidate exactly what factors generate susceptibility. Our results are compatible with at least two risk variants conferring susceptibility to SCZ, specifically both the common haplotypes of the four alleles in which associations have been previously reported at this gene.
Our family-based analyses detected significant transmission distortion incorporating all haplotypes. These observations were made using two different software programs, making it unlikely that they are due to idiosyncrasies in analytic software. The results could also not be attributed to deviations from Hardy-Weinberg Equilibrium in the parent population. We scrutinized these results and conducted additional analyses, all of which suggest that overtransmission of both of the two most common haplotypes appears to be the most parsimonious explanation for the results of the global tests.
Evaluation of the distribution of test statistics from individual samples also supported family-based associations, even after the most significant sample was excluded. These analyses would be particularly persuasive if consistent deviations from expected distributions were detected across multiple studies, rather than being attributable to few studies with large effect sizes. Indeed, inspection of transmission distortions at each sample revealed modest deviations from expectations in the global tests for most samples (Table 3).
Case-control analyses appear to support this conclusion. No individual risk haplotype was detected in cladistic analyses or assessment of the pooled Caucasian sample. Instead, the distribution of test statistics suggested associations with global haplotype tests across samples. If transmission to affected offspring was biased toward the two most common haplotypes, one would expect to detect this effect in a sufficiently powered case-control sample. We investigated this hypothesis in the Caucasian sample. Our results showed nonsignificant patterns similar to those of the family-based analyses of association with both common haplotypes compared with all other haplotypes. However, the differences between case and control haplotype frequencies were relatively small (<2%).
Collectively, our analyses point toward a modest association resulting from overtransmission of both of the common haplotypes to SCZ cases at the expense of other haplotypes. There are a number of possible explanations for the observed results, including biological, statistical, molecular, and population phenomena.
Is there a biologically plausible explanation why two common haplotypes, accounting for greater than 80% of all haplotypes, are overtransmitted to individuals with SCZ? Arguably, the simplest explanation is that the liability locus or loci remain undetected and are found more commonly (or exclusively) on these two haplotypes. Certainly recurrent mutations or recombinations that transfer liability alleles between haplotypes are likely to involve these common haplotypes. Our results could account for at least two different possibilities in such a scenario: allelic (intragenic) heterogeneity or the contribution of multiple individual loci to susceptibility. Similar results could also be obtained by the presence of a single, rare susceptibility variant occurring against the background of both common haplotypes. Evaluation of these explanations would require comprehensive sequencing through RGS4 and its surrounding regions in many individuals. The nebulous nature of what constitute the important elements for expression of RGS4 complicates this analysis. On the other hand, expression assays using RGS4 alleles and haplotypes are reasonably straightforward and of interest in light of past analyses (Mirnics et al 2001; Erdely et al 2004).
It is also possible, although more difficult to defend, that the haplotypes themselves have an impact on SCZ susceptibility. Notably, SNPs 1, 4, and 7 lay within the 5′ upstream region of the gene, and the haplotypes investigated here span the first exon of RGS4. The potential effect of these, or unknown variants as discussed above, on promoter activity and/or transcription is intriguing, given our results. The significance of our findings could also be rooted in phenotypic subgroups for which RGS4 may modulate expression of the disease phenotype. Seeking clinical subfeatures that may be significantly impacted by functional changes related to RGS4 could provide insight into the biological role of this gene on SCZ susceptibility.
Statistical phenomena may also contribute to our findings. One of the curious observations from the past studies of RGS4 is that one common haplotype would appear to be overtransmitted in one sample and yet the other common haplotype would appear to be overtransmitted in the next sample tested. Is this phenomenon compatible with our results, which suggest that both common haplotypes are overtransmitted? Recent simulation studies suggest that even when the liability locus is amongst the loci tested within a gene, the liability locus often does not produce the maximum test statistic (Roeder et al 2005). Instead, other loci in substantial LD with the liability locus yield the maximum test statistic. Moreover, haplotype analyses carry similar challenges, as simulations have shown that in the presence of a liability haplotype, multiple patterns of haplotype associations can be found (Seltman et al 2001). Seltman et al (2001) concluded that in many instances, cladograms and measured haplotype analyses, such as those conducted herein, can provide greater insight into what haplotype bears risk alleles. However, if the scenario revealed by our analyses is, in fact, true, then cladistic analyses are unlikely to yield much insight.
