

Abstract
Dioxins, including 2,3,7,8 tetrachlorodibenzo-p-dioxin (TCDD), produce a wide range of toxic effects in mammals. Most, if not all, of these toxic effects are regulated by the aryl hydrocarbon receptor (AHR). The AHR is a ligand activated transcription factor that has been shown to interact with numerous proteins capable of influencing the receptor's function. The ability of secondary proteins to alter AHR-mediated transcriptional events, a necessary step for toxicity, led us to determine whether additional interacting proteins could be identified. To this end, we have employed tandem affinity purification (TAP) of the AHR in Hepa1c1c7 cells. TAP of the AHR, followed by mass spectrometry (MS) identified ATP5α1, a subunit of the ATP synthase complex, as a strong AHR interactor in the absence of ligand. The interaction was lost upon exposure to TCDD. The association was confirmed by co-immunoprecipitation in multiple cell lines. In addition, cell fractionation experiments showed that a fraction of the AHR is found in the mitochondria. To ascribe a potential functional role to the AHR:ATP5α1 interaction, TCDD was shown to induce a hyperpolarization of the mitochondrial membrane in an AHR-dependent and transcription-independent manner. These results suggest that a fraction of the total cellular AHR pool is localized to the mitochondria and contributes to the organelle's homeostasis.
Introduction
2,3,7,8 tetrachlordibenzo-p-dioxin (TCDD) is a prototypical dioxin and a member of the halogenated aromatic hydrocarbons (HAH) (Poland and Glover, 1980; Bradfield and Poland, 1988; Mandal, 2005). Dioxins are pervasive, being found in soil and at every level of the food chain (Kearney et al., 1973; Poland and Knutson, 1982). These compounds are highly stable, lipophilic molecules that induce a battery of toxic endpoints in mammals (Poland et al., 1979). Among the adverse effects attributed to dioxin exposure are compromised immune system responses, wasting disease, hyperplasia of the liver, birth defects, metabolic syndrome, and an increased risk of diabetes and cancer (Schmidt and Bradfield, 1996; Mandal, 2005). The cellular mechanisms that govern these toxic endpoints have not yet been fully elucidated; however; most, if not all, involve the aryl hydrocarbon receptor (AHR).
TCDD and other planar HAHs have been characterized as exogenous ligands for the AHR (Poland et al., 1976; Poland and Glover, 1977). In the absence of ligand, the AHR is found in the cytosol, complexed with the aryl-hydrocarbon associated protein nine (ARA9, also known as XAP2 and AIP), and a homodimer of heat shock protein 90 (Hsp90)(Perdew, 1988; Carver et al., 1998). Upon ligand binding, the AHR translocates into the nucleus, and becomes an active transcription factor via heterodimerization with the aryl hydrocarbon receptor nuclear translocator (ARNT) (Prokipcak et al., 1990). The AHR/ARNT dimer binds to xenobiotic response element (XRE) regions of DNA to regulate gene expression (Fujisawa-Sehara et al., 1987; Denison et al., 1988). XREs are found in a wide battery of genes, including those that encode cytochrome P450s, such as CYP1A1, the canonical marker of dioxin exposure (Poland and Kende, 1977). Though AHR-regulated gene expression is necessary for toxicity, a complete understanding of the relationship between AHR cellular signaling, gene regulation, and the pleiotropic toxicity observed in dioxin exposure has not been achieved.
The AHR cytosolic complex has been previously reported to influence a host of proteins that are intimately involved with cell cycle, cellular homeostasis, mitochondrial function, disease, and cell death (Schmidt and Bradfield, 1996; Tian et al., 2002; Barhoover et al., 2009). The objective of the present investigation was to identify novel protein interactions with the AHR using tandem affinity purification (TAP) and mass spectrometry (MS). Using this approach, a physical interaction between the AHR and ATP5α1, a subunit of the ATP synthase complex, was demonstrated. The AHR/ATP5α1 interaction was further verified by co-immunoprecipitation followed by Western blotting and cellular fractionation studies also identified the AHR in the mitochondria. Interestingly, TCDD treatment of several different cell lines which vary in their level of AHR expression demonstrated a hyperpolarization of the mitochondrial inner membrane that was only observed in AHR expressing cells. Although the physiological role of the AHR within the mitochondria compartment is not fully understood, it is tempting to speculate that the metabolic disorders, including the enigmatic wasting syndrome, produced in some animal species by TCDD may be, in part, related to disruption of AHR function within the mitochondria. Collectively our findings suggest that the AHR plays a direct role in mitochondrial function and cellular energetics through protein-protein interactions with ATP5α1.
