

Abstract
We used one-dimensional photochemical and radiative transfer models to study the potential of organic sulfur compounds (CS2, OCS, CH3SH, CH3SCH3, and CH3S2CH3) to act as remotely detectable biosignatures in anoxic exoplanetary atmospheres. Concentrations of organic sulfur gases were predicted for various biogenic sulfur fluxes into anoxic atmospheres and were found to increase with decreasing UV fluxes. Dimethyl sulfide (CH3SCH3, or DMS) and dimethyl disulfide (CH3S2CH3, or DMDS) concentrations could increase to remotely detectable levels, but only in cases of extremely low UV fluxes, which may occur in the habitable zone of an inactive M dwarf. The most detectable feature of organic sulfur gases is an indirect one that results from an increase in ethane (C2H6) over that which would be predicted based on the planet's methane (CH4) concentration. Thus, a characterization mission could detect these organic sulfur gases—and therefore the life that produces them—if it could sufficiently quantify the ethane and methane in the exoplanet's atmosphere. Key Words: Exoplanets—Biosignatures—Anoxic atmospheres—Planetary atmospheres—Remote life detection—Photochemistry. Astrobiology 11, 419–441.
1. Introduction
The search for life may soon expand beyond the boundaries of our solar system via the detection of spectral features of “biosignature” gases on extrasolar planets (European Space Agency, 2010; Jet Propulsion Laboratory, 2010; New Worlds Observer Team, 2010). For a gas to be a biosignature it must have a biological production rate that far outpaces abiotic sources and an atmospheric lifetime that allows it to build up to detectable levels. To be detectable, the biosignature gas must have spectral features that are (1) within a wavelength region that can be covered by instrumentation, (2) larger than the signal-to-noise ratio (S/N) for these instruments, and (3) distinguishable from other spectral features.
For biospheres in which primary productivity is dominated by oxygenic photosynthesis (henceforth referred to as “oxic” biospheres), a number of gases have been identified that meet these criteria: oxygen (O2), ozone (O3), or both in the presence of reduced species such as methane (CH4) (Lovelock, 1965; Des Marais et al., 2002); nitrous oxide (N2O) (Sagan et al., 1993); and methyl chloride (CH3Cl) (Segura et al., 2005). The latter two gases are more difficult to detect in Earth's present atmosphere than are the first two; however, they might be more visible in the atmospheres of oxic Earth-like planets orbiting M stars due to longer atmospheric lifetimes resulting from lower photolysis rates (Segura et al., 2005).
Other biosignatures are needed for detection of “anoxic biospheres” that harbor life but not detectable amounts of atmospheric O2 and O3. Biogenic CH4 could be abundant enough to be detectable in such an atmosphere (Kasting et al., 1983, 2001; Kasting, 2005; Kharecha et al., 2005; Kaltenegger et al., 2007), but its interpretation would be ambiguous because abiotic processes such as serpentinization can also produce CH4 (Berndt et al., 1996; Kasting and Catling, 2003).
From the early history of life on Earth, we know that anoxic biospheres are possible. Studies of early Earth suggest that life was present well before significant O2 accumulated in the atmosphere (Schopf, 1983; Holland, 1984; Farquhar and Wing, 2003; Westall, 2005; Farquhar et al., 2007). This period had vigorous biological activity without significant O2 buildup and may have lasted as long as 1.5 billion years, approximately one-third of Earth's history. This suggests that planets with life, but without O2/O3, could represent a large fraction of inhabited planets. Thus, the absence of O2/O3 should not be taken as evidence that life does not exist on a planet's surface.
Furthermore, some planets and biospheres will not exhibit the more general feature of photochemical disequilibrium previously proposed as a universal biosignature (Lederberg, 1965; Lovelock, 1965; Des Marais et al., 2002). Unlike Earth's modern-day ecosystem, global anoxic ecosystems may drive an atmosphere toward equilibrium. For example, in the anoxic Archean biospheres considered by Kharecha et al. (2005), methanogens and acetogens combine H2 and CO with CO2 and H2O to produce CH4. They can make a metabolic living by doing this because CH4 has a lower Gibbs free energy and hence is thermodynamically stable in such a system. The biogenic gases released from such a biosphere result from a drive toward equilibrium, not disequilibrium. Because cases like these could complicate interpretation, it is important to identify additional biosignature gases that might be signs of anoxic biospheres. In this paper, we test the ability of various gases with carbon-sulfur bonds to act as remotely detectable biosignatures for anoxic, inhabited surface environments.
The biosignature potential of S-bearing gases was reviewed by Pilcher (2003), who focused on gases with bonds between methyl groups (−CH3) and sulfur: methanethiol (CH3SH, also known as methyl mercaptan), dimethyl sulfide (CH3SCH3 or DMS), and dimethyl disulfide (CH3S2CH3 or DMDS). More recently, Vance et al. (2011) suggested that CH3SH could be used as an in situ signature for life on Mars. On modern Earth, the production of these species is dominated by biota, but they are rapidly destroyed by photolysis and by reaction with hydroxyl (OH) radicals (Kettle et al., 2001), and do not build up to concentrations detectable across interstellar distances. In this work, we consider these gases, along with carbon disulfide (CS2) and carbonyl sulfide (OCS, sometimes abbreviated in other work as COS), two other biogenic gases that contain carbon-sulfur bonds. These two species—particularly OCS—also have volcanic and photochemical sources, but they are far smaller than biological fluxes. We henceforth use the term “Sorg” as shorthand to refer to the entire suite of biologically produced species with carbon-sulfur bonds (DMS, DMDS, CH3SH, CS2, and OCS). Although hydrogen sulfide (H2S) is another S-bearing gas produced by biota, large quantities of this species enter the atmosphere via volcanism. Thus, we do not consider it here as a biosignature. However, we do consider the possibility that volcanic H2S could act as a “false positive” for biogenic Sorg, as this abiotic H2S could react in the atmosphere to form Sorg species. Other work has explored the spectral signatures of sulfur dioxide (SO2) and H2S in detail (Kaltenegger and Sasselov, 2010), so we limit our discussion to their potential to be false positives for biological Sorg production. No study to date has predicted the concentrations of all the Sorg species in an anoxic atmosphere, nor has any study predicted the spectral features associated with these gases. We used a photochemical model to calculate vertical profiles of these gases for a variety of astronomical and biological contexts, and used a radiative transfer model to predict the spectral features consistent with those profiles.
2. Methods
2.1. Photochemical code
We modified the one-dimensional (altitude), low-O2 photochemical code originally developed by Kasting et al. (1979) to study the anoxic early Earth. The numerics of this model are described by Kasting and Ackerman (1985), and the chemistry was most recently modified by Pavlov et al. (2001). We have updated this code, adding seven long-lived chemical species that have lifetimes longer than the time scale for vertical mixing: CH3SH, DMS, DMDS, OCS, CS2, methylthiol (CH3S), and carbon monosulfide (CS). We also added three short-lived species, which are solved in photochemical equilibrium without considering vertical transport: excited-state CS2, OCS2, and HCS. These 10 species were incorporated into the chemical scheme by adding 73 chemical reactions. The current model contains 83 chemical species, 46 of which are long lived, connected by 433 chemical reactions. Additionally, many of the 360 reactions from prior work have updated reaction rate constants. A complete list of model reactions, reaction rate constants, and references can be found in Table 1.
Table 1.
List of Reactions in the Photochemical Code, along with the Reaction Rate Constants Used and a Source for the Reaction Rate Constant
| Rxn. # | Reaction | Reaction rate constant | Reference |
|---|---|---|---|
| 1 | OCS+CH→CO+HCS | 1.99·10−10×e−190/T | Zabarnick et al.,1989 |
| 2 | OCS+H→CO+HS | 9.07·10−12×e−1940/T | Lee et al.,1977 |
| 3 | OCS+O→S+CO2 | 8.3·10−11×e−5530/T | Singleton and Cvetanovic, 1988 |
| 4 | OCS+O→SO+CO | 2.1·10−11×e−2200/T | Toon et al.,1987 |
| 5 | OCS+OH→CO2+HS | 1.1·10−13×e−1200/T | Atkinson et al.,2004 |
| 6 | OCS+OH→HS+CO2 | 1.1·10−13×e−1200/T | Atkinson et al.,2004 |
| 7 | OCS+S→CO+S2 | 1.5·10−10×e−1830/T | Schofield, 1973 |
| 8 | OCS+S+M→OCS2+M | 8.3·10−33×den | Basco and Pearson, 1967 |
| 9 | OCS2+CO→OCS+OCS | 3.0·10−12 | Zahnle et al.,2006 |
| 10 | OCS2+S→OCS+S2 | 2.0·10−11 | Zahnle et al.,2006 |
| 11 | C2H6S+CH3→CH4+C2H4+HS | 6.92·10−13×e−4610/T | Arthur and Lee, 1976 |
| 12 | C2H6S+H→C2H5+H2S | 8.49·10−12×e−1200/T | Lam et al.,1989 |
| 13 | C2H6S+H→CH3SH+CH3 | 4.81·10−12×e−1100/T×(T/300)1.7 | Zhang et al.,2005 |
| 14 | C2H6S+H→H2+C2H4+HS | 8.34·10−12×e−2212/T×(T/300)1.6 | Zhang et al.,2005 |
| 15 | C2H6S+OH→H2O+C2H4+HS | 1.13·10−11×e−253/T | Atkinson et al.,2004 |
| 16 | C2H6S2+H→CH3SH+CH3S | 9.47·10−12×e−50/T | Ekwenchi et al.,1980 |
| 17 | CH3+HS→CH3SH | 1.66·10−11 | Shum and Benson, 1985 |
| 18 | CH3S+CH3S→C2H6S2 | 4.00·10−11 | Anastasi et al.,1991 |
| 19 | CH3S+CO→CH3+OCS | 2.6·10−11×e−5940/T | Assumed same as k(CH3O+CO) |
| 20 | CH3S+CS→CH3+CS2 | 2.6·10−11×e−5940/T | Assumed same as k(CH3O+CO) |
| 21 | CH3S+H2O2→CH3SH+H2O | 3.01·10−13 | Turnipseed et al.,1996 |
| 22 | CH3S+HCS→CH3SH+CS | 1.18·10−12×e−910/T×(T/300)0.65 | Liu et al.,2006 |
| 23 | CH3S+HS→CH3SH+S | 1.66·10−11 | Assumed same as k(CH3+HS) |
| 24 | CH3SH+CH3→CH4+CH3S | 2.99·10−31 | Kerr and Trotman-Dickenson, 1957 |
| 25 | C2H6S+O→CH3+CH3+SO | 1.30·10−11×e−410/T×(T/298)1.1 | Sander et al.,2006 |
| 26 | CH3SH+O→CH3+HSO | 1.30·10−11×e−410/T×(T/298)1.1 | Assumed same as k(C2H6S+O) |
| 27 | C2H6S2+O→CH3+CH3S+SO | 3.90·10−11×e290/T×(T/298)1.1 | Sander et al.,2006 |
| 28 | 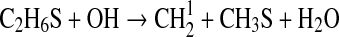 |
1.10·10−11×e−240/T×(T/298)1.1 | Sander et al.,2006 |
| 29 | C2H6S2+OH→CH3+CH3SH+SO | 6.00·10−11×e400/T×(T/298)1.2 | Sander et al.,2006 |
| 30 | CH3SH+OH→CH3S+H2O | 9.90·10−12×e360/T×(T/298)1.07 | Sander et al.,2006 |
| 31 | C2H6S+O→CH3+CH3+SO | 1.30·10−11×e−410/T×(T/298)1.1 | Sander et al.,2006 |
| 32 | CH3SH+O→CH3+HSO | 1.30·10−11×e−410/T×(T/298)1.1 | Assumed same as k(C2H6S+O) |