By our analysis plan, we first performed global tests of association using bootstrap testing and permutation tests; if these tests were significant, we then would explore the data to determine what was generating the significant findings. In fact, our global tests were significant, and our conclusions are based on our subsequent exploratory analyses. It is noteworthy, however, that even if we were to correct for our exploratory analyses by a conservative Bonferroni-type correction for the number of SNPs (4), common haplotypes (6), and study designs tested (2, family-based and case-control), our results would still exceed the significance threshold (p < .001) for significant transmission distortion.
It is possible that the overtransmission of the two common haplotypes results from technical issues of little interest to the genetics of SCZ, such as population heterogeneity or molecular analysis. Due to the use of transmission tests, confounding due to population heterogeneity is of little concern. We conducted analyses to assess population heterogeneity in our case-control sample, and our results suggest heterogeneity is relatively minor across samples of European ancestry. However, technical molecular issues could explain the result. Due to the retrospective nature of the analyses, uniform quality control in genotyping measures could not be imposed. Notably, we scrutinized quality control and found that rigorous checks were used in genotyping assays. Still, it is well known that genotyping errors can mimic biased transmissions (see Gordon et al 1999; Mitchell et al 2003 for review), and that bias is most likely to present itself as the overtransmission of common alleles/haplotypes. Countering this concern, somewhat, are shared observations: case-control analyses suggest similar overrepresentation of common haplotypes in SCZ cases, and rarer haplotypes showed similar frequencies in the singleton cases that could not be evaluated for Mendelian transmission (or in the control samples) than in the family-based probands that did have Mendelian checks. Still, potential confounds due to assay variation could impact on our results and warrant consideration.
In summary, we report a meta-analysis of RGS4 polymorphisms with schizophrenia. Genotype data from 13,807 individuals were analyzed collaboratively by 13 independent groups. To our knowledge, this is the largest such study to date in SCZ research. Future studies may require sequencing across the risk haplotypes in a large number of patients. Similar methodology to that presented here may help resolve some of the other controversial associations reported for psychiatric and nonpsychiatric genetically complex disorders.
Acknowledgments
This work was supported by grants from the National Institute of Mental Health (NIMH) (MH56242 and MH53459 to VLN), (K02 MH070786 to KM), (MH62440 to LMB), the Indo-US Project Agreement (#N-443-645 to VLN and BKT), an NIMH Conte Center for the Neuroscience of Mental Disorders (MH051456 to DAL), The Intramural Research Program of NIMH (DRW, RES, MFE), United Kingdom MRC (MJO, GK, MCO, and NMW), Science Foundation of Ireland (MG, DWM, APC), The National Science Foundation of China (TL), Welcome Trust (TL and DAC), NARSAD (TL), The Schizophrenia Research Fund (TL and DAC), GSK (FZ and DS), The Canadian Institutes of Health Research (ASB), The Bill Jefferies Schizophrenia Endowment Fund (ASB), Canada Research Chair in Schizophrenia Genetics (ASB), NARSAD (ASB), and Janssen Research Foundation funded family recruitment in Bulgaria.
We thank Shawn Wood for his help with the cladistic analyses.
Appendix 1. Electronic Database Information
Online Mendelian Inheritance in Man (OMIM), http://www.biomed.curtin.edu.au/genepop/index.html.