Materials and Methods
Materials
Oligonucleotide synthesis was performed at Macromolecule Synthesis Facility at Michigan State University. pTarget and pGEM-T Easy vectors were obtained from Promega (Madison, WI) and pZome1C vector was obtained from Cellzome (Cambridge, UK). The Phoenix-eco cell line was a generous gift from Dr. Garry Nolan (Stanford University, Palo Alto, CA) and the AHR−/− mouse embryonic fibroblast cell line and rabbit polyclonal anti-AHR BEAR3 were generous gifts from Dr. Christopher Bradfield (University of Wisconsin-Madison). The other antibodies used were obtained from the following sources: mouse monoclonal anti-ATP5α1 (cat #ab14748) and rabbit polyclonal anti-Hsp90 (cat #ab19021) were obtained from Abcam (Cambridge, MA), mouse monoclonal anti-COXIV (cat #A21349) was obtained from Invitrogen, and rabbit anti-LDH was a generous gift from John Wang (Michigan State University). Goat polyclonal anti-SODII (cat #sc18503), donkey anti-goat (cat #sc2033), goat-anti rabbit (cat # sc2004) and mouse (cat #sc2005), normal mouse IgG (cat #sc2025) and protein G resin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal Mouse Anti-Rabbit IgG, Light Chain Specific, DyLight 488 confugated (cat #211-482-171) and goat Anti-Mouse IgG, Light Chain Specific, DyLight 594 conjugated (cat#151-515-174) were obtained from Jackson Immunoresearch (West Grove, PA). Protease inhibitor tablets (cat # 11836170001) were purchased from Roche (Indianapolis, IN). All other chemicals were reagent grade and purchased from Sigma Aldrich (St. Louis, MO).
Plasmids
The cDNAs for the murine AHR and green fluorescent protein (GFP) were amplified using the following primers: AHR: 5'-ggatccccaccatgagcagcggcgccaacatcacc-3' and 5'-ggatcctgcactctgcaccttgcttagg-3' GFP: 5'-gggggatccaccatggtgagcaagggcgac-3' and 5'-gtggatccccgggcccgcggtaccgtcgactgc-3' The amplicons were cloned into p-Target and pGEM-T Easy, respectively. Each cDNA was subcloned into pZome1C vector. Use of the pZome1C vector places the TAP-tag at the C terminal of both the AHR and GFP gene. Clones were isolated with ampicillin selection and sequence verified.
Cell Culture and Dosing Regime
The Phoenix-eco and Hepa1c1c7 cells were maintained in DMEM with L-glutamate and supplemented with 10% cosmic calf serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 1 mM sodium pyruvate. Hepa C12 cells were maintained in DMEM with L-glutamate and supplemented with 10% cosmic calf serum and 1 mM sodium pyruvate. Tissue culture media and supplements were purchased from Invitrogen (Carlsbad, CA) and cosmic calf serum was purchased from HyClone (Waltman, MA). Hepa1c1c7 TAP-AHR and TAP-GFP were grown in the above media, supplemented with puromycin (2 μg/mL) (US Biological, Swampscott, MA). The AHR−/− mouse embryonic fibroblast (MEF) cell line was maintained in DMEM with L-glutamate and supplemented with 10% fetal calf serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium pyruvate, and 1× non-essential amino acids. AHR−/− MEF cell lines with TAP-AHR or TAP-GFP were grown in the above media and supplements with puromycin (3 μg/mL). The CH12.LX B cell line derived from the murine CH12 B-cell lymphoma (Arnold et al., 1983), which arose in B10.H-2aH-4bp/Wts mice (B10.A × B10.129), has been previously characterized by Bishop et al. (Bishop and Haughton, 1986) and was a generous gift from Dr. Geoffrey Haughton (University of North Carolina). The BCL-1 B-cell line was derived from a murine B-cell lymphoma that spontaneously arose in a BALB/c mouse (Slavin and Strober, 1978) and was generously provided by Dr. Kathryn H. Brooks (Michigan State University). CH12.LX and BCL-1 cells are mature B cells (surface Ig+) and were grown in RPMI-1640 (Gibco BRL, Grand Island, NY) supplemented with heat-inactivated 10% bovine calf serum (Hyclone, Logan, UT), 13.5 mM HEPES, 23.8 mM sodium bicarbonate, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate, and 50 μM b-mercaptoethanol. Cells were maintained at 37°C in an atmosphere of 5% CO2.