| 33 | CH3SH+H→CH3+H2S | 1.5·10−11×e−840/T | Amano et al.,1983 |
| 34 | CH3SH+H→H2+CH3S | 4.82·10−11×e−1310/T | Amano et al.,1983 |
| 35 | CH3SH+OH→H2O+CH3S | 9.9·10−12×e360/T | DeMore and Yung, 1982 |
| 36 | CH+CS2→HCS+CS | 3.49·10−10×e−40/T | Zabarnick et al.,1989 |
| 37 | CS+HS→CS2+H | 1.5·10−13×(1+0.6×den) | Assumed same as k(CO+OH) |
| 38 | CS+O→CO+S | 2.7·10−10×e−760/T | Atkinson et al.,2004 |
| 39 | CS+O2→CO+SO | 5·10−20 | Wine et al.,1981 |
| 40 | CS+O2→OCS+O | 4·10−19 | Wine et al.,1981 |
| 41 | CS+O3→CO+SO2 | 3·10−12 | Wine et al.,1981 |
| 42 | CS+O3→OCS+O2 | 3·10−12 | Wine et al.,1981 |
| 43 | CS+O3→SO+CO2 | 3·10−12 | Wine et al.,1981 |
| 44 | CS2+O→CO+S2 | 5.81·10−14 | Singleton and Cvetanovic, 1988 |
| 45 | CS2+O→OCS+S | 3·10−12×e−650/T | Toon et al.,1987 |
| 46 | CS2+O→SO+CS | 3.2·10−11×e−650/T | Toon et al.,1987 |
| 47 | CS2+OH→OCS+HS | 2·10−15 | Atkinson et al.,2004 |
| 48 | CS2+S→CS+S2 | 1.9·10−14×e−580/T×(T/300)3.97 | Woiki and Roth, 1995 |
| 49 | CS2+SO→OCS+S2 | 2.4·10−13×e−2370/T | Assumed same as k(SO*+O2) |
| 50 | 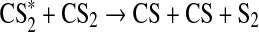 |
1·10−12 | Assumed same as 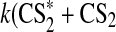
|
| 51 |  |
2.5·10−11 | Wine et al.,1981 |
| 52 | 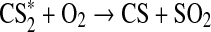 |
1·10−12 | Wine et al.,1981 |
| 53 | C+HS→CS+H | 4·10−11 | Assumed same as k(C+OH) |
| 54 | C+S2→CS+S | 3.3·10−11 | Assumed same as k(C+O2) |
| 55 | C2+S→C+CS | 5·10−11 | Assumed same as k(C2+O) |
| 56 | C2+S2→CS+CS | 1.5·10−11×e−550/T | Assumed same as k(C2+O2) |
| 57 | CH+S→CS+H | 9.5·10−11 | Assumed same as k(CH+CS2) |
| 58 | CH+S2→CS+HS | 5.9·10−11 | Assumed same as k(CH+O2) |
| 59 | 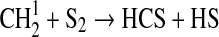 |
3·10−11 | Assumed same as 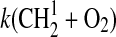
|
| 60 | CH3+HCS→CH4+CS | 8.2·10−11 | Assumed same as k(CH3+HCO) |
| 61 | H+CS+M→HCS+M | 2.0·10−33×e−850/T×den | Assumed same as k(H+CO) |
| 62 | H+HCS→H2+CS | 1.2·10−10 | Assumed same as k(H+HCO) |
| 63 | HS+CO→OCS+H | 4.2·10−14×e−7650/T | Kurbanov and Mamedov, 1995 |
| 64 | HS+HCS→H2S+CS | 5.0·10−11 | Assumed same as k(HS+HCO) |
| 65 | OCS+CH→CO+HCS | 1.99·10−10×e−190/T | Zabarnick et al.,1989 |
| 66 | S+CO+M→OCS+M | 6.5·10−33×e−2180/T×den | Assumed same as k(CO+O) |
| 67 | S+HCS→H+CS2 | 1.0·10−10 | Assumed same as k(O+HCO→H+CO2) |
| 68 | S+HCS→HS+CS | 5.0·10−11 | Assumed same as k(O+HCO→HS+CO) |
| 69 | 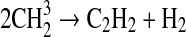 |
5.3·10−11 | Braun et al.,1970 |
| 70 | 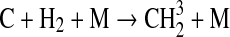 |
k0=8.75·10−31×e524/T | Zahnle, 1986 |
| k∞=8.3·10−1 | |||
| 71 | C+O2→CO+O | 3.3·10−11 | Donovan and Husain, 1970 |
| 72 | C+OH→CO+H | 4·10−11 | Giguere and Huebner, 1978 |
| 73 | C2+CH4→C2H+CH3 | 5.05·10−11×e−297/T | Pitts et al.,1982 |
| 74 | C2+H2→C2H+H | 1.77·10−10×e−1469/T | Pitts et al.,1982 |
| 75 | C2+O→C+CO | 5·10−11 | Prasad and Huntress, 1980 |
| 76 | C2+O2→CO+CO | 1.5·10−11×e−550/T | Baughcum and Oldenborg, 1984 |
| 77 | C2H+C2H2→HCAER+H | 1.5·10−10 | Stephens et al.,1987 |
| 78 | C2H+C2H6→C2H2+C2H5 | 3.6·10−11 | Lander et al.,1990 |
| 79 | C2H+C3H8→C2H2+C3H7 | 1.4·10−11 | Okabe, 1983 |
| 80 | C2H+CH2CCH2→HCAER+H | 1.5·10−10 | Pavlov et al.,2001 |
| 81 | C2H+CH4→C2H2+CH3 | 6.94·10−12×e−250/T | Allen et al.,1992; Lander et al.,1990 |
| 82 | C2H+H+M→C2H2+M | k0=2.64·10−26×e−721/T×(T/300)−3.1 | Tsang and Hampson, 1986 |
| k∞=3.0·10−10 | |||
| 83 | C2H+H2→C2H2+H | 5.58·10−11×e−1443/T | Allen et al.,1992; Stephens et al.,1987 |
| 84 | C2H+O→CO+CH | 1·10−10×e−250/T | Zahnle, 1986 |
| 85 | C2H+O2→CO+HCO | 2·10−11 | Brown and Laufer, 1981 |
| 86 | C2H2+H+M→C2H3+M | k0=2.6·10−31 | Romani et al.,1993 |
| k∞=8.3·10−11×e−1374/T | |||
| 87 |  |
2.9·10−11×e−1600/T | Zahnle, 1986 |
| 88 | C2H2+OH+M→C2H2OH+M | k0=5.5·10−30 | Sander et al.,2006 |
| k∞=8.3·10−13×(T/300)−2 | |||
| 89 | C2H2+OH+M→CH2CO+H+M | k0=5.8·10−31×e1258/T | Perry and Williamson, 1982 |
| k∞=1.4·10−12×e388/T | |||
| 90 | C2H2+OH→CO+CH3 | 2·10−12×e−250/T | Hampson and Garvin, 1977 |
| 91 | C2H2OH+H→H2+CH2CO | 3.3·10−11×e−2000/T | Miller et al.,1982 |
| 92 | C2H2OH+H→H2O+C2H2 | 5·10−11 | Miller et al.,1982 |
| 93 | C2H2OH+O→OH+CH2CO | 3.3·10−11×e−2000/T | Miller et al.,1982 |
| 94 | C2H2OH+OH→H2O+CH2CO | 1.7·10−11×e−1000/T | Miller et al.,1982 |
| 95 | C2H3+C2H3→C2H4+C2H2 | 2.4·10−11 | Fahr et al.,1991 |
| 96 | C2H3+C2H5→C2H4+C2H4 | 3·10−12 | Laufer et al.,1983 |
| 97 | C2H3+C2H5+M→CH3+C3H5+M | k0=1.9·10−27 | Romani et al.,1993 |
| k∞=2.5·10−11 | |||
| 98 | C2H3+C2H6→C2H4+C2H5 | 3·10−13×e−5170/T | Kasting et al.,1983 |
| 99 | C2H3+CH3→C2H2+CH4 | 34·10−11 | Fahr et al.,1991 |
| 100 | C2H3+CH3+M→C3H6+M | k0=1.3·10−22 | Raymond et al.,2006 |
| k∞=1.2·10−10 | |||
| 101 | C2H3+CH4→C2H4+CH3 | 2.4·10−24×e−2754/T×T4.02 | Tsang and Hampson, 1986 |
| 102 | C2H3+H→C2H2+H2 | 3.3·10−11 | Warnatz, 1984 |
| 103 | C2H3+H2→C2H4+H | 2.6·10−13×e−2646/T | Allen et al.,1992 |
| 104 | C2H3+O→CH2CO+H | 5.5·10−11 | Hoyermann et al.,1981 |
| 105 | C2H3+OH→C2H2+H2O | 8.3·10−12 | Benson and Haugen, 1967 |
| 106 | C2H4+H+M→C2H5+M | k0=2.15·10−29×e−349/T | Lightfoot and Pilling, 1987 |
| k∞=4.95·10−11×e−1051/T | |||
| 107 | C2H4+O→HCO+CH3 | 5.5·10−12×e−565/T | Hampson and Garvin, 1977 |
| 108 | C2H4+OH+M→C2H4OH+M | k0=1.0·10−28×(T/300)4.5 | Sander et al.,2006 |
| k∞=8.8·10−12×(T/300)0.85 | |||
| 109 | C2H4+OH→H2CO+CH3 | 2.2·10−12×e385/T | Hampson and Garvin, 1977 |
| 110 | C2H4OH+H→H2+CH3CHO | 3.3·10−11×e−2000/T | Zahnle and Kasting, 1986 |
| 111 | C2H4OH+H→H2O+C2H4 | 5·10−11 | Miller et al.,1982 |
| 112 | C2H4OH+O→OH+CH3CHO | 3.3·10−11×e−2000/T | Zahnle and Kasting, 1986 |
| 113 | C2H4OH+OH→H2O+CH3CHO | 1.7·10−11×e−1000/T | Zahnle and Kasting, 1986 |
| 114 | C2H5+C2H3→C2H6+C2H2 | 6·10−12 | Laufer et al.,1983 |
| 115 | C2H5+C2H5→C2H6+C2H4 | 2.3·10−12 | Tsang and Hampson, 1986 |
| 116 | C2H5+CH3→C2H4+CH4 | 1.88·10−12×(T/300)−0.5 | Romani et al.,1993 |
| 117 | C2H5+CH3+M→C3H8+M | k0=3.9·10−10×(T/300)2.5 | Romani et al.,1993 |
| k∞=1.4·10−8×(T/300)0.5 | |||
| 118 | C2H5+H→C2H4+H2 | 3·10−12 | Tsang and Hampson, 1986 |
| 119 | C2H5+H+M→C2H6+M | k0=5.5·10−23×e−1040/T | Gladstone et al.,1996 |
| k∞=1.5·10−10 | |||
| 120 | C2H5+H→CH3+CH3 | 7.95·10−11 | Gladstone et al.,1996 |
| 121 | C2H5+HCO→C2H6+CO | 5·10−11 | Pavlov et al.,2001 |
| 122 | C2H5+HNO→C2H6+NO | 3·10−14 | Pavlov et al.,2001 |
| 123 | C2H5+O→CH3+HCO+H | 1.1·10−10 | Pavlov et al.,2001 |
| 124 | C2H5+O→CH3CHO+H | 1.33·10−10 | Tsang and Hampson, 1986 |
| 125 | C2H5+O2+M→CH3+HCO+OH+M | k0=1.5·10−28×(T/300)3.0 | Sander et al.,2006 |
| k∞=8·10−12 | |||
| 126 | C2H5+OH→CH3CHO+H2 | 1·10−10 | Pavlov et al.,2001 |
| 127 | C2H5+OH→C2H4+H2O | 4.0·10−11 | Pavlov et al.,2001 |
| 128 | C2H6+O→C2H5+OH | 8.62·10−12×e−2920/T×(T/300)1.5 | Baulch et al.,1994 |
| 129 | C2H6+O1D→C2H5+OH | 6.29·10−10 | Matsumi et al.,1993 |
| 130 | C2H6+OH→C2H5+H2O | 8.54·10−12×e−1070/T | Sander et al.,2006 |
| 131 | C3H2+H+M→C3H3+M | k0=1.7·10−26 | Yung et al.,1984 |
| k∞=1.5·10−10 | |||
| 132 | C3H3+H+M→CH2CCH2+M | k0=1.7·10−26 | Yung et al.,1984 |
| k∞=1.5·10−10 | |||
| 133 | C3H3+H+M→CH3C2H+M | k0=1.7·10−26 | Yung et al.,1984 |
| k∞=1.5·10−10 | |||
| 134 | C3H5+CH3→CH2CCH2+CH4 | 4.5·10−12 | Yung et al.,1984 |
| 135 | C3H5+CH3→CH3C2H+CH4 | 4.5·10−12 | Yung et al.,1984 |
| 136 | C3H5+H+M→C3H6+M | k0=1.0·10−28 | Yung et al.,1984 |
| k∞=1.0·10−11 | |||
| 137 | C3H5+H→CH2CCH2+H2 | 1.5·10−11 | Yung et al.,1984 |
| 138 | C3H5+H→CH3C2H+H2 | 1.5·10−11 | Yung et al.,1984 |
| 139 | C3H5+H→CH4+C2H2 | 1.5·10−11 | Yung et al.,1984 |
| 140 | C3H6+H+M→C3H7+M | k0=2.15·10−29×e−349/T | Pavlov et al.,2001 |
| k∞=4.95·10−11×e−1051/T | Assumed same as k(C2H4+H) | ||
| 141 | C3H6+O→CH3+CH3CO | 4.1·10−12×e−38/T | Hampson and Garvin, 1977 |
| 142 | C3H6+OH→CH3CHO+CH3 | 4.1·10−12×e540/T | Hampson and Garvin, 1977 |
| 143 | C3H7+CH3→C3H6+CH4 | 2.5·10−12×e−200/T | Yung et al.,1984 |
| 144 | C3H7+H→CH3+C2H5 | 7.95·10−11×e−127/T | Pavlov et al.,2001 |
| 145 | C3H7+O→C2H5CHO+H | 1.1·10−10 | Pavlov et al.,2001 |
| 146 | C3H7+OH→C2H5CHO+H2 | 1.1·10−10 | Pavlov et al.,2001 |
| 147 | C3H8+O+M→C3H7+OH+M | k0=1.6·10−11×e−2900/T | Hampson and Garvin, 1977 |
| k∞=2.2·10−11×e−2200/T | |||
| 148 | C3H8+O1D→C3H7+OH | 1.4·10−10 | Pavlov et al.,2001 |
| 149 | C3H8+OH→C3H7+H2O | 8.6·10−12×e−615/T | Sander et al.,2006 |
| 150 | CH+C2H2+M→C3H2+H+M | k0=2.15·10−29×e−349/T | Romani et al.,1993 |
| k∞=4.95·10−11×e−1051/T | |||
| 151 | CH+C2H4+M→CH2CCH2+H+M | k0=1.75·10−10×e61/T | Romani et al.,1993 |
| k∞=5.3·10−10 | |||
| 152 | CH+C2H4+M→CH3C2H+H+M | k0=1.75·10−10×e61/T | Romani et al.,1993 |
| k∞=5.3·10−10 | |||
| 153 | CH+CH4+M→C2H4+H+M | k0=2.5·10−11×e200/T | Romani et al.,1993 |
| k∞=1.7·10−10 | |||
| 154 | CH+CO2→HCO+CO | 5.9·10−12×e−350/T | Berman et al.,1982 |
| 155 | CH+H→C+H2 | 1.4·10−11 | Becker et al.,1989 |
| 156 |  |
2.38·10−10×e−1760/T | Zabarnick et al.,1986 |
| 157 | CH+H2+M→CH3+M | k0=8.75·10−31×e524/T | Romani et al.,1993 |
| k∞=8.3·10−11 | |||
| 158 | CH+O→CO+H | 9.5·10−11 | Messing et al.,1981 |
| 159 | CH+O2→CO+OH | 5.9·10−11 | Butler et al.,1981 |
| 160 |  |
7.14·10−12×e−5050/T | Böhland et al.,1985 |
| 161 |  |
1·10−12 | Zahnle, 1986 |
| 162 | 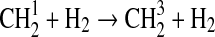 |
1.26·10−11 | Romani et al.,1993 |
| 163 | 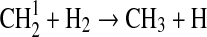 |
5·10−15 | Tsang and Hampson, 1986 |
| 164 | 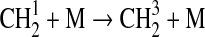 |
8.8·10−12 | Ashfold et al.,1981 |
| 165 | 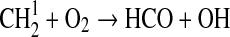 |
3·10−11 | Ashfold et al.,1981 |
| 166 | 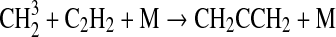 |
k0=3.8·10−25 | Laufer, 1981; Laufer et al.,1983 |
| k∞=3.7·10−12 | |||
| 167 | 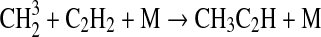 |
k0=3.8·10−25 | Laufer, 1981; Laufer et al.,1983 |
| k∞=2.2·10−12 | |||
| 168 | 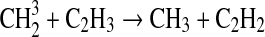 |
3·10−11 | Tsang and Hampson, 1986 |
| 169 | 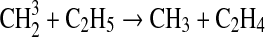 |
3·10−11 | Tsang and Hampson, 1986 |
| 170 | 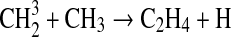 |
7·10−11 | Tsang and Hampson, 1986 |
| 171 | 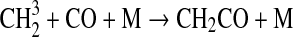 |
k0=1.0·10−28 | Yung et al.,1984 |
| k∞=1.0·10−15 | |||
| 172 | 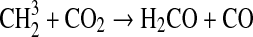 |
3.9·10−14 | Laufer, 1981 |
| 173 | 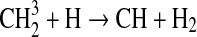 |
4.7·10−10×e−370/T | Zabarnick et al.,1986 |
| 174 | 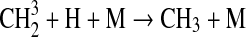 |
k0=3.1·10−30×e457/T | Gladstone et al.,1996 |
| k∞=1.5·10−10 | |||
| 175 | 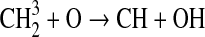 |
8·10−12 | Huebner and Giguere, 1980 |
| 176 | 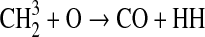 |
8.3·10−11 | Homann and Wellmann, 1983 |
| 177 | 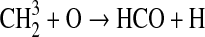 |
1·10−11 | Huebner and Giguere, 1980 |
| 178 | 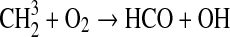 |
4.1·10−11×e −750/T | Baulch et al.,1994 |