FSTAT: http://www2.unil.ch/popgen/softwares/fstat.htm
GENEHUNTER: http://www.fhcrc.org/labs/kruglyak
FBAT: http://www.biostat.harvard.edu/~fbat
EHAP: http://wpicr.wpic.pitt.edu/WPICCompGen/
References
- Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS. Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science. 2000;288:678–682. doi: 10.1126/science.288.5466.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzustowicz LM, Simone J, Mohseni P, Hayter JE, Hodgkinson KA, Chow EW, Basset AS. Linkage disequilibrium mapping of schizophrenia susceptibility to the CAPON region of chromosome 1q22. Am J Hum Genet. 2004;74:1057–1063. doi: 10.1086/420774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R. Analysis of genetic structure of populations: Meaning, methods, and implications. In: Majumder PP, editor. Human Population Genetic. New York: Plenum Press; 1993. pp. 189–206. [Google Scholar]
- Chen X, Dunham C, Kendler S, Wang X, O’Neill FA, Walsh D, et al. Regulator of G-protein signaling 4 (RGS4) gene is associated with schizophrenia in Irish high density families. Am J Med Genet B Neuropsychiatr Genet. 2004;129:23–26. doi: 10.1002/ajmg.b.30078. [DOI] [PubMed] [Google Scholar]
- Chen X, Levin L, Kwok PY. Fluorescence ploarization in homogeneous nucleic acid analysis. Genome Res. 1999;9:492–498. [PMC free article] [PubMed] [Google Scholar]
- Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, et al. Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet. 2002;11:1373–1380. doi: 10.1093/hmg/11.12.1373. [DOI] [PubMed] [Google Scholar]
- Clayton D, Jones H. Transmission/disequilibrium tests for extended marker haplotypes. Am J Hum Genet. 1999;65:1161–1169. doi: 10.1086/302566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham CC, Weir BS. Estimation of inbreeding parameters in stratified populations. Am Hum Genet. 1986;50:271–281. doi: 10.1111/j.1469-1809.1986.tb01048.x. [DOI] [PubMed] [Google Scholar]
- Cordeiro Q, Talkowski ME, Chowdari KV, Wood J, Nimgaonkar V, Vallada H. Association and linkage analysis of RGS4 polymorphisms with schizophrenia and bipolar disorder in Brazil. Genes Brain Behav. 2005;4:45–50. doi: 10.1111/j.1601-183x.2004.00096.x. [DOI] [PubMed] [Google Scholar]
- Corvin AP, Morris DW, McGhee K, Schwaiger S, Scully P, Quinn J, et al. Confirmation and refinement of an ‘at-risk’ haplotype for schizophrenia suggests the EST cluster, Hs.97362, as a potential susceptibility gene at the Neuregulin-1 locus. Mol Psychiatry. 2004;9:208–213. doi: 10.1038/sj.mp.4001412. [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K, Bacanu S-A. Unbiased methods for population-based association studies. Genet Epidemiol. 2001;21:273–284. doi: 10.1002/gepi.1034. [DOI] [PubMed] [Google Scholar]
- De Vries L, Zheng B, Fischer T, Elenko E, Farquhar MG. The regulator of G protein signaling family. Annu Rev Pharmacol Toxicol. 2000;40:235–271. doi: 10.1146/annurev.pharmtox.40.1.235. [DOI] [PubMed] [Google Scholar]
- Dubertret C, Gorwood P, Ades J, Feingold J, Schwartz JC, Sokoloff P. Meta-analysis of DRD3 gene and schizophrenia: Ethnic heterogeneity and significant association in Caucasians. Am J Med Genet. 1998;81:318–322. doi: 10.1002/(sici)1096-8628(19980710)81:4<318::aid-ajmg8>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Gscheidle T, Weirich M, Bigelow LB, Weinberger DR. Relative risk of attention deficits in siblings of patients with schizophrenia. Am J Psychiatry. 2000;157:1309–1316. doi: 10.1176/appi.ajp.157.8.1309. [DOI] [PubMed] [Google Scholar]
- Erdely HA, Lahti RA, Lopez MB, Myers CS, Roberts RC, Tamminga CA, et al. Regional expression of RGS4 mRNA in human brain. Eur J Neurosci. 2004;19:3125–3128. doi: 10.1111/j.0953-816X.2004.03364.x. [DOI] [PubMed] [Google Scholar]
- Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfeld M, Cohen D, et al. Genetic analysis of case/control data using estimated haplotype frequencies: Application to APOE locus variation and Alzheimer’s disease. Genome Res. 2001;11:143–151. doi: 10.1101/gr.148401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamma F, Faraone SV, Glatt SJ, Yeh YC, Tsuang MT. Meta-analysis shows schizophrenia is not associated with the 40-base-pair repeat polymorphism of the dopamine transporter gene. Schizophr Res. 2005;73:55–58. doi: 10.1016/j.schres.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Glatt SJ, Faraone SV, Tsuang MT. Association between a functional catechol O-methyltransferase gene polymorphism and schizophrenia: Meta-analysis of case-control and family-based studies. Am J Psychiatry. 2003a;160:469–476. doi: 10.1176/appi.ajp.160.3.469. [DOI] [PubMed] [Google Scholar]