Transfection/Stable Cell Line Infection
The TAP-AHR and TAP-GFP retroviral vectors were transfected into Phoenix-eco (P-eco) cells for packaging using Lipofectamine (Invitrogen) via manufacturer's instructions. After the initial incubation (5 to 8 h, 37°C) in the presence of DNA, the media was removed and replaced with Phoenix cell growth media containing chloroquine (25 μM). Cells were incubated for 24 h (37°C) and media was replaced with fresh Phoenix cell growth media and incubated for an additional 24 h (32°C). Cellular debris was removed from the media by centrifugation (3 min, 67 × g, in RT7, Sorvall, Rockford, IL), and purified by filtration through a 0.45 μm membrane filter (Millipore, Billerica, MA). Hepa1c1c7 wild type and MEF AHR −/− target cell lines were exposed to this virus containing media for 3 h at 32°C and 5% CO2. After this incubation, Hepa media containing 15 μg/mL polybrene was added and target cells were incubated for 24 h (32°C). Media was replaced and the target cells were cultured at 37°C to approximately 80% confluence. Cells were then passaged and selected using Hepa media containing 2 μg/mL puromycin.
Western Blot Analysis
Total protein samples were prepared and concentrations determined as previously described (Lowry et al., 1951; Lee et al., 2006). Proteins were separated on 4–12% Nu-Page Bis-Tris gels (Invitrogen), transferred to nitrocellulose membranes, and probed with various antibodies as previously described (Vengellur and LaPres, 2004). Westerns blots were visualized using ECL Western blotting substrate (Pierce, Rockford, IL)
Quantitative Real-time PCR Analysis
The AHR-TAP and GFP-TAP MEF AHR−/− cell lines were used for functional analysis of the TAP-AHR. CYP1A1 gene expression was examined after exposure to vehicle (0.01% dimethyl sulfoxide, DMSO) or TCDD (10 nM) for 6h. Total RNA was extracted from the cells using TRIzol (Invitrogen) per manufacturer's protocol. RNA concentrations were determined by UV spectrophotometry (Nanodrop, Wilminton, DE). Total RNA (1 μg) was reverse-transcribed using anchored oligo (dT)18 primers and M-MLV reverse transcriptase (Invitrogen). Reactions were carried out according to manufacturer's protocol. CYP1A1 gene expression was measured by quantitative real-time PCR (qRT-PCR) using the SYBR-Green system and an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA) (Vengellur and LaPres, 2004). The primers used were: CYP1A1, 5'-AAGTGCAGATGCGGTCTTCT-3' and 5'-AAAGTAGGAGGCAGGCACAA-3'; Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) 5'-AAGCCTAAGATGAGCGCAAG-3' and 3'-TTACTAGGCAGATGGCCACA-5'.
Tandem Affinity Purification
Approximately 2.5 × 109 Hepa1c1c7 TAP-AHR and TAP-GFP cell were treated with vehicle (0.01% DMSO) or TCDD (10 nM) for 30, 120, or 240 min. Dosing media was removed and cells were washed three times with phosphate buffered saline (PBS) and harvested in TAP-tag lysis buffer (TTLB) (5% glycerol, 50 mM Tris, pH 7.5, 50 mM MgCl2, 100 mM NaCl, 0.1% NP40, 1 mM dithiothreitol (DTT), 1 mM Na3VO4, 25 mM NaF, protease inhibitor tablets). Cells were disrupted by two cycles of freeze/thaw, and insoluble material was removed by centrifugation (7000 × g, 20 min). Samples were then incubated with 200 μL of IgG Sepharose beads (GE Healthcare, Waukesha, WI) for 4 h, at 4°C with rotation. Beads were collected by centrifugation (45 × g, 30 sec) and transferred to Poly-Prep Chromatography columns (BioRad, Hercules, CA). Columns were extensively washed with TTLB, followed by washing with tobacco etch virus (TEV) cleavage buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 0.1% NP40, 0.5 mM EDTA, 1 mM DTT). Beads were incubated (18 h, 4°C) in 1.5 mL TEV cleavage buffer containing AcTEV protease (Invitrogen) (450 units). Eluates were collected by gravity flow. IgG sepharose resin was washed with 1.5 mL of TEV buffer and 9 mL of Calmodulin binding buffer (CBB) (10 mM β-mercaptoethanol, 10 mM Tris, pH 7.5, 150 mM NaCl, 1 mM MgOAc, 1 mM imidazole, 0.1% NP40, 2 mM DTT). Washes were combined with eluate. Samples were then incubated with 200 μL Calmodulin affinity resin (Stratagene, La Jolla, CA) (4 h, 4°C). Calmodulin resin was transferred to new Ploy-Prep Chromatography columns, supernatant was removed and resin was washed extensively with CBB. Warm 3× sodium dodecyl sulfate (SDS) buffer (4.8% SDS, 100mM Tris pH 6.8, 16% glycerol, 8% β-mercaptoethanol, 0.4% bromophenol blue) was added to resin to remove protein complexes.