| 179 | CH2CCH2+H→C3H5 | k0=8.9·10−29×e−1225/T×(T/300)−2.0 | Yung et al.,1984 |
| k∞=1.4·10−11×e−1000/T | |||
| 180 | CH2CCH2+H→CH3+C2H2 | k0=8.9·10−29×e−1225/T×(T/300)−2.0 | Yung et al.,1984 |
| k∞=9.7·10−13×e−1550/T | |||
| 181 | CH2CCH2+H→CH3C2H+H | 1·10−11×e−1000/T | Yung et al.,1984 |
| 182 | CH2CO+H→CH3+CO | 1.9·10−11×e−1725/T | Michael et al.,1979 |
| 183 | CH2CO+O→H2CO+CO | 3.3·10−11 | Lee, 1980; Miller et al.,1982 |
| 184 | CH3+C2H3→C3H5+H | 2.4·10−13 | Romani et al.,1993 |
| 185 | CH3+CH3+M→C2H6+M | k0=4.0·10−24×e−1390/T×(T/300)−7.0 | Wagner and Wardlaw, 1988 |
| k∞=1.79·10−10×e−329/T | |||
| 186 | CH3+CO+M→CH3CO+M | 1.4·10−32×e−3000/T×den | Watkins and Word, 1974 |
| 187 | CH3+H+M→CH4+M | k0=6.0·10−28×(T/298)−1.80 | Baulch et al.,1994; Tsang and Hampson, 1986 |
| k∞=2.0·10−10×(T/298)−0.40 | |||
| 188 | CH3+H2CO→CH4+HCO | 1.60·10−16×e899/T×(T/298)6.10 | Baulch et al.,1994 |
| 189 | CH3+HCO→CH4+CO | 2.01·10−10 | Tsang and Hampson, 1986 |
| 190 | CH3+HNO→CH4+NO | 1.85·10−11×e−176/T×(T/298)0.6 | Choi and Lin, 2005 |
| 191 | CH3+O→H2CO+H | 1.1·10−10 | Sander et al.,2006 |
| 192 | CH3+O2→H2CO+OH | k0=4.0·10−31×(T/300)−3.6 | Sander et al.,2006 |
| k∞=1.2·10−12×(T/300)−1.1 | |||
| 193 | CH3+O3→H2CO+HO2 | 5.4·10−12×e−220/T | Sander et al.,2006 |
| 194 | CH3+OH→CH3O+H | 9.3·10−11×e−1606/T×(T/298) | Jasper et al.,2007 |
| 195 | CH3+OH→CO+H2+H2 | 6.7·10−12 | Fenimore, 1969 |
| 196 | CH3C2H+H+M→C3H5+M | k0=8.88·10−29×e−1225/T×(T/300)−2 | Yung et al.,1984 |
| k∞=9.7·10−12×e−1550/T | |||
| 197 | CH3C2H+H→CH3+C2H2 | k0=8.88·10−29×e−1225/T×(T/300)−2 | Whytock et al.,1976 |
| k∞=9.7·10−12×e−1550/T | |||
| 198 | CH3CHO+CH3→CH3CO+CH4 | 2.8·10−11×e−1540/T | Zahnle, 1986 |
| 199 | CH3CHO+H→CH3CO+H2 | 2.8·10−11×e−1540/T | Zahnle, 1986 |
| 200 | CH3CHO+O→CH3CO+OH | 5.8·10−13 | Washida, 1981 |
| 201 | CH3CHO+OH→CH3CO+H2O | 1.6·10−11 | Niki et al.,1978 |
| 202 | CH3CO+CH3→C2H6+CO | 5.4·10−11 | Adachi et al.,1981 |
| 203 | CH3CO+CH3→CH4+CH2CO | 8.6·10−11 | Adachi et al.,1981 |
| 204 | CH3CO+H→CH4+CO | 1·10−10 | Zahnle, 1986 |
| 205 | CH3CO+O→H2CO+HCO | 5·10−11 | Zahnle, 1986 |
| 206 | CH3O+CO→CH3+CO2 | 2.6·10−11×e−5940/T | Wen et al.,1989 |
| 207 | CH3O2+H→CH4+O2 | 1.6·10−10 | Tsang and Hampson, 1986 |
| 208 | CH3O2+H→H2O+H2CO | 1·10−11 | Zahnle et al.,2006 |
| 209 | CH3O2+O→H2CO+HO2 | 1·10−11 | Vaghjiani and Ravishankara, 1990 |
| 210 | CH4+HS→CH3+H2S | 2.99·10−31 | Kerr and Trotman-Dickenson, 1957 |
| 211 | CH4+O→CH3+OH | 8.75·10−12×e−4330/T×(T/298)1.5 | Tsang and Hampson, 1986 |
| 212 | CH4+O1D→CH3+OH | 1.28·10−10 | Sander et al.,2006 |
| 213 | CH4+O1D→H2CO+H2 | 2.25·10−11 | Sander et al.,2006 |
| 214 | CH4+OH→CH3+H2O | 2.45·10−12×e−1775/T | Sander et al.,2006 |
| 215 | CO+O+M→CO2+M | 1.7·10−33×e−1515/T×den | Tsang and Hampson, 1986 |
| 216 | CO+OH→CO2+H | 1.5·10−13×(1+0.6×den) | Sander et al.,2006 |
| 217 | H+CO+M→HCO+M | 5.29·10−34×e−100/T×den | Baulch et al.,1994 |
| 218 | H+H+M→H2+M | 8.85·10−33×(T/298)−0.6×den | Baulch et al.,1994 |
| 219 | H+HCO→H2+CO | 1.5·10−10 | Baulch et al.,1992 |
| 220 | H+HNO→H2+NO | 3.01·10−11×e500/T | Tsang and Herron, 1991 |
| 221 | H+HO2→H2+O2 | 6.9·10−12 | Sander et al.,2006 |
| 222 | H+HO2→H2O+O | 1.62·10−12 | Sander et al.,2006 |
| 223 | H+HO2→OH+OH | 7.29·10−11 | Sander et al.,2006 |
| 224 | H+NO+M→HNO+M | 2.1·10−32×(T/298)1.00×den | Hampson and Garvin, 1977 |
| 225 | H+O2+M→HO2+M | 5.7·10−32×7.5·10−11×(T/298)1.6 | Sander et al.,2006 |
| 226 | H+O3→OH+O2 | 1.4·10−10×e−470/T | Sander et al.,2006 |
| 227 | H+OH+M→H2O+M | 6.8·10−31×(T/300)−2×den | McEwan and Phillips, 1975 |
| 228 | H+SO+M→HSO+M | k0=5.7·10−32×(T/298)1.6 | Kasting, 1990 |
| k∞=7.5·10−11 | |||
| 229 | H2+O→OH+H | 1.34·10−15×e−1460/T×(T/298)6.52 | Robie et al.,1990 |
| 230 | H2+O1D→OH+H | 1.1·10−11 | Sander et al.,2006 |
| 231 | H2+OH→H2O+H | 5.5·10−12×e−2000/T | Sander et al.,2006 |
| 232 | H2CO+H→H2+HCO | 2.14−12×e−1090/T×(T/298)1.62 | Baulch et al.,1994 |
| 233 | H2CO+O→HCO+OH | 3.4·10−11×e−1600/T | Sander et al.,2006 |
| 234 | H2CO+OH→H2O+HCO | 5.5·10−12×e125/T | Sander et al.,2006 |
| 235 | H2O+O1D→OH+OH | 2.2·10−10 | Sander et al.,2006 |
| 236 | H2O2+O→OH+HO2 | 1.4·10−12×e−2000/T | Sander et al.,2006 |
| 237 | H2O2+OH→HO2+H2O | 2.9·10−12×e−160/T | Sander et al.,2006 |
| 238 | H2S+H→H2+HS | 3.66·10−12×e−455/T×(T/298)1.94 | Peng et al.,1999 |
| 239 | H2S+O→OH+HS | 9.2·10−12×e−1800/T | Sander et al.,2006 |
| 240 | H2S+OH→H2O+HS | 6.0·10−12×e−70/T | Sander et al.,2006 |
| 241 | HCO+H+M→CO+M | 6.0·10−11×e−7721/T×den | Krasnoperov et al.,2004 |
| 242 | HCO+H2CO→CH3O+CO | 3.8·10−17 | Wen et al.,1989 |
| 243 | HCO+HCO→H2CO+CO | 3.0·10−11 | Tsang and Hampson, 1986 |
| 244 | HCO+NO→HNO+CO | 1.2·10−11 | Tsang and Hampson, 1986 |
| 245 | HCO+O2→HO2+CO | 5.2·10−12 | Sander et al.,2006 |
| 246 | HNO+NO+M→H+M | 1.04·10−6×e25618/T×(T/298)−1.61×den | Tsang and Hampson, 1986 |
| 247 | HNO2+OH→H2O+NO2 | 1.8·10−11×e−390/T | Sander et al.,2006 |
| 248 | HNO3+OH→H2O+NO2+O | 7.2·10−15×e−785/T+ | Sander et al.,2006 |
| (1.9·10−33×e725/T×den)/ | |||
| (1+4.6·10−16×e−715/T×den) | |||
| 249 | HO2+HO2→H2O2+O2 | k0=2.3·10−13×e590/T | Sander et al.,2006 |
| k∞=1.7·10−33×e1000/T | |||
| 250 | HO2+O→OH+O2 | 3.0·10−11×e200/T | Sander et al.,2006 |
| 251 | HO2+O3→OH+O2+O2 | 1.1·10−14×e−490/T | Sander et al.,2006 |
| 252 | HS+H→H2+S | 3.0·10−11 | Schofield, 1973 |
| 253 | HS+H2CO→H2S+HCO | 1.7·10−11×e−800/T | Sander et al.,2006 |
| 254 | HS+HCO→H2S+CO | 5.0·10−11 | Kasting, 1990 |
| 255 | HS+HO2→H2S+O2 | 1.0·10−11 | Stachnik and Molina, 1987 |
| 256 | HS+HS→H2S+S | 1.5−11 | Schofield, 1973 |
| 257 | HS+NO2→HSO+NO | 2.9·10−11×e240/T | Sander et al.,2006 |
| 258 | HS+O→H+SO | 1.6·10−10 | Sander et al.,2006 |
| 259 | HS+O2→OH+SO | 4.0·10−19 | Sander et al.,2006 |
| 260 | HS+O3→HSO+O2 | 9.0·10−12×e−280/T | Sander et al.,2006 |
| 261 | HS+S→H+S2 | 2.2·10−11×e−120/T | Kasting, 1990 |
| 262 | HSO+H→H2+SO | 6.48·10−12 | Sander et al.,2006 |
| 263 | HSO+H→HS+OH | 7.29·10−11 | Sander et al.,2006 |
| 264 | HSO+HS→H2S+SO | 1·10−12 | Kasting, 1990 |
| 265 | HSO+NO→HNO+SO | 1.0·10−15 | Atkinson et al.,2004 |
| 266 | HSO+O→OH+SO | 3.0·10−11×e−200/T | Kasting, 1990 |
| 267 | HSO+OH→H2O+SO | 5.2·10−12 | Sander et al.,2006 |
| 268 | HSO+S→HS+SO | 1·10−11 | Kasting, 1990 |
| 269 | HSO3+H→H2+SO3 | 1.0·10−11 | Kasting, 1990 |
| 270 | HSO3+O→OH+SO3 | 1.0·10−11 | Kasting, 1990 |
| 271 | HSO3+O2→HO2+SO3 | 1.3·10−12×e−330/T | Sander et al.,2006 |
| 272 | HSO3+OH→H2O+SO3 | 1.0·10−11 | Kasting, 1990 |
| 273 | N+NO→N2+O | 2.1·10−11×e−100/T | Sander et al.,2006 |
| 274 | N+O2→NO+O | 1.5·10−12×e−3600/T | Sander et al.,2006 |
| 275 | N+OH→NO+H | 3.8·10−11×e85/T | Atkinson et al.,1989 |
| 276 | N2H3+H→NH2+NH2 | 2.7·10−12 | Gehring et al.,1971 |
| 277 | N2H3+N2H3→N2H4+N2+H2 | 6·10−11 | Kuhn and Atreya, 1979 |
| 278 | N2H4+H→N2H3+H2 | 9.9·10−12×e−1200/T | Stief and Payne, 1976 |
| 279 | NH+H+M→NH2+M | (6·10−30×den)/(1+3·10−20×den) | Kasting, 1982 |
| 280 | NH+NO→N2+OH | 4.9·10−11 | Sander et al.,2006 |
| 281 | NH+O→N+OH | 1·10−11 | Kasting, 1982 |
| 282 | NH+O→NH2+CO | 1·10−11 | Pavlov et al.,2001 |
| 283 | NH2+H+M→NH3+M | (6·10−30×den)/(1+3·10−20×den) | Gordon et al.,1971 |
| 284 | NH2+HCO→NH3+CO | 1·10−11 | Pavlov et al.,2001 |
| 285 | NH2+NH2→N2H4 | 1·10−10 | Gordon et al.,1971 |
| 286 | NH2+NO→N2+H2O | 3.8·10−12×e450/T | Sander et al.,2006 |
| 287 | NH2+O→HNO+H | 5·10−12 | Albers et al.,1969 |
| 288 | NH2+O→NH+OH | 5·10−12 | Albers et al.,1969 |
| 289 | 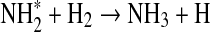 |
3·10−11 | Kasting, 1982 |
| 290 |  |
3·10−11 | Kasting, 1982 |
| 291 | NH3+O1D→NH2+OH | 2.5·10−10 | Sander et al.,2006 |
| 292 | NH3+OH→NH2+H2O | 1.7·10−12×e−710/T | Sander et al.,2006 |
| 293 | NO+HO2→NO2+OH | 3.5·10−12×e250/T | Sander et al.,2006 |
| 294 | NO+O+M→NO2+M | 9·10−31×3·10−11×(T/298)1.5 | Sander et al.,2006 |
| 295 | NO+O3→NO2+O2 | 2.0·10−12×e−1400/T | Sander et al.,2006 |
| 296 | NO+OH+M→HNO2+M | k0=7·10−31×(T/298)2.6 | Sander et al.,2006 |
| k∞=3.6·10−11×(T/298)0.1 | |||
| 297 | NO2+H→NO+OH | 4·10−10×e−340/T | Sander et al.,2006 |
| 298 | NO2+O→NO+O2 | 5.6·10−12×e180/T | Sander et al.,2006 |
| 299 | NO2+OH+M→HNO3+M | k0=2.0·10−30×(T/298)3.0 | Sander et al.,2006 |
| k∞=2.5·10−11 | |||
| 300 | O+HCO→H+CO2 | 5.0·10−11 | Tsang and Hampson, 1986 |
| 301 | O+HCO→OH+CO | 1.0·10−10 | Hampson and Garvin, 1977 |
| 302 | O+HNO→OH+NO | 5.99·10−11 | Tsang and Hampson, 1986 |
| 303 | O+O+M→O2+M | 9.46·10−34×e480/T×den | Campbell and Gray, 1973 |
| 304 | O+O2+M→O3+M | 6·10−34×3·10−11×(T/298)2.40 | Sander et al.,2006 |
| 305 | O+O3→O2+O2 | 8.0·10−12×e−2060/T | Sander et al.,2006 |
| 306 | O1D+M→O+M | 1.8·10−11×e110/T | Sander et al.,2006 |
| 307 | O1D+O2→O+O2 | 3.2·10−11×e70/T | Sander et al.,2006 |
| 308 | OH+HCO→H2O+CO | 1.7·10−10 | Baulch et al.,1992 |
| 309 | OH+HNO→H2O+NO | 5·10−11 | Sun et al.,2001 |
| 310 | OH+HO2→H2O+O2 | 4.8·10−11×e250/T | Sander et al.,2006 |
| 311 | OH+O→H+O2 | 2.2·10−11×e120/T | Sander et al.,2006 |
| 312 | OH+O3→HO2+O2 | 1.6·10−12×e−940/T | Sander et al.,2006 |
| 313 | OH+OH→H2O+O | 4.2·10−12×e−240/T | Sander et al.,2006 |
| 314 | OH+OH→H2O2 | 6.9·10−31×1.5·10−11×(T/298)0.80 | Sander et al.,2006 |
| 315 | S+CO2→SO+CO | 1.0·10−20 | Yung and Demore, 1982 |
| 316 | S+HCO→HS+CO | 5.0·10−11 | Kasting, 1990 |
| 317 | S+HO2→HS+O2 | 1.5·10−11 | Kasting, 1990 |
| 318 | S+HO2→SO+OH | 1.5·10−11 | Kasting, 1990 |
| 319 | S+O2→SO+O | 2.3·10−12 | Sander et al.,2006 |
| 320 | S+O3→SO+O2 | 1.2·10−11 | Sander et al.,2006 |
| 321 | S+OH→SO+H | 6.6·10−11 | Sander et al.,2006 |
| 322 | S+S+M→S2+M | 1.98·10−33×e−206/T×den | Du et al.,2008 |
| 323 | S+S2+M→S3+M | 2.8·10−32×den | Kasting, 1990 |
| 324 | S+S3+M→S4+M | 2.8·10−31×den | Kasting, 1990 |
| 325 | S2+O→S+SO | 1.1·10−11 | Hills et al.,1987 |
| 326 | S2+S2+M→S4+M | 2.8·10−31×den | Baulch et al.,1976 |
| 327 | S4+S4+M→S8AER+M | 2.8·10−31×den | Kasting, 1990 |
| 328 | SO+HCO→HSO+CO | 5.6·10−12×(T/298)−0.4 | Kasting, 1990 |
| 329 | SO+HO2→SO2+OH | 2.8·10−11 | Kasting, 1990 |
| 330 | SO+NO2→SO2+NO | 1.4·10−11 | Sander et al.,2006 |
| 331 | SO+O+M→SO2+M | 6.0·10−31×den | Sander et al.,2006 |
| 332 | SO+O2→O+SO2 | 2.4·10−13×e−2370/T | Sander et al.,2006 |
| 333 | SO+O3→SO2+O2 | 4.5·10−12×e−1170/T | Atkinson et al.,2004 |
| 334 | SO+OH→SO2+H | 8.6·10−11 | Sander et al.,2006 |
| 335 | SO+SO→SO2+S | 3.5·10−15 | Martinez and Herron, 1983 |
| 336 | SO2+HO2→SO3+OH | 8.63·10−16 | Lloyd, 1974 |
| 337 | SO2+O+M→SO3+M | k0=1.3·10−33×(T/298)−3.6 | Sander et al.,2006 |
| k∞=1.5·10−11 | |||
| 338 | SO2+OH+M→HSO3+M | k0=3·10−31×(T/298)3.3 | Sander et al.,2006 |
| k∞=1.5·10−12 | |||
| 339 | 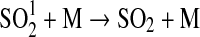 |
1.0·10−11 | Turco et al.,1982 |
| 340 | 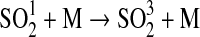 |
1.0·10−12 | Turco et al.,1982 |