- Glatt SJ, Faraone SV, Tsuang MT. Meta-analysis identifies an association between the dopamine D2 receptor gene and schizophrenia. Mol Psychiatry. 2003b;8:911–915. doi: 10.1038/sj.mp.4001321. [DOI] [PubMed] [Google Scholar]
- Glatt SJ, Wang RS, Yeh YC, Tsuang MT, Faraone SV. Five NOTCH4 polymorphisms show weak evidence for association with schizophrenia: Evidence from meta-analyses. Schizophr Res. 2005;73:281–290. doi: 10.1016/j.schres.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Gordon D, Matise TC, Heath SC, Ott J. Power loss for multiallelic transmission/disequilibrium test when errors introduced: GAW11 simulated data. Genet Epidemiol. 1999;17(suppl 1):S587–S592. doi: 10.1002/gepi.1370170795. [DOI] [PubMed] [Google Scholar]
- Goudet J. Fstat version 1.2: a computer program to calculate Fstatistics. J Heredity. 1995;86:485–486. [Google Scholar]
- Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- Iwata N, Suzuki T, Ikeda M, Kitajima T, Yamanouchi Y, Inada T, et al. No association with the neuregulin 1 haplotype to Japanese schizophrenia. Mol Psychiatry. 2004;9:126–127. doi: 10.1038/sj.mp.4001456. [DOI] [PubMed] [Google Scholar]
- Jonsson EG, Flyckt L, Burgert E, Crocq MA, Forslund K, Mattila-Evenden M, et al. Dopamine D3 receptor gene Ser9Gly variant and schizophrenia: Association study and meta-analysis. Psychiatr Genet. 2003;13:1–12. doi: 10.1097/00041444-200303000-00001. [DOI] [PubMed] [Google Scholar]
- Jonsson EG, Kaiser R, Brockmoller J, Nimgaonkar VL, Crocq MA. Meta-analysis of the dopamine D3 receptor gene (DRD3) Ser9Gly variant and schizophrenia. Psychiatr Genet. 2004;14:9–12. doi: 10.1097/00041444-200403000-00002. [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and non-parametric linkage analysis: A unified multipoint approach. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- Laird N, Horvath S, Xu X. Implementing a unified approach to family based tests of association. Genet Epidemiol. 2000;19(suppl 1):536–542. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Li T, Stefansson H, Gudfinnsson E, Cai G, Liu X, Murray RM, et al. Identification of a novel neuregulin 1 at-risk haplotype in Han schizophrenia Chinese patients, but no association with the Icelandic/Scottish risk haplotype. Mol Psychiatry. 2004;9(7):698–704. doi: 10.1038/sj.mp.4001485. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001;6:293–301. doi: 10.1038/sj.mp.4000866. [DOI] [PubMed] [Google Scholar]
- Mitchell AA, Cutler DJ, Chakravarti A. Undetected genotyping errors cause apparent overtransmission of common alleles in the transmission/disequilibrium test. Am J Hum Genet. 2003;72:598–610. doi: 10.1086/368203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DW, McGhee KA, Schwaiger S, Scully P, Quinn J, Meagher D, et al. No evidence for association of the dysbindin gene [DTNBP1] with schizophrenia in an Irish population-based study. Schizophr Res. 2003;60:167–172. doi: 10.1016/s0920-9964(02)00527-3. [DOI] [PubMed] [Google Scholar]
- Morris DW, Rodgers A, McGhee KA, Schwaiger S, Scully P, Quinn J, et al. Confirming RGS4 as a susceptibility gene for schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2004;125:50–53. doi: 10.1002/ajmg.b.20109. [DOI] [PubMed] [Google Scholar]
- Morton NE. Genetic structure of forensic populations. Proc Natl Acad Sci U S A. 1992;89:2556–2560. doi: 10.1073/pnas.89.7.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. PedCheck: A program for identification of genotype incompatibilties in linakge analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryshen TL, Middleton FA, Kirby A, Aldinger KA, Purcell S, Tahl AR, et al. Support for involvement of neuregulin 1 in schizophrenia pathophysiology. Mol Psychiatry. 2005;10(4):366–374. 328. doi: 10.1038/sj.mp.4001608. [DOI] [PubMed] [Google Scholar]
- Roeder K, Bacanu SA, Sonpar V, Zhang X, Devlin B. Analysis of single-locus tests to detect gene/disease associations. Genet Epidemiol. 2005;28(3):207–219. doi: 10.1002/gepi.20050. [DOI] [PubMed] [Google Scholar]