Gel Electrophoresis and Mass Spectrometry
Samples were separated on a 4–12% Bis-Tris gradient gel (NuPage, Invitrogen) by electrophoresis. Gels were silver stained using SilverSNAP staining kit (Pierce, Rockford, IL) according to manufacturer's protocol and the gel was photographed. Specific bands were excised from the matrix and destained with SilverSNAP destaining kit and subjected to in-gel tryptic digestion as previously described (Shevchenko et al., 1996). The extracted peptides were then automatically injected by a Waters nanoAcquity Sample Manager (Milford, MA) loaded for 5 min onto a Waters Symmetry C18 peptide trap (5 μm, 180 μm × 20 mm) at 4 μL/min in 5% acetonitrile /0.1% formic acid. The bound peptides were then eluted onto a Waters BEH C18 nanoAcquity column (1.7 μm, 100 μm × 100 mm) over 35 min with a gradient of 2% B to 35% B in 21 min, 90% B from 21–24 min and back to constant 5% B at 24.1 min using a Waters nanoAcquity UPLC (Buffer A = 99.9% Water/0.1% Formic Acid, Buffer B = 99.9% Acetonitrile/0.1% Formic Acid) with an initial flow rate of 600 nL/min, ramping to 700 nL/min at 80 min and back to 600 nL/min at 86min. Eluted peptides were sprayed into a ThermoFisher LTQ Linear Ion trap mass spectrometer outfitted with a MICHROM Bioresources ADVANCE nano-spray source. The top five ions in each survey scan were then subjected to data-dependant zoom scans followed by low energy collision induced dissociation (CID) and the resulting MS/MS spectra were converted to peak lists using BioWorks Browser v 3.3.1 (ThermoFisher, Rockford, IL) using the default LTQ instrument parameters. Peak lists were searched against all mouse sequences available in the NCBI nr database, downloaded on 11-16-2008 from NCBI, using the Mascot searching algorithm, v2.2 (www.matrixscience.com). The Mascot output was then analyzed using Scaffold (www.proteomesoftware.com) to probabilistically validate protein identifications using the ProteinProphet computer algorithm. Assignments validated above the Scaffold 95% confidence filter are considered true.
Co-immunoprecipitation
Hepa1c1c7 and CH12.LX wild type cells were grown to confluence, washed with cold PBS and harvested in TAP-tag lysis buffer. Equal amounts of the Hepa1c17 and CH12.LX cellular supernatant were incubated with 2 μg/uL of normal mouse IgG or ATP5α1 antibody for 90 min and then Protein G beads (30 μL) were added to the samples and incubated for an additional 90 min. The supernatant was removed and the beads were washed in TAP-tag lysis buffer. Samples were eluted from the beads using 3× SDS buffer and separated by Nu-PAGE Bis-Tris 4–12% gradient gel matrix, transferred to nitrocellulose membrane, and probed with primary AHR antibody.