| 341 | 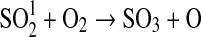 |
1.0·10−16 | Turco et al.,1982 |
| 342 | 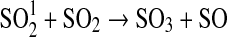 |
4.0·10−12 | Turco et al.,1982 |
| 343 | 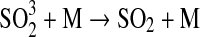 |
1.5·10−13 | Turco et al.,1982 |
| 344 | 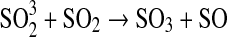 |
7.0·10−14 | Turco et al.,1982 |
| 345 | SO3+H2O→H2SO4 | 1.2·10−15 | Sander et al.,2006 |
| 346 | SO3+SO→SO2+SO2 | 2.0·10−15 | Chung et al.,1975 |
| 347 | 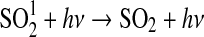 |
2.2·10+4 | Turco et al.,1982 |
| 348 | 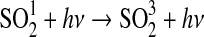 |
1.5·10+3 | Turco et al.,1982 |
| 349 | 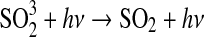 |
1.13·10+3 | Turco et al.,1982 |
| 350 | O2+hν→O+O1D | 1.51·10+02 | |
| 351 | O2+hν→O+O | 2.90·10+00 | |
| 352 | H2O+hν→H+OH | 1.65·10−01 | |
| 353 | O3+hν→O2+O1D | 6.44·10−04 | |
| 354 | O3+hν→O2+O | 1.64·10−04 | |
| 355 | H2O2+hν→OH+OH | 2.79·10−14 | |
| 356 | CO2+hν→CO+O | 2.50·10+01 | |
| 357 | H2CO+hν→H2+CO | 7.71·10−01 | |
| 358 | H2CO+hν→HCO+H | 9.33·10−01 | |
| 359 | CO2+hν→CO+O1D | 2.73·10+03 | |
| 360 | HO2+hν→OH+O | 0.00·10+00 | |
| 361 | 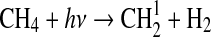 |
1.75·10+00 | |
| 362 | 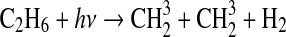 |
0.00 | |
| 363 | 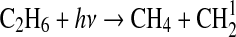 |
1.48·10−05 | |
| 364 | HNO2+hν→NO+OH | 8.68·10−22 | |
| 365 | HNO3+hν→NO2+OH | 2.74·10−28 | |
| 366 | NO+hν→N+O | 2.04·10−10 | |
| 367 | NO2+hν→NO+O | 4.40·10−14 | |
| 368 | 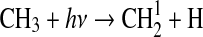 |
6.67·10−04 | |
| 369 | SO+hν→S+O | 0.00·10+00 | |
| 370 | SO2+hν→SO+O | 1.37·10−10 | |
| 371 | H2S+hν→HS+H | 1.00·10−23 | |
| 372 | 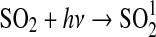 |
1.52·10−09 | |
| 373 | 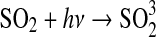 |
8.14·10−13 | |
| 374 | S2+hν→S+S | 5.94·10−42 | |
| 375 | S2+hν→S2 | 0.00·10+00 | |
| 376 | H2SO4+hν→SO2+OH+OH | 1.66·10−13 | |
| 377 | SO3+hν→SO2+O | 0.00·10+00 | |
| 378 | 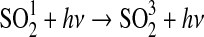 |
9.70·10−11 | |
| 379 | 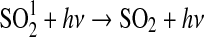 |
1.42·10−09 | |
| 380 | 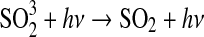 |
9.78·10−11 | |
| 381 | HSO+hν→HS+O | 7.19·10−17 | |
| 382 | S4+hν→S2+S2 | 0.00·10+00 | |
| 383 | S3+hν→S2+S | 4.22·10−72 | |
| 384 | NH3+hν→NH2+H | 6.00·10−34 | |
| 385 | N2H4+hν→N2H3+H | 9.75·10−93 | |
| 386 | NH+hν→N+H | 3.99·10−35 | |
| 387 | NH2+hν→NH+H | 7.49·10−37 | |
| 388 | 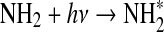 |
3.99·10−35 | |
| 389 |  |
3.99·10−35 | |
| 390 | C2H2+hν→C2H+H | 5.51·10−07 | |
| 391 | C2H2+hν→C2+H2 | 4.09·10−07 | |
| 392 | C2H4+hν→C2H2+H2 | 5.51·10−07 | |
| 393 | C3H8+hν→C3H6+H2 | 1.45·10−12 | |
| 394 | 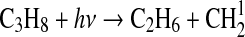 |
2.49·10−13 | |
| 395 | C3H8+hν→C2H4+CH4 | 1.08·10−12 | |
| 396 | C3H8+hν→C2H5+CH3 | 5.88·10−13 | |
| 397 | C2H6+hν→C2H2+H2+H2 | 1.80·10−05 | |
| 398 | C2H6+hν→C2H4+H+H | 1.93·10−05 | |
| 399 | C2H6+hν→C2H4+H2 | 5.29·10−07 | |
| 400 | C2H6+hν→CH3+CH3 | 4.79·10−06 | |
| 401 | C2H4+hν→C2H2+H+H | 5.29·10−07 | |
| 402 | C3H6+hν→C2H2+CH3+H | 5.26·10−16 | |
| 403 | 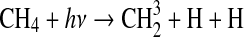 |
1.42·10+00 | |
| 404 | CH4+hν→CH3+H | 2.91·10+00 | |
| 405 | CH+hν→C+H | 9.52·10−06 | |
| 406 | 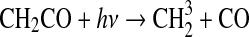 |
8.21·10−10 | |
| 407 | CH3CHO+hν→CH3+HCO | 1.14·10−08 | |
| 408 | CH3CHO+hν→CH4+CO | 1.14·10−08 | |
| 409 | C2H5CHO+hν→C2H5+HCO | 6.42·10−07 | |
| 410 | C3H3+hν→C3H2+H | 6.88·10−07 | |
| 411 | CH3C2H+hν→C3H3+H | 6.42·10−07 | |
| 412 | CH3C2H+hν→C3H2+H2 | 2.41·10−07 | |
| 413 | CH3C2H+hν→CH3+C2H | 3.21·10−08 | |
| 414 | CH2CCH2+hν→C3H3+H | 6.49·10−13 | |
| 415 | CH2CCH2+hν→C3H2+H2 | 2.43·10−13 | |
| 416 | 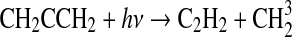 |
9.73·10−14 | |
| 417 | C3H6+hν→CH2CCH2+H2 | 8.81·10−16 | |
| 418 | 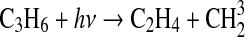 |
3.09·10−17 | |
| 419 | C3H6+hν→C2H+CH4+H | 1.43·10−10 | |
| 420 | OCS+hν→CO+S | 2.67·10−36 | |
| 421 | CS2+hν→CS+S | 5.40·10−47 | |
| 422 | CH3SH+hν→H+CH3S | 1.48·10−30 | |
| 423 | CH3SH+hν→HS+CH3 | 1.11·10−31 | |
| 424 | C2H6S+hν→CH3S+CH3 | 4.01·10−93 | |
| 425 | C2H6S2+hν→CH3S+CH3S | 1.65·10−34 | |
| 426 | 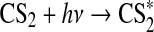 |
6.57·10−48 |
For photolysis reactions (bottom of table), the “Reaction rate constant” column shows the reaction rate (not the rate constant) at the top of the atmosphere during our “standard” simulation, the modern-day fluxes of CH4, H2S, and the Sorg species on a planet orbiting the Sun. For more on how to calculate reaction rates, see Sander et al. (2006).
The model grid is composed of 100 plane-parallel layers that are each 1 km thick in altitude. We did not perform climate calculations for this work; instead, we assumed a temperature profile for an aerosol-free, ozone-free atmosphere. This profile had a surface temperature of 278 K that decreased to 180 K at the tropopause and was isothermal through the stratosphere. The relatively low surface temperature was picked for consistency with previous Archean photochemistry and climate models (Haqq-Misra et al., 2008), and the isothermal stratosphere is consistent with the model's lack of O3. The code calculates the mixing ratios of each species in each layer by solving the coupled mass-continuity/flux equations with the reverse Euler method (appropriate for stiff systems) and a variable time-stepping algorithm. For further details on the photochemical code, see Pavlov et al. (2001) and references therein.
Unless otherwise stated, all model runs were for a 1-bar, N2-dominated atmosphere with 3% CO2 (30,000 ppmv, or ∼100 times the present level of CO2 in Earth's atmosphere) and CH4/CO2 ratios < 0.1. These boundary conditions prevent formation of a significant organic haze (Pavlov et al., 2001; Trainer et al., 2006; Domagal-Goldman et al., 2008). These concentrations and the model's other chemical boundary conditions are by no means unique; however, they were chosen for consistency with a methanogen-acetogen ecosystem (Kharecha et al., 2005). The modeling of haze-free atmospheres is, from a photochemical standpoint, conservative. Including haze in the model would shield the gases we are studying from UV radiation and thereby increase their mixing ratios.
2.2. Boundary conditions
At the top of the atmosphere we allowed H and H2 to escape at the diffusion-limited rate (Walker, 1977). We also applied a constant downward flux of CO and O at the top of our model atmosphere. This accounts for CO and O that is produced from CO2 photolysis above the top layer of our atmosphere and subsequently flows downward into the model grid. For all other species, we used a zero-flux boundary condition at the top of the atmosphere (i.e., no escape).
At the bottom of the atmosphere, we used constant deposition velocities (to account for reactions with surface rocks and for dissolution in the ocean) for all species except the Sorg species, CH4, and NH3. In addition to constant deposition velocities, H2S, SO2, and H2 had volcanic fluxes of 1×109 molecules/cm2/s, 1×1010 molecules/cm2/s, and 3×1010 molecules/cm2/s, respectively, consistent with past models of Archean Earth (Zahnle et al., 2006) that assume volcanism rates about 3 times modern-day values. These fluxes were distributed throughout the troposphere to simulate volcanism. CH4 was modeled with a constant flux of 200 Tg C/year (7×1010 molecules/cm2/s) into the bottom layer of the atmosphere, in line with estimates of modern-day non-anthropogenic fluxes on Earth (Intergovernmental Panel on Climate Change, 2007). [The total CH4 flux today is about 2 times higher; see the Intergovernmental Panel on Climate Change (2007)]. Despite this modern-day flux, the concentrations of CH4 in our models were much higher than they are today because the lack of atmospheric O2 allowed CH4 to accumulate. We imposed a constant mixing ratio of 10−10 for NH3. The corresponding surface flux needed to maintain this mixing ratio was 12.4 Tg N/year, slightly larger than the present-day non-anthropogenic NH3 flux, 10.5 Tg N/year (Intergovernmental Panel on Climate Change, 2007). All photochemical boundary conditions are listed in Table 2.
Table 2.
A List of Species in Our Photochemical Code along with the Lower Boundary Condition Type and Values, the Latter Given in cgs Units: cm/s for Deposition Velocity (Vdep), Dimensionless Mixing Ratio by Volume for Fixed Concentration (f0), and Molecules/cm2/s for Flux (flux)
| Species | Lower boundary type | Vdep/f0/flux |
|---|---|---|
| O | constant deposition velocity | 1 |
| O2 | constant deposition velocity | 1·10−04 |
| H2O | constant deposition velocity | 0 |
| H | constant deposition velocity | 1 |
| OH | constant deposition velocity | 1 |
| HO2 | constant deposition velocity | 1 |
| H2O2 | constant deposition velocity | 2·10−01 |
| H2 | constant deposition velocity* | 2.4·10−04 |
| CO | constant deposition velocity | 1.2·10−04 |
| HCO | constant deposition velocity | 1 |
| H2CO | constant deposition velocity | 2·10−01 |
| CH4 | constant flux | 7·10+10 |
| CH3 | constant deposition velocity | 1 |
| C2H6 | constant deposition velocity | 0 |
| NO | constant deposition velocity | 3·10−04 |
| NO2 | constant deposition velocity | 3·10−03 |
| HNO | constant deposition velocity | 1 |
| H2S | constant deposition velocity* | 2·10−02 |
| HS | constant deposition velocity | 1 |
| S | constant deposition velocity | 1 |
| SO | constant deposition velocity | 3·10−04 |
| SO2 | constant deposition velocity* | 1 |
| H2SO4 | constant deposition velocity | 1 |
| HSO | constant deposition velocity | 1 |
| S2 | constant deposition velocity | 0 |
| NH3 | constant mixing ratio | 1·10−10 |
| NH2 | constant deposition velocity | 1 |
| N2H3 | constant deposition velocity | 1 |
| N2H4 | constant deposition velocity | 2·10−01 |
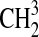 |
constant deposition velocity | 0 |
| C2H5 | constant deposition velocity | 0 |
| C2H2 | constant deposition velocity | 0 |
| C2H4 | constant deposition velocity | 0 |
| C3H8 | constant deposition velocity | 0 |
| C2H3 | constant deposition velocity | 0 |
| C3H6 | constant deposition velocity | 0 |
| C3H2 | constant deposition velocity | 0 |
| CH2CCH2 | constant deposition velocity | 0 |
| CH3C2H | constant deposition velocity | 0 |
| C2H6S2 (DMDS) | constant flux | 0 |
| C2H6S (DMS) | constant flux | 4.20·10+09 |
| CH3S | constant deposition velocity | 1·10−02 |
| CH3SH | constant flux | 8.3·10+08 |
| CS2 | constant flux | 1.4·10+07 |
| OCS | constant flux | 1.4·10+07 |
| CS | constant deposition velocity | 1·10−04 |
In addition to a constant deposition velocity, we also use a volcanic flux for these gases. Specifically, we used volcanic fluxes of 3·1010 molecules/cm2/s of H2, 1·1010 molecules/cm2/s of SO2, and 1·109 molecules/cm2/s of H2S.