- Schwab SG, Knapp M, Mondabon S, Hallmayer J, Borrmann-Hassenbach M, Albus M, et al. Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an additional sample of triad families. Am J Hum Genet. 2003;72:185–190. doi: 10.1086/345463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltman H, Roeder K, Devlin B. Transmission/disequilibrium test meets measured haplotype analysis: Family-based association analysis guided by evolution of haplotypes. Am J Hum Genet. 2001;68:1250–1263. doi: 10.1086/320110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltman H, Roeder K, Devlin B. Evolutionary-based association analysis using haplotype data. Genet Epidemiol. 2003;25:48–58. doi: 10.1002/gepi.10246. [DOI] [PubMed] [Google Scholar]
- Shirts BH, Nimgaonkar V. The genes for schizophrenia: Finally a breakthrough? Curr Psychiatry Rep. 2004;6:303–312. doi: 10.1007/s11920-004-0081-1. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Sarginson J, Kong A, Yates P, Steinthorsdottir V, Gudfinnsson E, et al. Association of neuregulin 1 with schizophrenia confirmed in a Scottish population. Am J Hum Genet. 2003;72:83–87. doi: 10.1086/345442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71:337–348. doi: 10.1086/341750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang JX, Chen WY, He G, Zhou J, Gu NF, Feng GY, et al. Polymorphisms within 5′ end of the Neuregulin 1 gene are genetically associated with schizophrenia in the Chinese population. Mol Psychiatry. 2004;9:11–12. doi: 10.1038/sj.mp.4001436. [DOI] [PubMed] [Google Scholar]
- Tang JX, Zhou J, Fan JB, Li WX, Shi YY, Gu NF, et al. Family-based association study of DTNBP1 in 6p22.3 and schizophrenia. Mol Psychiatry. 2003;8(12):1008. doi: 10.1038/sj.mp.4001287. [DOI] [PubMed] [Google Scholar]
- Van Den Bogaert A, Schumacher J, Schulze TG, Otte AC, Ohlraun S, Kovalenko S, et al. The DTNBP1 (dysbindin) gene contributes to schizophrenia, depending on family history of the disease. Am J Hum Genet. 2003;73:1438–1443. doi: 10.1086/379928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oord EJ, Jiang Y, Riley BP, Kendler KS, Chen X. FP-TDI SNP scoring by manual and statistical procedures: A study of error rates and types. Biotechniques. 2003a;34:610–616. 618–620. doi: 10.2144/03343dd04. 622 passim. [DOI] [PubMed] [Google Scholar]
- van den Oord EJ, Sullivan PF, Jiang Y, Walsh D, O’Neill FA, Kendler KS, et al. Identification of a high-risk haplotype for the dystrobrevin binding protein 1 (DTNBP1) gene in the Irish study of high-density schizophrenia families. Mol Psychiatry. 2003b;8:499–510. doi: 10.1038/sj.mp.4001263. [DOI] [PubMed] [Google Scholar]
- Weir BS, Hill WG. Estimating F-statistics. Annu Rev Genet. 2002;36:721–750. doi: 10.1146/annurev.genet.36.050802.093940. [DOI] [PubMed] [Google Scholar]
- Williams NM, Preece A, Morris DW, Spurlock G, Bray NJ, Stephens M, et al. Identification in 2 independent samples of a novel schizophrenia risk haplotype of the dystrobrevin binding protein gene (DTNBP1) Arch Gen Psychiatry. 2004a;61:336–344. doi: 10.1001/archpsyc.61.4.336. [DOI] [PubMed] [Google Scholar]
- Williams NM, Preece A, Spurlock G, Norton N, Williams HJ, McCreadie RG, et al. Support for RGS4 as a susceptibility gene for schizophrenia. Biol Psychiatry. 2004b;55:192–195. doi: 10.1016/j.biopsych.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Williams NM, Preece A, Spurlock G, Norton N, Williams HJ, Zammit S, et al. Support for genetic variation in neuregulin 1 and susceptibility to schizophrenia. Mol Psychiatry. 2003;8:485–487. doi: 10.1038/sj.mp.4001348. [DOI] [PubMed] [Google Scholar]
- Wright S. Genetical structure of populations. Nature. 1950;166:247–249. doi: 10.1038/166247a0. [DOI] [PubMed] [Google Scholar]
- Yang JZ, Si TM, Ruan Y, Ling YS, Han YH, Wang XL, et al. Association study of neuregulin 1 gene with schizophrenia. Mol Psychiatry. 2003;8:706–709. doi: 10.1038/sj.mp.4001377. [DOI] [PubMed] [Google Scholar]
- Zhao X, Shi Y, Tang J, Tang R, Yu L, Gu N, et al. A case control and family based association study of the neuregulin1 gene and schizophrenia. J Med Genet. 2004;41:31–34. doi: 10.1136/jmg.2003.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]