Cellular Fractionation
Hepa1c1c7 wild type cells were grown to near confluence and then treated with vehicle (0.01% DMSO) or TCDD (10 nM) for 2 h. Cells were harvested in fractionation buffer (25 mM sucrose, 20 mM Tris-HCl, 1 mM EDTA, pH7.4) and dounce homogenized (100 stokes). An aliquot of this was taken as the whole cell lysates (WLC) fraction. CH12.LX cells were harvested in fractionation buffer and dounce homogenized, as above. Liver tissue was homogenized with Tissue-Tearor (Biospec, Bartlesville, OK) in fractionation buffer and then dounce homogenized, as above. Insoluble materials were removed by centrifugation (400 × g, 10 min) and the supernatant was transferred to a new tube. The supernatant and organelle fractions were separated by centrifugation (4,500 × g, 10 min). The supernatant was removed and the organelle pellet was resuspended in 1 mL of fractionation buffer and dounce homogenized (14–20 strokes). An aliquot of the supernatant was taken as the cytosolic fraction (Cyto) and an aliquot of the pellet after resuspension and homogenization was taken as the impure mitochondrial fraction (MF1). Large debris was removed by centrifugation (400 × g, 10 min), the supernatant was transferred to a new tube and mitochondria were collected by centrifugation (4,500 × g, 10 min). The isolated mitochondrial pellet was resuspended in 250μL fractionation buffer and is the pure mitochondrial fraction (MF2). Mitoplasts were prepared using osmotic shock as previously described with minor modifications (Daum et al., 1982). The purified mitochondria pellet was resuspended in mitoplast buffer (4.2 mM sucrose, 20 mM Tris-HCl, 1 mM EDTA, pH 7.4) and incubated on ice, for 20 mins. Large debris was again removed by centrifugation (400 × g, 10 min), the supernatant was transferred to a new tube and mitoplasts were collected by centrifugation (4,500 × g, 20 min). Supernatant was collected and is innermembrane space fraction (MF3). The pellet was resuspended in 100μL fractionation buffer and is the isolated mitochondrial inner membrane fraction (MF4).
Immunohistochemistry
Hepa1c1c7 wild type cell were grown to 80% confluence and mitochondria were isolate as described above. Isolated mitochondria were resuspended in 150 μL of mitochondria buffer and 20 μL were placed on glass slide. Mitochondria were fixed in methanol/acetone (1:1 v/v, 20 min., −20°C). Samples were allowed to air dry at room temperature. Primary antibodies for AHR and ATP5α1 were diluted 1:10,000 in a 1 % BSA PBS solution, added to samples and incubated overnight at 4°C. Samples were washed 3× with ice cold PBS. Secondary Immunofluorescence antibodies were diluted 1:5000 in a 1% BSA in PBS, added to samples, and incubated for 20 min at 4°C. Samples were washed with ice cold PBS. A glass cover slip was placed onto the samples & then stored in the dark until imaging. Images were captured using an Olympus FluoView FV1000 (Olympus America, Inc, Center Valley, PA) configured on a fully automated IX81 inverted microscope at the Center for Advanced Microscopy, Michigan State University.
Mitochondrial Inner Membrane Potential Determination
CH12.LX and BCL-1 cells were grown in cell type appropriate media to a concentration of 1 × 106 cells/ml. Cells were then transferred into a 96 well culture plate at 1 × 105 cells per well. The culture plates were immediately centrifuged at 300 × g for 5 min (24 °C), media was decanted and culture plates were gently agitated to disperse the cell pellet. Replicate wells (8 replicates per treatment) were then exposed to control media, vehicle (0.01%, DMSO), carbonyl cyanide m-chlorophenylhydrazone (CCCP, 12.5 μM), or TCDD (3, 10, or 30 nM) for 6 h. Cells were exposed to mitomycin C where indicated. Culture plates were centrifuged as above, treatment media was decanted and 4 wells per treatment were exposed to 100 μl of high glucose normal buffer (HGNB, 130 mM NaCl, 5.5 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 25 mM glucose, 20 mM HEPES, pH 7.4) with either 500 nM tetramethylrhodamine methyl ester (TMRM) or 250 nM MitoTracker Green (MTG) for 30 mins. Culture plates were then centrifuged, decanted, agitated, and cells were resuspended in 150 μl of HGNB. This procedure was repeated two more times and then the culture plates were analyzed by flow cytometry (FCM). Hepa1c17 and Hepa C12 cells were grown to 70% confluence and dosed with vehicle or TCDD for 6h. Media were removed and the cells were exposed to TMRM or MTG for 30 mins in HGNB. Cells were then trypsinized, washed as above, and analyzed by FCM. Events, 35,000, were recorded per well on a BD FACSCanto II (BD Biosciences, San Jose, CA). Singlet gating was applied and the mean fluorescence intensity (MFI) for both TMRM and MTG fluorescence per cell was calculated for each well and then the average MTG across replicates was determined within a treatment group. Average TMRM per well was divided by average MTG per well per treatment to generate TMRM/MTG fluorescence.