We parameterized the biological production of Sorg. The modern-day Sorg fluxes, predominantly biological in source, are as follows (in units of molecules/cm2/s): 0 for DMDS, 4.2×109 for DMS, 0 for CH3S, 8.3×108 for CH3SH, 1.4×107 for CS2, 1.4×107 for OCS, and 0 for CS (Kettle et al., 2001). We will use “MDF” as a unit to represent these modern-day fluxes in the rest of this paper, such that 1 MDF Sorg is equivalent to an atmosphere that receives all Sorg species at the above fluxes. DMDS, CH3S, and CS have zero direct biological production but are produced photochemically from other Sorg species and are needed to ensure a comprehensive modeling of Sorg chemistry. To determine the effect of Sorg fluxes on Sorg mixing ratios and ultimately on disc-averaged planetary spectra, we parameterized Sorg flux rates by holding the ratios of these fluxes constant and multiplying each flux by a common factor.
Most Sorg species are produced via methylation of (addition of methyl groups to) CH3SH or dehydrogenation of (removal of H atoms from) CH3SH, or both. The main modern-day global source of CH3SH is the degradation of methionine, an amino acid that contains a terminal methio group (−SCH3), from eukaryotes. Based on the production rate of methionine, Pilcher (2003) estimated the flux of CH3SH during the Archean to be ∼3×109 mol/year, or about 0.01 MDF CH3SH. This estimate agrees with what one would get by simply scaling CH3SH production linearly with net primary productivity, as that is also estimated to have been ∼0.01 times the modern value (Kharecha et al., 2005). Because the Archean is our lone example of an anoxic planet, Pilcher's work serves as an estimate for the Sorg fluxes on extrasolar planets with anoxic surface conditions. However, these fluxes could vary if methionine (or some other S-containing amino acid) was more or less prevalent in the planet's biota or if the biospheric productivity was different. Thus, in our primary suite of model runs, we parameterize the Sorg fluxes from methionine degradation, using values from 0 to 3000 times those estimated by Pilcher (2003) (this is equivalent to 0–30 MDF Sorg).
The direct production of CH3SH for metabolic purposes could lead to higher Sorg fluxes. Methanosarcina acetivorans, a methanogen, can produce CH3SH via the metabolic reaction 3CO+H2S+H2O→CH3SH+2CO2 (Moran et al., 2008). In the rest of this manuscript, we will refer to this metabolism as “mercaptogenesis” and to the organisms that utilize it as “mercaptogens.” Assuming substrate-limited (CO-/H2S-limited) conditions with no competition for substrates places an upper limit on mercaptogenesis. CO should build up to extremely high levels on planets with anoxic atmospheres unless consumed by biota (Zahnle, 1986; Kharecha et al., 2005); thus, H2S is likely the limiting substrate on such planets. Estimates of the net primary productivity of S-consumers on Archean Earth vary by orders of magnitude, from 5×109 mol S/year (Kharecha et al., 2005) to 2×1014 mol S/year (Canfield, 2005). Both estimates have caveats: the lower estimate did not include a complete S cycle that allowed for recycling of S, and the upper estimate neglected inorganic sinks for S such as metal-sulfide deposition. The former omission likely has a larger impact, so we used Canfield's estimate as an upper limit to S utilization. If mercaptogens accounted for all H2S used by metabolism, the range of the above S consumption estimates would correspond to CH3SH fluxes of ∼3×109 to 1×1014 moles CH3SH/year, or 0.03–1000 MDF CH3SH. Thus, 1000 MDF CH3SH is an upper limit to the CH3SH produced by mercaptogens on early Earth. The actual CH3SH production was likely much lower than this, due to competition for CO and H2S from other metabolisms or from scavenging of S from the oceans by metal precipitates. On an extrasolar planet, the CH3SH production rate could be higher if the planet has larger volcanic H2S flux rates. Constraining such fluxes may be possible via absorption features of volcanic gases in planetary spectra (Kaltenegger et al., 2010).
Unfortunately, no anoxic ocean model currently exists that includes biological S recycling and a complete accounting of oceanic S sources. Furthermore, no code exists that can model mercaptogens in the context of CO-consuming methanogens and sulfur oxidizers that could compete for substrates. These problems might eventually be addressed by the development of ocean biogeochemistry codes with flexible chemistries and a wide variety of metabolisms. In the absence of such codes, we parameterized CH3SH fluxes from 1 to 100 MDF to simulate a biosphere with CO-consuming mercaptogens. Because the CO they consume would otherwise be used by methanogens, we decreased the biological CH4 flux in proportion to the biological CH3SH flux in these simulations. In this set of mercaptogenesis experiments with 1–100 MDF CH3SH, we held the fluxes of the other Sorg gases (DMS, DMDS, OCS, and CS2) constant at 1 MDF, because, unlike CH3SH, these gases are not directly produced by this metabolism. To distinguish between the two sets of experiments, we label model simulations where we changed the flux of all Sorg gases with “X MDF Sorg,” and label model simulations where we changed only the flux of CH3SH with “X MDF CH3SH.”
Each set of Sorg boundary conditions was applied to planets orbiting stars of three different spectral types, following Segura et al. (2005). Specifically, we used time-averaged spectra of the Sun, the active M dwarf AD Leo, and a model-generated M dwarf with a surface temperature of 3100 K and no chromosphere (Allard et al., 1997). This star, referred to as “T3100” in the remainder of this manuscript, is not presented as a physically meaningful case but rather as a low-UV-flux, end-member simulation. All stellar spectra were scaled such that the total energy flux at the top of the model planet's atmosphere was 1092 W/m2, including radiation outside the bounds of our photochemical model wavelength grid. This is 80% of the flux Earth currently receives from the Sun, which is in line with the amount of energy the anoxic, Archean Earth received. Because the total energy flux received by the planet is the same, this is equivalent to assuming that the planet orbits within the habitable zone of that star. The resulting scaled stellar spectra are plotted in Fig. 1, as binned for use in the photochemical code.
FIG. 1.

(a) The stellar energy distribution at a planet receiving the same amount of total energy flux that the Earth received ∼2.5 billion years ago for three different stars: the Sun, AD Leo, and T3100 (a model M dwarf that has no chromosphere). (b) The bottom panel is an expansion of the UV region of the top panel, with a logarithmic y axis. The bottom panel also shows the absorption cross section of CH3SH, units for which are on the right y axis (also logarithmic). Color images available online at www.liebertonline.com/ast
2.3. Radiative transfer code
We used the line-by-line Spectral Mapping Atmospheric Radiative Transfer model (Meadows and Crisp, 1996; Crisp, 1997) to generate synthetic planetary spectra of our model planets. Spectra were computed by using the vertical mixing ratio profiles of CH3SH, DMS, DMDS, OCS, CS2, SO2, H2S, CH4, C2H6, CO2, and H2O generated by the photochemical code. The underlying surface consisted of a 278 K global ocean with an emissivity of ∼1, and we used the same assumed temperature structure applied in the photochemical model. The input stellar spectra and molecular absorption data were obtained from the Virtual Planetary Laboratory's online database (http://vpl.astro.washington.edu/spectra/VPLSpectra/frontpage.htm) and include molecular line parameters from the HITRAN (Rothman et al., 2005) and PNNL databases (Sharpe et al., 2004).
We did not include any aerosols in our spectral model, so the model spectra shown here should be considered idealized “clear sky” simulations. However, we limited parameter space (see Boundary conditions, above) so that the atmospheric CH4/CO2 ratio was less than 0.1, a condition for which thick organic haze layers will not form (Trainer et al., 2006; Haqq-Misra et al., 2008). S8 and sulfate hazes were also limited by these conditions. Assuming Mie scattering, all S8, hydrocarbon, and sulfate particles in our simulations had extinction optical depths less than 0.05 within the “IR window” between 8.5 and 13 μm in which most of the absorption features explored here appear. While organo-sulfate particles can form in sulfur-rich anoxic atmospheres (DeWitt et al., 2010), the optical properties of these particles have not yet been explored. Water clouds may also impact the spectra simulated here. For more on the effects of water clouds, see Robinson et al. (2011). We leave the exploration of aerosol and cloud effects for future studies.
3. Results
The habitable-zone planets around stars with lower surface temperatures receive proportionally fewer UV photons and more long-wavelength, low-energy photons (Fig. 1). This leads to lower photolysis rates on these planets, as there are fewer photons with the requisite energy to dissociate molecules. Figure 1b illustrates this by showing the wavelength-dependent absorption cross section for CH3SH (Sharpe et al., 2004), along with the incident UV flux from the three different stars. Photolysis of CH3SH (and the other Sorg species) generally occurs at wavelengths <300 nm, where the fluxes from the Sun, AD Leo, and T3100 differ by orders of magnitude. Except below 170 nm, where the AD Leo habitable-zone planet receives the highest relative flux, the UV flux decreases dramatically going from the Sun to AD Leo to T3100. Because CH3SH photolysis occurs mostly in the 200–300 nm region, its photolysis rate follows this same pattern. The same holds true for other gases, for example, H2O, whose photolysis creates highly reactive radicals that destroy Sorg.
3.1. Production and loss of Sorg species
We define a standard Archean model with the general boundary conditions above along with 1 MDF Sorg and 1 MDF CH4. The largest Sorg sinks in this simulation were the following reactions:
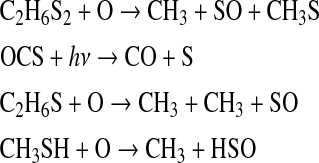 |
These reactions outpaced other net Sorg sinks by at least an order of magnitude. Thus, the major sink for Sorg in our model was reaction with O, and the major by-products were CH3 and oxidized sulfur species—SO and HSO. The major source of O atoms to the atmosphere was photolysis of major atmospheric components (in an anoxic atmosphere, CO2, SO2, and H2O), and the inventory of O atoms decreased when the flux of UV photons to the atmosphere was diminished.
On the model planet orbiting T3100, the biggest sinks for Sorg species were the following reactions:
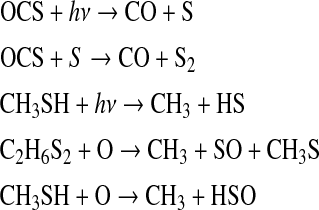 |
In the model simulations around these stars, the lack of UV photons entering the atmosphere led to a lack of O radicals in the atmosphere. This caused a slower destruction rate of the Sorg gases and shifted the main by-products of Sorg photochemistry to carbon monoxide (CO) and reduced sulfur species (S, S2, and H2S). The planets orbiting AD Leo were between these two end-member cases for atomic O production. As a result, the by-products of Sorg chemistry on planets around M dwarfs were a mix of oxidized and reduced sulfur species.
3.2. Atmospheric profiles
Results from nine photochemical model runs are shown in Figs. 2 and 3. Each figure contains a 3×3 grid of panels with decreasing UV flux (Sun, AD Leo, T3100) from left to right and increasing organic sulfur gases (0 MDF Sorg, our control; 1 MDF Sorg, the modern-day fluxes; and 10 MDF CH3SH, corresponding to a biosphere containing mercaptogens) from top to bottom. Figure 2 shows the calculated mixing ratio profiles of the major Sorg species along with SO2 and H2S, while Fig. 3 shows the calculated vertical profiles of H2O, CH4, C2H6, H2, and O2. The profiles generated with 0 MDF Sorg are our control experiments, as this boundary condition is equivalent to assuming no biological Sorg production. In these cases, the atmospheric mixing ratios of all Sorg gases were extremely low.
FIG. 2.

These nine panels each show model-predicted vertical profiles of the mixing ratios of the organic sulfur species. Panels toward the left are for planets orbiting stars with greater UV radiation, and panels toward the bottom are for planets with higher biological Sorg production. The Sorg mixing ratios increase with higher ground Sorg fluxes (bottom panels) and with lower UV radiation (right panels). Color images available online at www.liebertonline.com/ast
FIG. 3.

These nine panels each show model-predicted vertical profiles of the mixing ratios of the greenhouse gases in our climate and line-by-line radiative transfer models. Panels on the left are for planets orbiting stars with greater UV radiation, and panels on the bottom are for planets with higher biological Sorg production. H2O and CO2 concentrations are identical in all model runs, while CH4 concentrations vary only modestly between simulations. Note the increase in C2H6 concentrations on planets with higher Sorg fluxes or lower UV radiation, or both. Color images available online at www.liebertonline.com/ast
For models with the modern-day Sorg flux, near-surface mixing ratios of DMS built up to at least ∼10 ppt (10−11) for all three stellar types. These relatively low concentrations are due to higher photolysis rates in the absence of an O2/O3 UV shield. For the T3100 model planet, DMDS and CH3SH peaked above 100 ppb (10−7). The shapes of the Sorg profiles also changed as a function of star type, as the sulfur gases remained well mixed to higher altitudes in the low-UV-flux models, further increasing the total column depths of the Sorg species. C2H6 concentrations also increased when surface Sorg production was included, because of additional production of CH3 radicals (Fig. 3).
As expected, increasing the CH3SH flux to 10 MDF while keeping the rest of the Sorg gases at 1 MDF (the mercaptogen experiments) resulted in a further increase in all Sorg mixing ratios (Fig. 2). This increase was most pronounced in CH3SH and in DMDS and was greatest on planets receiving relatively low UV radiation. C2H6 concentrations also increased with these higher CH3SH fluxes (Fig. 3), despite the fact that we decreased the CH4 fluxes in these simulations so that the total flux of CH3 groups to the atmosphere remained constant.
3.3. Spectra
To illustrate where each of the gases plotted in Figs. 2 and 3 are spectrally active, we present sensitivity spectra of the model planet with 30 MDF CH3SH orbiting AD Leo (Fig. 4). We generated Fig. 4 by running a full spectral model (shown as a black curve) and then subsequent model runs with one gas removed in each run. These sensitivity spectra are not self-consistent atmospheres; rather, they are tools to determine what gases are causing certain absorption features in the full spectral model. The spectral regions in which a sensitivity spectrum for a particular gas differs from the planet's complete spectrum show where that gas absorbs. For example, the effects of H2O are clearly seen (difference between black and gray curves) from 5 to 7 μm and longward of 17 μm. Likewise, CO2 absorption features (difference between black and brown curves) are present from 9 to 11 μm and from 12 to 19 μm, CH4 absorption is present from 6 to 9 μm, and C2H6 has a deep absorption feature from 11 to 13 μm. The distinguishable Sorg absorption features include those caused by CH3SH from 9 to 11 μm and by DMDS from 10 to 11 μm.