ATP Determination
CH12.LX cells were seeded at 8 × 105 cells/well in a 6 well culture plate and grown to ~80% confluence. The cells were then treated with vehicle (0.01%, DMSO) or TCDD (10 nM) for 6 h. An aliquot of each sample was taken for cell counts. Samples were then precipitated with 1N perchloric acid, centrifuged (13,000 × g, 15 min) to pellet cellular debris, aqueous sample was then neutralized with equal volume of 1N NaOH. ATP levels were measured using ATP determination kit (Invitrogen) according to manufacturer's protocol.
Results
AHR-TAP and GFP-TAP tagged cell line expression and function
AHR−/− MEFs expressing AHR-TAP and GFP-TAP were created and tested for proper expression and function. Western blot analysis showed that the TAP-tagged versions of both proteins were expressed at high levels in the MEFs and Hepa1c1c7 cells (Fig. 1A and B). In addition, the transduced AHR-TAP protein was capable of rescuing dioxin-induced CYP1A1 expression in the AHR−/− MEF cell line (Fig. 1C). The expression of the AHR-TAP protein in the MEF cell line did not remain stable; therefore, AHR-TAP and GFP-TAP genes were transduced into wild type Hepa1c1c7 cells (Fig. 1B). It is hypothesized that the loss of stable expression of the AHR-TAP in the AHR−/− MEFs explains the decrease in TCDD-induced Cyp1A1 expression; however, a modest inhibition of the transactivation domain due to the TAP-tag cannot be ruled out.
Figure 1.

Western blot analysis with an AHR specific antibody was performed to confirm protein expression. Cell strains expressing TAP-tagged GFP (GFP-TAP) or murine AHR (AHR-TAP) were established in AHR null (−/−) MEFs. AHR +/+ (WT) MEFs are included as a positive control (A). GFP-TAP and AHR-TAP were also expressed in Hepa1c1c7 cells. The parental Hepa1c1c7 cells are included as a negative control (CNT). β-actin was used as loading control (B). Functional activity of AHR-TAP (C) was determined by qRT-PCR of Cyp1A1 induction in wild type (WT), AHR −/− (−/−), AHR −/− expressing AHR-TAP, and AHR −/− expressing GFP-TAP MEFs. Cell strains were exposed to DMSO (0.01% vehicle control) and TCDD (10nM) for 6 h, RNA was isolated and quantitated for Cyp1A1 levels using SYBR green. Cyp1A1 levels were normalized to HPRT. * = p < 0.05 when compared to DMSO treated cells within the same cell line.
Identification of AHR complex proteins
Initially, the AHR-TAP and GFP-TAP expressing Hepa1c1c7 cell strains were used to verify the specificity of known AHR-interacting proteins, Hsp90 and ARA9. TAP of the AHR, followed by MS identified the AHR, Hsp90, ARA9, and ARNT at the correct molecular weights only in the AHR-TAP expressing cells (Fig. 2A). Hsp90 and ARA9were identified with greater than 95% confidence using MASCOT and Scaffold data analysis software and ARNT was identified consistently, but with less confidence (Fig. 2B). In the TCDD (10nM) dosed time course experiment data sets, these protein interactions were decreased, in accordance with disassociation of the AHR from its cytosolic complex in the presence of ligand (Fig. 2C). ARNT was also identified in three of four time points including the 240 min sample. The number of ARNT peptides identified was modest compared to Hsp90 and ARA9. This apparent lack of affinity is attributed to the transient nature of the AHR-ARNT interaction and TCDD-induced degradation of the AHR.
Figure 2.

Silver stain gel of TAP samples. TAP final elute samples of AHR-TAP and GFP-TAP Hepa1c1c7 were separated on a 4–12% gradient SDS-Page gel and visualized using silver stain (A). Gel slices (black boxes) were analyzed as described in materials and methods. (B) MS identification of AHR and cytosolic complex proteins Hsp90 and ARA9. Highlighted regions show identified peptide sequences. All were identified at a >95% confidence using the ProteinProfit algorithm. (C) Total numbers of peptides identified by mass spectrometry of known interactors following TAP of the AHR.
Identification of ATP5α1 protein interaction with the AHR
A novel interaction between the AHR and ATP5α1 was identified by TAP/MS method (Fig. 3A). ATP5α1, a catalytic subunit of ATP synthase, was identified at a confidence level of greater than 95% using the MASCOT and Scaffold analysis software (Fig. 3B). Western blot analysis of the purified AHR complex, after TAP, confirmed the identity of ATP5α1 (Fig. 4A). The interaction between the AHR and ATP5α1 was lost upon TCDD treatment (10 ηM, 120 min), suggesting that activation of the AHR leads to its dissociation from the ATP synthase complex (Fig. 4A). Given that the AHR:ATP5α1 interaction was identified using an ectopic expression system, co-immunoprecipitation from wild type Hepa1c1c7 cells was performed. The AHR:ATP5α1 interaction was further confirmed at endogenous protein levels (Fig. 4B). To verify that the interaction was not unique to Hepa1c1c7 cells, co-immunoprecipitation experiments were performed in the AHR expressing murine B cell lymphoma, CH12.LX line. Once again, the AHR/ATP5α1 interaction was observed, this time in a mouse immune cell line (Fig. 4C).
Figure 3.

Silver stained gel of TCDD time course experiment, lanes represent GFP-TAP, AHR-TAP (no dose, 30, 120, 240 min) 10nM TCDD exposure. Black box denotes gel slice from which ATP5α1 was only one of the identified proteins (A). ATP5α1 mass spectrometry data following AHR TAP, including error probability, signal intensity measurements and peptide sequence coverage (highlighted) aligned to the protein sequence (B).
Figure 4.

Co-Immunoprecipitation of the AHR and ATP5α1. Extracts from DMSO (0.01% vehicle control) or TCDD treated (10 ηM, 120 min) AHR-TAP Hepa1c1c7 cells were immunoprecipitated with an AHR specific antibody or IgG control. The resulting precipitate was separated by gel electrophoresis. Western blotting was performed and probed with an ATP5α1 specific antibody (A). Extracts from WT Hepa1c17 (B) or CH12.LX (C) were immunoprecipitated with an ATP5α1 specific antibody or IgG control. Protein separation and Western blot analysis were performed on isolated protein samples with αAHR.
AHR mitochondrial localization
As discussed above, ATP5α1 functions as part of the multisubunit ATP synthase, raising the possibility that a portion of the cellular pool of AHR may localize to the mitochondria. To investigate this possibility, cellular fractionation studies were performed. Indeed, the AHR co-purified with the mitochondrial-specific protein cytochrome c oxidase IV (COX IV), suggesting that a fraction of the cellular AHR pool resides in the mitochondria (Fig. 5A). TCDD treatment (10 ηM, 120 min) appeared to diminish the amount of AHR localized to the mitochondrial compartment; however, this decrease is concomitant with a decrease in the total cellular amount of the receptor (Fig 5A). Mitochondrial localization of the AHR was also observed in CH12LX cells (Fig. 5B). In both cases, ATP5α1 was predominantly identified in the mitochondria with a modest amount present in the cytoplasmic fraction. Additional confirmatory experiments were performed on cellular fractionations from livers of C57BL/6 mice. These liver-derived cellular fractions similarly showed that a portion of the cellular AHR pool was in the mitochondria, and with a small fraction of ATP5α1 present in the cytoplasm (Fig. 5C). To rule out the possibility that binding of the AHR occurred nonspecifically to the outer mitochondrial membrane mitoplasts were prepared (Fig. 5D). These experiments demonstrated the presence of AHR in the mitoplast fraction. Finally, isolated mitochondria were analyzed by immunohistochemistry using AHR- and ATP5α1-specific antibodies and demonstrated a colocalization of the two proteins in the purified organelle (Fig. 5E). Collectively, these data strongly suggest that a portion of the cellular pool of AHR is localized to the matrix of the mitochondria, the same compartment in which ATP5α1 is most abundant.
Figure 5.


Cell fractionation was performed on WT Hepa1c1c7 exposed to DMSO or TCDD (10 ηM, 120 min) (A), CH12LX cells (B), or samples from a C57Bl/6 mouse liver (C). Whole cell lysates (WCL), cytosolic (Cyto), crude mitochondrial (MF1), and pure mitochondrial (MF2), mitochondrial innermembrane space (MF3), and mitoplast (MF4) fractions were prepared as described in materials and methods section Mitoplast were prepared from naïve WT Hepa1c1c7 (D). Cellular fractionation samples were analyzed by Western blot using antibodies specific to the AHR (top panel), ATP5α1 (second panel from top), lactate dehydrogenase (LDH, third panel from top), and Cytochrome C oxidase subunit IV (COXIV, bottom panel). Immunofluorescence imaging on isolated mitochondria using an AHR specific anitbody (Left panel), an ATP5α1 specific antibody (Center panel) shows AHR and ATP5α1 localization to the isolated organelles and an overlay image (Right Panel) shows the two proteins co-localized in the isolated organelles (E).