FIG. 4.

The top panel shows the absorption cross sections for the gases included in our spectral model. The middle and bottom panels show the simulated spectra for a simulation of a planet with 30 MDF Sorg orbiting AD Leo. The black line shows the full model spectrum, including the influence of all the gases in our line-by-line radiative transfer model. The colored lines show model spectra in which one gas is removed from the line-by-line radiative transfer model, with lines of the same color showing the absorption cross-section spectrum for that gas in the top pane. For example, the gray line shows the spectrum with the radiative influence of H2O removed from the model. The bottom panel shows a zoom-in on the “infrared window” between 8.5 and 11 μm.
Model spectra from 4 to 20 μm are presented at a spectral resolution of R (λ/Δλ)∼50 in Fig. 5. This resolution is consistent with the requirement goal for the Terrestrial Planet Finder Interferometer (TPF-I), a first-generation thermal-IR planet characterization mission (Lawson et al., 2007). For the simulations of a mercaptogen biosphere on a planet with a spectrum of the Sun or AD Leo, the greatest remotely observable difference was the C2H6 absorption feature between 11 and 13 μm, the strength of which increases at higher CH3SH fluxes (30×modern CH3SH flux). This feature became more prevalent if we increased the flux of the other Sorg gases (30×modern Sorg flux) or if we decreased the UV radiation reaching the planet (bottom panel). The model simulations with these deeper C2H6 features also exhibited enhanced absorption features from 8.5 to 11 μm caused by DMDS.
FIG. 5.

Spectra for planets around the Sun (top panel), AD Leo (middle panel), and T3100, a model M dwarf with no chromosphere (bottom panel), all at a spectral resolution of λ/Δλ∼50. The black curve is a spectrum for a planet with 0 Sorg flux. The red and blue lines show model spectra for planets with 1 and 30 times the modern Sorg fluxes. The purple lines show spectra for planets with 30 times the modern day flux of CH3SH and 0.65 times the modern day flux of CH4. The cyan lines show spectra with 2 times the modern day flux of CH4 and 0 Sorg flux. The green lines show spectra with 1000 times the modern day flux of H2S and 0 Sorg flux. The goldenrod, orange, and brown dashed lines represent the Planck function for an object at 278 K (the surface temperature), 276 or 273 K (the highest “color temperature” for the “1 modern Sorg flux” spectrum), and 180 K (the stratospheric temperature).
H2S fluxes are unlikely to cause false positives. H2S had a large spectral influence only on planets with extremely large H2S fluxes (1000×H2S MDF) orbiting stars with extremely low UV radiation (T3100). Except for these end-member cases, we do not expect H2S to provide a false negative for the other absorption features discussed here.
4. Discussion
Several trends from our photochemical simulations (Figs. 2 and 3) have implications for the interpretation of future exoplanetary spectra. As the stellar UV flux to the planet decreases, the ground-level mixing ratios and altitudinal extent of Sorg species increase. The same effects can also be caused by increases to the Sorg surface fluxes. Both trends can be explained by an increase in the ratio of Sorg sources to Sorg sinks. The main sources of Sorg to the atmosphere are the biogenic surface fluxes; an increase in these raises the source/sink ratio. The two main sinks of Sorg species are direct photolysis and reaction with radicals such as OH and O that themselves are by-products of photochemical reactions. The decrease in UV radiation slows all photolysis and therefore decreases the sinks for Sorg species.
The other robust trend in the photochemical simulations is an increase in C2H6 with increasing Sorg fluxes and with decreasing UV radiation. Increasing Sorg fluxes increases the source of CH3 radicals that combine to form C2H6. Decreases in UV fluxes lead to lower C2H6 photolysis rates, lower concentrations of C2H6-destroying radicals, and smaller sinks for C2H6.
C2H6 has not previously been identified as a potential biosignature for anoxic atmospheres, although most concepts for mid-IR exoplanet characterization missions already include plans to detect CH4 by looking for its absorption feature centered near 7.7 μm (Lawson et al., 2007). According to our model simulations, C2H6 detection would require an interferometer with a spectral resolution of λ/Δλ∼20 and a S/N ∼ 15 in the 11–13 μm range to resolve the distinctive band profile for this gas. Such a mission could discriminate at a 3σ level between C2H6 produced by the model with the modern-day Sorg flux and the model with no Sorg flux, for a planet around an M dwarf similar to AD Leo.
C2H6 concentrations can be enhanced both by increased Sorg concentrations and by increased CH4. Because CH4 can have an abiogenic source, CH4-derived C2H6 could be abiogenic in origin. Figure 5 shows low-resolution (R∼50) spectra with high C2H6 concentrations arising from either high Sorg fluxes or high CH4 fluxes. Models that have higher Sorg fluxes have higher C2H6 concentrations and a deeper C2H6 absorption feature between 11 and 13 μm. Similarly, models that have higher CH4 fluxes also have increased C2H6 concentrations and more absorption between 11 and 13 μm. However, models that achieve C2H6 buildup through increased CH4 fluxes also exhibit a detectable increase in the CH4 concentrations in the atmosphere: there was a doubling in the near-surface CH4 mixing ratios when the CH4 fluxes were increased to 1.5 MDF, and another doubling when the CH4 fluxes were increased to 2.0 MDF. These increased CH4 concentrations caused significantly more absorption between 8 and 9 μm. In other words, changes in the absorption by CH4 could potentially allow us to discriminate between the spectra with “abiogenic, CH4-derived C2H6” and the spectra with “biogenic, Sorg-derived C2H6.” Thus, an exoplanet characterization mission that can measure the depths of the CH4 and C2H6 absorption features accurately enough to estimate the C2H6/CH4 ratio may be able to determine whether biological Sorg production contributes to the source of C2H6.
These above differences in CH4 absorption depths in biological and abiological model simulations are the result of higher C2H6/CH4 ratios in models with biological Sorg fluxes. These fluxes caused an increase in atmospheric CH3 groups, which in turn increased the atmospheric C2H6/CH4 ratio. Thus, for a given amount of C2H6, the CH4 concentrations were lower in models with higher Sorg fluxes. (The converse is also true; for a given CH4 concentration, models with higher Sorg fluxes exhibited higher C2H6 concentrations.) This effect could be augmented by inclusion of other biological CH3X species, such as CH3Cl, that were not included in these simulations.
In addition to the influence of Sorg species on the C2H6 feature, several other features were caused directly by the presence of the Sorg in the model atmospheres: absorption just shortward of 7 μm by DMS, absorption just longward of 7 μm by DMDS, absorption from 8.5 to 9.5 μm by DMDS, and absorption between 9 and 11 μm by DMDS and CH3SH. When present, these features created a continuous, but not constant, increase in absorption from 6 μm all the way to the C2H6 feature at 11 μm. Thus, they have a significant impact across a wide wavelength range. However, these features only appeared in model simulations with extremely low UV fluxes (the T3100 case) or in simulations with at least 30-fold increases in the flux rate of all Sorg gases. On planets around more active stars, these features would only be detectable if the biosphere is much more productive than Earth's biosphere or if the organisms living on the planet have high concentrations of sulfur in their proteins. Even planets with an active mercaptogen community would not produce these features unless that community produces CH3SH at a rate that is greater than 30 times the modern-day CH3SH flux from the oceans.
Additional confusion in interpreting potential arises from the influence of surface temperature. Discriminating between planets with absorption by Sorg species and planets with lower surface temperatures may prove problematic, as the Sorg gases all absorb in the 8–12 μm “atmospheric window” wavelength region. This is a part of the spectrum that some have suggested could be used to discern surface temperatures, because on modern-day Earth that region is the most transparent to the IR radiation emitted by the surface of Earth. However, an increase in greenhouse gases that absorb photons in this region (including Sorg species) will increase its opacity, thereby decreasing the effectiveness with which the surface temperature can be ascertained.
The quantitative effect of Sorg absorption on inferred planetary temperature is shown by the dashed curves in Fig. 5. Here the model spectra, which are cloud free, have been degraded to the spectral resolution goal for TPF-I and are shown with blackbody spectra at three temperatures: (1) 180 K, the stratospheric temperature in our model (drawn in brown); (2) 278 K, the surface temperature in our model (drawn in goldenrod); and (3) either 276 K (top, middle) or 273 K (bottom), the maximum temperature derived for 1 MDF Sorg case within the window region of the model spectrum (drawn in orange). Figure 5 shows that the Sorg gas absorption, in addition to weak water vapor absorption, increases the opacity of the atmosphere in the atmospheric window sufficiently that the majority of the radiation sensed comes from higher, colder regions of the planet's troposphere. The discrepancy between actual surface temperature (278 K) and maximum observed temperature is as much as 8 K for the highest Sorg fluxes and lowest UV fluxes. This will increase the planet's greenhouse effect but decrease the effectiveness with which the surface temperature can be sensed remotely. This effect is from atmospheric absorption alone and does not account for the atmospheric column-truncating effects of clouds or hazes, which for an unresolved Earth-like planet can further reduce the measured brightness temperature in the window region.
Obtaining the best possible estimates of planetary surface temperatures for extrasolar planets of unknown composition will therefore require sufficient spectral wavelength range and resolution to identify non-Earth-like atmospheric window regions, and good estimates of planetary composition and the presence of cloud or aerosol cover. These measurements, combined with atmospheric modeling, will be crucial for understanding limitations on planetary temperature retrieval from MIR spectra for planets with atmospheric characteristics unlike those of modern Earth. For anoxic atmospheres, it is important to be able to detect Sorg absorption features at wavelengths shortward of the window region. Absorption by DMS and DMDS between 6 and 9 μm provides an extra constraint on the abundance of these gases. Similarly, the C2H6 feature could be used in conjunction with photochemical models to further constrain the Sorg flux rates. The atmospheric Sorg inventory could then be input to a climate model to calculate self-consistent surface temperatures and spectra. A fairly comprehensive characterization of an anoxic atmosphere could therefore be achieved with spectra from 6 to 13 μm (and preferably down to 5 μm and out to 20 μm to help constrain water abundances) at a spectral resolution of at least 20 and a S/N greater than 15. These baseline parameters are consistent with the current requirement goals for the TPF-I mission concept.
5. Conclusions
In this paper, we have shown that an anoxic biosphere could be detected over interstellar distances by searching for organic S species produced by biology. On planets orbiting Sun-type stars, Sorg fluxes at 30 times modern-day levels could be detected in the form of elevated C2H6/CH4 ratios that are a photochemical by-product of Sorg gases. On planets around M dwarfs such as AD Leo, detection of heightened C2H6/CH4 ratios is possible at present-day Sorg fluxes. Features caused directly by Sorg gases may be observable on planets that have much higher Sorg fluxes or on planets orbiting M dwarfs that exhibit low amounts of stellar activity, or both. An important caveat to this work is that aerosols, including water clouds, hydrocarbon aerosols, sulfate aerosols, and S8 particles, were not considered in the spectral portion of this study but may impact the ability to detect these species.
The detection of any of these features will require an instrument with spectral resolution R>20, broad coverage of the IR spectrum (6–14 μm), and low total noise levels (S/N>15 or noise < 1 W/m2/μm). Current expected performance levels for TPF-I meet these requirements (Lawson et al., 2007). The use of models to interpret the spectra will also be required in order to separate the effects of surface temperature, organic sulfur gases, and other atmospheric constituents on the planetary spectrum.
Despite the difficulties involved, the benefits offered by such a search are considerable. By including organic sulfur species in our repertoire of remotely detectable biosignatures, the detection of life on some planets may also come with rudimentary lessons on the composition of that planet's biosphere. Thus, this work supports exoplanet characterization missions with a wavelength range and spectral resolution sufficient to detect C2H6 and the organic sulfur gases discussed above.
Acknowledgments
The quality of this manuscript was improved by the useful critiques of three reviewers. This work was performed as part of the NASA Astrobiology Institute's Virtual Planetary Laboratory, supported by the National Aeronautics and Space Administration through the NASA Astrobiology Institute under solicitation No. NNH05ZDA001C. Additional support was provided by the Pennsylvania State Astrobiology Research Center, supported by the National Aeronautics and Space Administration through the NASA Astrobiology Institute under grant number NNA09DA76A. S.D.D.-G. acknowledges additional support from the Astrobiology NSF IGERT grant at the University of Washington. M.C. and S.D.D.-G. acknowledge additional support from the NASA Postdoctoral Program. J.F.K. acknowledges additional support from NASA's Exobiology and Evolutionary Biology Program.
Abbreviations
DMS, dimethyl sulfide; DMDS, dimethyl disulfide; MDF, modern-day flux; S/N, signal-to-noise ratio; TPF-I, Terrestrial Planet Finder Interferometer.
References
- Adachi H. Basco N. James D.G.L. The acetyl radicals CH3CO− and CD3CO− studied by flash photolysis and kinetic spectroscopy. International Journal of Chemical Kinetics. 1981;13:1251–1276. [Google Scholar]
- Albers E.A. Hoyermann K. Wagner H.G. Wolfrum J. Study of the reaction of ammonia with oxygen atoms. Symposium (International) on Combustion. 1969;12:313–321. [Google Scholar]
- Allard F. Hauschildt P.H. Alexander D.R. Starrfield S. Model atmospheres of very low mass stars and brown dwarfs. Annu Rev Astron Astrophys. 1997;35:137–177. [Google Scholar]
- Allen M. Yung Y.L. Gladstone G.R. The relative abundance of ethane to acetylene in the jovian stratosphere. Icarus. 1992;100:527–533. doi: 10.1016/0019-1035(92)90115-n. [DOI] [PubMed] [Google Scholar]
- Amano A. Yamada M. Hashimoto K. Sugiura K. Kinetic feature of the reaction between methanethiol and hydrogen atoms. Nippon Kagaku Kaishi. 1983;12:385–393. [Google Scholar]
- Anastasi C. Broomfield M. Nielsen O.J. Pagsberg P. Ultraviolet absorption spectra and kinetics of CH3S and CH2SH radicals. Chem Phys Lett. 1991;182:643–648. [Google Scholar]
- Arthur N.L. Lee M. Reactions of methyl radicals. I. Hydrogen abstraction from dimethyl sulphide. Aust J Chem. 1976;29:1483–1492. [Google Scholar]
-
Ashfold M.N.R. Fullstone M.A. Hancock G. Ketley G.W. Singlet methylene kinetics: direct measurements of removal rates of ã1A1 and
 CH2 and CD2. Chem Phys. 1981;55:245–257. [Google Scholar]
CH2 and CD2. Chem Phys. 1981;55:245–257. [Google Scholar] - Atkinson R. Baulch D.L. Cox R.A. Hampson J.R.F. Kerr J.A. Troe J. Evaluated kinetic and photochemical data for atmospheric chemistry: supplement III. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry. Journal of Physical and Chemical Reference Data. 1989;18:881–1097. [Google Scholar]
- Atkinson R. Baulch D.L. Cox R.A. Crowley J.N. Hampson R.F. Hynes R.G. Jenkin M.E. Rossi M.J. Troe J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I—gas phase reactions of OX, HOX, NOX and SOX species. Atmos Chem Phys. 2004;4:1461–1738. [Google Scholar]
- Basco N. Pearson A.E. Reactions of sulphur atoms in presence of carbon disulphide, carbonyl sulphide, and nitric oxide. Transactions of the Faraday Society. 1967;63:2684–2694. [Google Scholar]
-
Baughcum S.L. Oldenborg R.C. Measurement of the C2(a3Πu) and
 Disappearance Rates with O2 from 298 to 1300 Kelvin. Oxford University Press; Cary, NC: 1984. [Google Scholar]
Disappearance Rates with O2 from 298 to 1300 Kelvin. Oxford University Press; Cary, NC: 1984. [Google Scholar] - Baulch D.L. Drysdale D.D. Home D.G. Evaluated Kinetic Data for High Temperature Reactions. Butterworths; London: 1976. [Google Scholar]
- Baulch D.L. Cobos C.J. Cox R.A. Esser C. Frank P. Just T. Kerr J.A. Pilling M.J. Troe J. Walker R.W. Warnatz J. Evaluated kinetic data for combustion modelling. Journal of Physical and Chemical Reference Data. 1992;21:411–734. [Google Scholar]
- Baulch D.L. Cobos C.J. Cox R.A. Frank P. Hayman G. Just T. Kerr J.A. Murrells T. Pilling M.J. Troe J. Walker R.W. Warnatz J. Evaluated kinetic data for combustion modeling. Supplement I. Journal of Physical and Chemical Reference Data. 1994;23:847–848. [Google Scholar]
- Becker K.H. Engelhardt B. Wiesen P. Bayes K.D. Rate constants for CH(X2Π) reactions at low total pressures. Chem Phys Lett. 1989;154:342–348. [Google Scholar]
- Benson S.W. Haugen G.R. Mechanism of the high-temperature reactions between acetylene and hydrogen. The Journal of Physical Chemistry. 1967;71:4404–4411. doi: 10.1021/j100865a029. [DOI] [PubMed] [Google Scholar]
- Berman M.R. Fleming J.W. Harvey A.B. Lin M.C. Temperature dependence of CH radical reactions with O2, NO, CO and CO2. Symposium (International) on Combustion. 1982;19:73–79. [Google Scholar]
- Berndt M.E. Allen D.E. Seyfried W.E. Reduction of CO2 during serpentinization of olivine at 300 degrees C and 500 bar. Geology. 1996;24:351–354. [Google Scholar]
-
Böhland T. Dõbe S. Temps F. Wagner H.G. Kinetics of the reactions between
 -radicals and saturated hydrocarbons in the temperature range 296 K≤T≤707 K. Berichte der Bunsengesellschaft für physikalische Chemie. 1985;89:1110–1116. [Google Scholar]
-radicals and saturated hydrocarbons in the temperature range 296 K≤T≤707 K. Berichte der Bunsengesellschaft für physikalische Chemie. 1985;89:1110–1116. [Google Scholar] - Braun W. Bass A.M. Pilling M. Flash photolysis of ketene and diazomethane: the production and reaction kinetics of triplet and singlet methylene. J Chem Phys. 1970;52:5131–5143. [Google Scholar]
- Brown R.L. Laufer A.H. Calculation of activation energies for hydrogen-atom abstractions by radicals containing carbon triple bonds. The Journal of Physical Chemistry. 1981;85:3826–3828. [Google Scholar]
- Butler J.E. Fleming J.W. Goss L.P. Lin M.C. Kinetics of CH radical reactions with selected molecules at room temperature. Chem Phys. 1981;56:355–365. [Google Scholar]
- Campbell I.M. Gray C.N. Rate constants for O(3P) recombination and association with N(4S) Chem Phys Lett. 1973;18:607–609. [Google Scholar]
- Canfield D.E. The early history of atmospheric oxygen: homage to Robert A. Garrels. Annu Rev Earth Planet Sci. 2005;33:1–36. [Google Scholar]
- Choi Y.M. Lin M.C. Kinetics and mechanisms for reactions of HNO with CH3 and C6H5 studied by quantum-chemical and statistical-theory calculations. International Journal of Chemical Kinetics. 2005;37:261–274. [Google Scholar]
- Chung K. Calvert J.G. Bottenheim J.W. The photochemistry of sulfur dioxide excited within its first allowed band (3130 Å) and the “forbidden” band (3700–4000 Å) International Journal of Chemical Kinetics. 1975;7:161–182. [Google Scholar]
- Crisp D. Absorption of sunlight by water vapor in cloudy conditions: a partial explanation for the cloud absorption anomaly. Geophys Res Lett. 1997;24:571–574. [Google Scholar]
- DeMore W.B. Yung Y.L. Catalytic processes in the atmospheres of Earth and Venus. Science. 1982;217:1209–1213. doi: 10.1126/science.217.4566.1209. [DOI] [PubMed] [Google Scholar]
- Des Marais D.J. Harwit M.O. Jucks K.W. Kasting J.F. Lin D.N.C. Lunine J.I. Schneider J. Seager S. Traub W.A. Woolf N.J. Remote sensing of planetary properties and biosignatures on extrasolar terrestrial planets. Astrobiology. 2002;2:153–181. doi: 10.1089/15311070260192246. [DOI] [PubMed] [Google Scholar]
- DeWitt H.L. Hasenkopf C.A. Trainer M.G. Farmer D.K. Jimenez J.L. McKay C.P. Toon O.B. Tolbert M.A. The formation of sulfate and elemental sulfur aerosols under varying laboratory conditions: implications for early Earth. Astrobiology. 2010;10:773–781. doi: 10.1089/ast.2009.9455. [DOI] [PubMed] [Google Scholar]
- Domagal-Goldman S.D. Kasting J.F. Johnston D.T. Farquhar J. Organic haze, glaciations and multiple sulfur isotopes in the Mid-Archean era. Earth Planet Sci Lett. 2008;269:29–40. [Google Scholar]
- Donovan R.J. Husain D. Recent advances in the chemistry of electronically excited atoms. Chem Rev. 1970;70:489–516. [Google Scholar]
- Du S. Francisco J.S. Shepler B.C. Peterson K.A. Determination of the rate constant for sulfur recombination by quasiclassical trajectory calculations. J Chem Phys. 2008:128. doi: 10.1063/1.2919569. [DOI] [PubMed] [Google Scholar]
- Ekwenchi M.M. Jodhan A. Strausz O.P. Reaction of hydrogen atoms with dimethyldisulfide. International Journal of Chemical Kinetics. 1980;12:431–438. [Google Scholar]
- European Space Agency. ESA—Space Science—Darwin. European Space Agency; Paris: 2010. [Google Scholar]
- Fahr A. Laufer A. Klein R. Braun W. Reaction rate of determinations of vinyl radical reactions with vinyl, methyl, and hydrogen atoms. The Journal of Physical Chemistry. 1991;95:3218–3224. [Google Scholar]
- Farquhar J. Wing B.A. Multiple sulfur isotopes and the evolution of the atmosphere. Earth Planet Sci Lett. 2003;213:1–13. [Google Scholar]
- Farquhar J. Peters M. Johnston D.T. Strauss H. Masterson A. Wiechert U. Kaufman A.J. Isotopic evidence for Mesoarchaean anoxia and changing atmospheric sulphur chemistry. Nature. 2007;449:706–709. doi: 10.1038/nature06202. [DOI] [PubMed] [Google Scholar]
- Fenimore C.P. Destruction of methane in water gas by reaction of CH3 and OH radicals. Symposium (International) on Combustion. 1969;12:463–467. [Google Scholar]
- Gehring Mv. Hoyermann K. Wagner H.G. Wolfrum J. Die reaktion von atomarem wasserstoff mit hydrazin. Berichte der Bunsengesellschaft für physikalische Chemie. 1971;75:1287–1294. [Google Scholar]
- Giguere P.T. Huebner W.F. A model of comet comae. I—Gas-phase chemistry in one dimension. Astrophys J. 1978;223:638–654. [Google Scholar]
- Gladstone G.R. Allen M. Yung Y.L. Hydrocarbon photochemistry in the upper atmosphere of Jupiter. Icarus. 1996;119:1–52. doi: 10.1006/icar.1996.0001. [DOI] [PubMed] [Google Scholar]
- Gordon S. Mulac W. Nangia P. Pulse radiolysis of ammonia gas. II. Rate of disappearance of the NH2(X2B1) radical. The Journal of Physical Chemistry. 1971;75:2087–2093. [Google Scholar]
- Hampson R.F. Garvin D. Evaluation and compilation of reaction rate data. The Journal of Physical Chemistry. 1977;81:2317–2319. [Google Scholar]
- Haqq-Misra J.D. Domagal-Goldman S.D. Kasting P.J. Kasting J.F. A revised, hazy methane greenhouse for the Archean Earth. Astrobiology. 2008;8:1127–1137. doi: 10.1089/ast.2007.0197. [DOI] [PubMed] [Google Scholar]
- Hills A.J. Cicerone R.J. Calvert J.G. Birks J.W. Kinetics of the reactions of diatomic sulfur with atomic oxygen, molecular oxygen, ozone, nitrous oxide, nitric oxide, and nitrogen dioxide. The Journal of Physical Chemistry. 1987;91:1199–1204. [Google Scholar]
- Holland H.D. The Chemical Evolution of the Atmosphere and Oceans. Princeton University Press; Princeton, NJ: 1984. [Google Scholar]
- Homann K.H. Wellmann C. Kinetics and mechanism of hydrocarbon formation in the system C2H2/O/H at temperatures up to 1300 K. Berichte der Bunsengesellschaft für physikalische Chemie. 1983;87:609–616. [Google Scholar]
- Hoyermann K. Loftfield N.S. Sievert R. Wagner H.G. Mechanisms and rates of the reactions of CH3O and CH2OH radicals with H atoms. Symposium (International) on Combustion. 1981;18:831–842. [Google Scholar]
- Huebner W.F. Giguere P.T. A model of comet comae. II—Effects of solar photodissociative ionization. Astrophys J. 1980;238:753–762. [Google Scholar]
- Intergovernmental Panel on Climate Change. Climate Change 2007: Synthesis Report. Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. In: Pachauri R.K., editor; Reisinger A., editor. Intergovernmental Panel on Climate Change; Geneva: 2007. and the Core Writing Team. [Google Scholar]
- Jasper A.W. Klippenstein S.J. Harding L.B. Ruscic B. Kinetics of the reaction of methyl radical with hydroxyl radical and methanol decomposition. J Phys Chem A. 2007;111:3932–3950. doi: 10.1021/jp067585p. [DOI] [PubMed] [Google Scholar]
- Jet Propulsion Laboratory. Planet Quest: Missions—Terrestrial Planet Finder. Jet Propulsion Laboratory; Pasadena, CA: 2010. [Google Scholar]
- Kaltenegger L. Sasselov D. Detecting planetary geochemical cycles on exoplanets: atmospheric signatures and the case of SO2. Astrophys J. 2010;708:1162–1167. [Google Scholar]
- Kaltenegger L. Traub W.A. Jucks K.W. Spectral evolution of an Earth-like planet. Astrophys J. 2007;658:598–616. [Google Scholar]
- Kaltenegger L. Henning W.G. Sasselov D. Detecting volcanism on extrasolar planets. Astron J. 2010;140:1370–1380. [Google Scholar]
- Kasting J.F. Stability of ammonia in the primitive terrestrial atmosphere. J Geophys Res. 1982;87:3091–3098. [Google Scholar]
- Kasting J.F. Bolide impacts and the oxidation state of carbon in the Earth's early atmosphere. Orig Life Evol Biosph. 1990;20:199–231. doi: 10.1007/BF01808105. [DOI] [PubMed] [Google Scholar]
- Kasting J.F. Methane and climate during the Precambrian era. Precambrian Res. 2005;137:119–129. [Google Scholar]
- Kasting J.F. Ackerman T.P. High atmospheric NOX levels and multiple photochemical steady-states. J Atmos Chem. 1985;3:321–340. [Google Scholar]
- Kasting J.F. Catling D. Evolution of a habitable planet. Annu Rev Astron Astrophys. 2003;41:429–463. [Google Scholar]
- Kasting J.F. Liu S.C. Donahue T.M. Oxygen levels in the prebiological atmosphere. J Geophys Res. 1979;84:3097–3107. [Google Scholar]
- Kasting J.F. Zahnle K.J. Walker J.C.G. Photochemistry of methane in the Earth's early atmosphere. Precambrian Res. 1983;20:121–148. [Google Scholar]
- Kasting J.F. Pavlov A.A. Siefert J.L. A coupled ecosystem-climate model for predicting the methane concentration in the archean atmosphere. Orig Life Evol Biosph. 2001;31:271–285. doi: 10.1023/a:1010600401718. [DOI] [PubMed] [Google Scholar]
- Kerr J.A. Trotman-Dickenson A.F. The reactions of methyl radicals with thiols. The Journal of the Chemical Society. 1957;79:3322. [Google Scholar]
- Kettle A.J. Rhee T.S. von Hobe M. Poulton A. Aiken J. Andreae M.O. Assessing the flux of different volatile sulfur gases from the ocean to the atmosphere. J Geophys Res. 2001;106:12193–12209. [Google Scholar]
- Kharecha P. Kasting J.F. Siefert J.L. A coupled atmosphere-ecosystem model of the early Archean Earth. Geobiology. 2005;3:53–76. [Google Scholar]
- Krasnoperov L.N. Chesnokov E.N. Stark H. Ravishankara A.R. Unimolecular dissociation of formyl radical, HCO→H+CO, studied over 1–100 bar pressure range. J Phys Chem A. 2004;108:11526–11536. [Google Scholar]
- Kuhn W.R. Atreya S.K. Ammonia photolysis and the greenhouse effect in the primordial atmosphere of the earth. Icarus. 1979;37:207–213. [Google Scholar]
- Kurbanov M.A. Mamedov K.H.F. The Role of the Reaction of CO+SH→COS+H in Hydrogen Formation in the Course of Interaction between CO and H2S. Maik Nauka/Interperiodica; Moscow: 1995. [Google Scholar]
- Lam W.W. Yokota T. Safarik I. Strausz O.P. Photolysis of and reactions of hydrogen atoms with ethanethiol. J Photochem Photobiol A Chem. 1989;47:47–63. [Google Scholar]
- Lander D.R. Unfried K.G. Glass G.P. Curl R.F. Rate Constant Measurements of C2H with CH4, C2H6, C2H4, D2, and CO. American Chemical Society; Washington DC: 1990. [Google Scholar]
- Laufer A. Kinetics of gas phase reactions of methylene. Research on Chemical Intermediates. 1981;4:225–257. [Google Scholar]
- Laufer A.H. Gardner E.P. Kwok T.L. Yung Y.L. Computations and estimates of rate coefficients for hydrocarbon reactions of interest to the atmospheres of outer Solar System. Icarus. 1983;56:560–567. [Google Scholar]
- Lawson P.R. Lay O.P. Johnston K.J. Beichman C.A. Terrestrial Planet Finder Interferometer Science Working Group Report. Jet Propulsion Laboratory, California Institute of Technology; Pasadena, CA: 2007. [Google Scholar]
- Lederberg J. Signs of life: criterion-system of exobiology. Nature. 1965;207:9–13. doi: 10.1038/207009a0. [DOI] [PubMed] [Google Scholar]
- Lee J.H. Stief L.J. Timmons R.B. Absolute rate parameters for the reaction of atomic hydrogen with carbonyl sulfide and ethylene episulfide. J Chem Phys. 1977;67:1705–1709. [Google Scholar]
- Lee L.C. CN(A2Πi→X2Σ+) and CN(B2Σ+→X2Σ+) yields from HCN photodissociation. J Chem Phys. 1980;72:6414–6421. [Google Scholar]
- Lightfoot P.D. Pilling M.J. Temperature and pressure dependence of the rate constant for the addition of hydrogen atoms to ethylene. The Journal of Physical Chemistry. 1987;91:3373–3379. [Google Scholar]
- Liu Y. Wang W.L. Wang W.N. Luo Q. Li Q.S. Density functional theory study on the biradical reaction between CH3S and HCS. Acta Chimica Sinica. 2006;17:1785–1792. [Google Scholar]
- Lloyd A.C. Evaluated and estimated kinetic data for phase reactions of the hydroperoxyl radical. International Journal of Chemical Kinetics. 1974;6:169–228. [Google Scholar]
- Lovelock J.E. A physical basis for life detection experiments. Nature. 1965;207:568–570. doi: 10.1038/207568a0. [DOI] [PubMed] [Google Scholar]
- Martinez R.I. Herron J.T. Methyl thiirane: kinetic gas-phase titration of sulfur atoms in SXOY systems. International Journal of Chemical Kinetics. 1983;15:1127–1132. [Google Scholar]
- Matsumi Y. Tonokura K. Inagaki Y. Kawasaki M. Isotopic branching ratios and translational energy release of hydrogen and deuterium atoms in reaction of oxygen (1D) atoms with alkanes and alkyl chlorides. The Journal of Physical Chemistry. 1993;97:6816–6821. [Google Scholar]
- McEwan M.J. Phillips L.F. Chemistry of the Atmosphere. Wiley/Halsted Press; New York: 1975. [Google Scholar]
- Meadows V.S. Crisp D. Ground-based near-infrared observations of the Venus nightside: the thermal structure and water abundance near the surface. J Geophys Res. 1996;101:4595–4622. [Google Scholar]
- Messing I. Filseth S.V. Sadowski C.M. Carrington T. Absolute rate constants for the reactions of CH with O and N atoms. J Chem Phys. 1981;74:3874–3881. [Google Scholar]
- Michael J.V. Nava D.F. Payne W.A. Stief L.J. Absolute rate constants for the reaction of atomic hydrogen with ketene from 298 to 500 K. J Chem Phys. 1979;70:5222–5227. [Google Scholar]
- Miller J.A. Mitchell R.E. Smooke M.D. Kee R.J. Toward a comprehensive chemical kinetic mechanism for the oxidation of acetylene: comparison of model predictions with results from flame and shock tube experiments. Symposium (International) on Combustion. 1982;19:181–196. [Google Scholar]
- Moran J.J. House C.H. Vrentas J.M. Freeman K. Methyl sulfide production by a novel carbon monoxide metabolism in Methanosarcina acetivorans. Appl Environ Microbiol. 2008;74:540–542. doi: 10.1128/AEM.01750-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki H. Maker P.D. Savage C.M. Breitenbach L.P. Relative rate constants for the reaction of hydroxyl radical with aldehydes. The Journal of Physical Chemistry. 1978;82:132–134. [Google Scholar]
- Okabe H. Photochemistry of acetylene at 1849 Å. J Chem Phys. 1983;78:1312–1317. [Google Scholar]
- Pavlov A.A. Brown L.L. Kasting J.F. UV shielding of NH3 and O2 by organic hazes in the Archean atmosphere. J Geophys Res. 2001;106:23267–23287. [Google Scholar]
- Peng J. Hu X. Marshall P. Experimental and ab initio investigations of the kinetics of the reaction of H atoms with H2S. J Phys Chem A. 1999;103:5307–5311. [Google Scholar]
- Perry R.A. Williamson D. Pressure and temperature dependence of the OH radical reaction with acetylene. Chem Phys Lett. 1982;93:331–334. [Google Scholar]
- Pilcher C.B. Biosignatures of early earths. Astrobiology. 2003;3:471–486. doi: 10.1089/153110703322610582. [DOI] [PubMed] [Google Scholar]
-
Pitts W.M. Pasternack L. McDonald J.R. Temperature dependence of the
 reaction with H2 and CH4 and
reaction with H2 and CH4 and  and a3Πu equilibrated states) with O2. Chem Phys. 1982;68:417–422. [Google Scholar]
and a3Πu equilibrated states) with O2. Chem Phys. 1982;68:417–422. [Google Scholar] - Prasad S.S. Huntress W.T., Jr A model for gas phase chemistry in interstellar clouds. I—The basic model, library of chemical reactions, and chemistry among C, N, and O compounds. Astrophys J Suppl Ser. 1980;43:1–35. [Google Scholar]
- Raymond S.N. Mandell A.M. Sigurdsson S. Exotic Earths: forming habitable worlds with giant planet migration. Science. 2006;313:1413–1416. doi: 10.1126/science.1130461. [DOI] [PubMed] [Google Scholar]
- Robie D.C. Arepalli S. Presser N. Kitsopoulos T. Gordon R.J. The intramolecular kinetic isotope effect for the reaction O(3P)+HD. J Chem Phys. 1990;92:7382–7393. [Google Scholar]
- Robinson T.D. Meadows V.S. Crisp D. Deming D. A'Hearn M.F. Charbonneau D. Livengood T.A. Seager S. Barry R.K. Hearty T. Hewagama T. Lisse C.M. McFadden L.A. Wellnitz D.D. Earth as an extrasolar planet: Earth model validation using EPOXI Earth observations. Astrobiology. 2011:11. doi: 10.1089/ast.2011.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani P.N. Bishop J. Bezard B. Atreya S. Methane photochemistry on Neptune—ethane and acetylene mixing ratios and haze production. Icarus. 1993;106:442–463. [Google Scholar]
- Rothman L.S. Jacquemart D. Barbe A. Chris Benner D. Birk M. Brown L.R. Carleer M.R. Chackerian J.C. Chance K. Coudert L.H. Dana V. Devi V.M. Flaud J.-M. Gamache R.R. Goldman A. Hartmann J.-M. Jucks K.W. Maki A.G. Mandin J.-Y. Massie S.T. Orphal J. Perrin A. Rinsland C.P. Smith M.A.H. Tennyson J. Tolchenov R.N. Toth R.A. Vander Auwera J. Varanasi P. Wagner G. The HITRAN 2004 molecular spectroscopic database. J Quant Spectrosc Radiat Transf. 2005;96:139–204. [Google Scholar]
- Sagan C. Thompson W.R. Carlson R. Gurnett D. Hord C. A search for life on Earth from the Galileo spacecraft. Nature. 1993;365:715–721. doi: 10.1038/365715a0. [DOI] [PubMed] [Google Scholar]
- Sander S.P. Friedl R.R. Golden D.M. Kurylo M.J. Moortgat G.K. Keller-Rudek H. Wine P.H. Ravishankara A.R. Kolb C.E. Molina M.J. Finlayson-Pitts B.J. Huie R.E. Orkin V.L. Chemical kinetics and photochemical data for use in atmospheric studies : evaluation number 15, JPL Publication 06-2, Jet Propulsion Laboratory. California Institute of Technology; Pasadena, CA: 2006. [Google Scholar]
- Schofield K. Evaluated chemical kinetic rate constants for various gas phase reactions. Journal of Physical and Chemical Reference Data. 1973;2:25–84. [Google Scholar]
- Schopf J.W., editor. Earth's Earliest Biosphere: Its Origin and Evolution. Princeton University Press; Princeton: 1983. [Google Scholar]
- Segura A. Kasting J.F. Meadows V. Cohen M. Scalo J. Crisp D. Butler R.A.H. Tinetti G. Biosignatures from Earth-like planets around M dwarfs. Astrobiology. 2005;5:706–725. doi: 10.1089/ast.2005.5.706. [DOI] [PubMed] [Google Scholar]
- Sharpe S.W. Johnson T.J. Sams R.L. Chu P.M. Rhoderick G.C. Johnson P.A. Gas-phase databases for quantitative infrared spectroscopy. Appl Spectrosc. 2004;58:1452–1461. doi: 10.1366/0003702042641281. [DOI] [PubMed] [Google Scholar]
- Shum L.G.S. Benson S.W. The pyrolysis of dimethyl sulfide, kinetics and mechanism. International Journal of Chemical Kinetics. 1985;17:749–761. [Google Scholar]
- Singleton D.L. Cvetanovic R.J. Evaluated chemical kinetic data for the reactions of atomic oxygen O(3P) with sulfur containing compounds. Journal of Physical and Chemical Reference Data. 1988;17:1377–1437. [Google Scholar]
- Stachnik R.A. Molina M.J. Kinetics of the reactions of mercapto radicals with nitrogen dioxide and oxygen. The Journal of Physical Chemistry. 1987;91:4603–4606. [Google Scholar]
- Stephens J.W. Hall J.L. Solka H. Yan W.B. Curl R.F. Glass G.P. Rate constant measurements of reactions of ethynyl radical with hydrogen, oxygen, acetylene and nitric oxide using color center laser kinetic spectroscopy. The Journal of Physical Chemistry. 1987;91:5740–5743. [Google Scholar]
- Stief L.J. Payne W.A. Absolute rate parameters for the reaction of atomic hydrogen with hydrazine. J Chem Phys. 1976;64:4892–4896. [Google Scholar]
- Sun F. DeSain J.D. Scott G. Hung P.Y. Thompson R.I. Glass G.P. Curl R.F. Reactions of NH2 with NO2 and of OH with NH2O. J Phys Chem A. 2001;105:6121–6128. [Google Scholar]
- New Worlds Observer Team. New Worlds. New Worlds Observer Team; Boulder, CO: 2010. [Google Scholar]
- Toon O.B. Kasting J.F. Turco R.P. Liu M.S. The sulfur cycle in the marine atmosphere. J Geophys Res. 1987;92:943–963. [Google Scholar]
- Trainer M.G. Pavlov A.A. DeWitt H.L. Jimenez J.L. McKay C.P. Toon O.B. Tolbert M.A. Organic haze on Titan and the early Earth. Proc Natl Acad Sci USA. 2006;103:18035–18042. doi: 10.1073/pnas.0608561103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W. Hampson R.F. Chemical kinetic data base for combustion chemistry. Part I. Methane and related compounds. Journal of Physical and Chemical Reference Data. 1986;15:1087–1279. [Google Scholar]
- Tsang W. Herron J.T. Chemical kinetic data base for propellant combustion I. Reactions involving NO, NO2, HNO, HNO2, HCN and N2O. Journal of Physical and Chemical Reference Data. 1991;20:609–663. [Google Scholar]
- Turco R.P. Whitten R.C. Toon O.B. Stratospheric aerosols: observation and theory. Reviews of Geophysics and Space Physics. 1982;20:233–280. [Google Scholar]
- Turnipseed A.A. Barone S.B. Ravishankara A.R. Reaction of OH with dimethyl sulfide. 2. Products and mechanisms. The Journal of Physical Chemistry. 1996;100:14703–14713. [Google Scholar]
- Vance S. Christensen L.E. Webster C.R. Sung K. Volatile organic sulfur compounds as biomarkers complementary to methane: infrared absorption spectroscopy of CH3SH enables in-situ measurements on Earth and Mars. Planet Space Sci. 2011;59:299–303. [Google Scholar]
- Vaghjiani G.L. Ravishankara A.R. Photodissociation of H2O2 and CH3OOH at 248 nm and 298 K—quantum yields for OH, O(3P) and H(2S) J Chem Phys. 1990;92:996–1003. [Google Scholar]
- Wagner A.F. Wardlaw D.M. Study of the recombination reaction methyl+methyl→ethane. 2. Theory. The Journal of Physical Chemistry. 1988;92:2462–2471. [Google Scholar]
- Walker J.C.G. Evolution of the Atmosphere. Macmillan; New York: 1977. [Google Scholar]
- Warnatz J. Rate coefficients in the C/H/O system. In: Gadner W.C. Jr., editor. Combustion Chemistry. Springer-Verlag; New York: 1984. pp. 197–360. [Google Scholar]
- Washida N. Reaction of ethanol and CH3CH(OH) radicals with atomic and molecular oxygen. J Chem Phys. 1981;75:2715–2722. [Google Scholar]
- Watkins K.W. Word W.W. Addition of methyl radicals to carbon monoxide: chemically and thermally activated decomposition of acetyl radicals. International Journal of Chemical Kinetics. 1974;6:855–873. [Google Scholar]
- Wen J.-S. Pinto J.P. Yung Y.L. Photochemistry of CO and H2O—analysis of laboratory experiments and applications to the prebiotic Earth's atmosphere. J Geophys Res. 1989;94:14957–14970. doi: 10.1029/jd094id12p14957. [DOI] [PubMed] [Google Scholar]
- Westall F. Evolution: life on the early Earth: a sedimentary view. Science. 2005;308:366–367. doi: 10.1126/science.1107227. [DOI] [PubMed] [Google Scholar]
- Whytock D.A. Payne W.A. Stief L.J. Rate of the reaction of atomic hydrogen with propyne over an extended pressure and temperature range. J Chem Phys. 1976;65:191–195. [Google Scholar]
- Wine P.H. Chameides W.L. Ravishankara A.R. Potential role of CS2 photooxidation in tropospheric sulfur chemistry. Geophys Res Lett. 1981;8:543–546. [Google Scholar]
- Woiki D. Roth P. Oxidation of S and SO by O2 in high-temperature pyrolysis and photolysis reaction systems. International Journal of Chemical Kinetics. 1995;27:59–71. [Google Scholar]
- Yung Y.L. Demore W.B. Photochemistry of the stratosphere of Venus—implications for atmospheric evolution. Icarus. 1982;51:199–247. [Google Scholar]
- Yung Y.L. Allen M. Pinto J.P. Photochemistry of the atmosphere of Titan—comparison between model and observations. Astrophys J Suppl Ser. 1984;55:465–506. doi: 10.1086/190963. [DOI] [PubMed] [Google Scholar]
- Zabarnick S. Fleming J.W. Lin M.C. Kinetic study of the reaction CH(X2Π)+H2↔CH2(X3B1)+H in the temperature range 372 to 675 K. J Chem Phys. 1986;85:4373–4376. [Google Scholar]
- Zabarnick S. Fleming J.W. Lin M.C. Kinetics of CH radical reactions with N2O, SO2, OCS, CS2, and SF6. International Journal of Chemical Kinetics. 1989;21:765–774. [Google Scholar]
- Zahnle K.J. Photochemistry of methane and the formation of hydrocyanic acid (HCN) in the Earth's early atmosphere. J Geophys Res. 1986;91:2819–2834. [Google Scholar]
- Zahnle K.J. Kasting J.F. Mass fractionation during transonic escape and implications for loss of water from Mars and Venus. Icarus. 1986;68:462–480. [Google Scholar]
- Zahnle K.J. Claire M.W. Catling D.C. The loss of mass-independent fractionation in sulfur due to a Palaeoproterozoic collapse of atmospheric methane. Geobiology. 2006;4:271–283. [Google Scholar]
- Zhang Q. Sun T. Zhou X. Wang W. Rate parameters and branching ratios for the multiple-channel reaction of dimethyl sulfide DMS with atomic H. Chem Phys Lett. 2005;414:316–321. [Google Scholar]