AHR influence on mitochondrial function
To investigate the functional relevance of AHR:ATP5α1 protein interactions, the mitochondrial inner membrane potential was indirectly measured using tetramethylrhodamine methyl ester (TMRM) by flow cytometry in several cell lines which differed in AHR expression levels. Comparison of murine B cell lymphoma lines, CH12.LX (AHR+/+) and BCL1 (AHR−/−) cells revealed a concentration-dependent hyperpolarization of the mitochondrial inner membrane in CH12.LX cells with TCDD treatment, when compared to the vehicle control (Fig. 6A). In contrast, BCL1 cells demonstrates a basal inner membrane polarization that was 1.5-fold higher in magnitude compared to the basal level observed in CH12.LX cells but only exhibited a modest hypopolarization of the inner membrane with TCDD treatment (Fig 6B). Additional experiments were conducted to investigate the influence transcription may have on the AHR-dependent TCDD-induced hyperpolarization of the mitochondrial inner membrane by pretreatment of CH12.LX cells with mitomycin C. A comparable level of hyperpolarization of the inner membrane was also observed, in the presence of mitomycin C; approximately 3.5–4 fold with 30 nM TCDD treated. These results show that TCDD-mediate mitochondrial hyperpolarization occurs independently of AHR-mediated transcription (Fig 6C).
Figure 6.


Flow cytometric analysis of mitochondrial membrane potential was performed on CH12.LX (AHR+/+) cell lines exposed to DMSO (0.01% vehicle control) or TCDD (3, 10, and 30 nM) for 6h. TMRM intensity showed a dose dependent shift to the right when compared to vehicle control (Shaded portion) (A). Hyperpolarization was only observed in the CH12.LX cells (AHR+/+) and not observed in the BCL1 cell line, an AHR−/− murine B cell line. CCCP was used as a positive control, showing effect of electron transport chain uncoupling on membrane polarization. The values were normalized to the DMSO treated CH12.LX signal (B). The hyperpolarization was not dependent upon AHR-mediated transcription as it was observed in both the presence and absence of the transcriptional inhibitor, mitomycin C. Again, CCCP was used as a positive control and the values were normalized to DMSO treated control (C).
Because CH12.LX and BCL1 cells do not share a common parental lineage, similar experiments were performed in the genetically related Hepa1c1c7 (AHR+/+) and HepaC12 (AHR deficient) cell lines (Van Gurp and Hankinson, 1984). Initially, the level of AHR in the Hepa1c1c7 and HepaC12 was confirmed by Western blot. The HepaC12 cells express negligible amount of AHR compared to Hepa1c1c7 (Fig. 7A). TCDD induced a significant hyperpolarization of the mitochondrial inner membrane in the Hepa1c1c7 cells, similar to the CH12.LX cells. In contrast, the HepaC12 cell line showed no change in inner membrane potential when treated with TCDD (Fig. 7B and C). These results further support that TCDD-induced hyperpolarization is an AHR-dependent event. It is noteworthy that siRNA experiments were also performed to knockdown AHR in the Hepa1c1c7 cells to confirm AHR dependence. Although a significant and reproducible AHR knockdown could be achieved, the transfected cells displayed little or no TCDD-induced hyperpolarization. The mock transfected cells (i.e. lipofectamine alone) and siRNA control (scrambled sequence) also displayed no TCDD-inducible hyperpolarization, demonstrating that the transfection reagent alone disrupts mitochondrial function. This result is not surprising since the function of lipofectamine is to make membranes more fluid and permeable, in this case disrupting the mitochondrial membrane as well.
Figure 7.

Equal amounts of whole cell lysate from Hepa1c1c7 and Hepa C12 cell lines were examined for AHR expression levels by western blotting (A) Flow cytometric analysis of mitochondrial membrane potential were performed on Hepa1c1c7 (AHR+/+) and Hepa C12 (AHR deficient) cell lines exposed to DMSO (0.01% vehicle control) or TCDD (30 nM) for 6h. TMRM intensity showed a TCDD-induced shift to the right when compared to vehicle control in the Hepa1c1c7 cell line. This shift was not observed in the HepaC12 (AHR deficient) cell line (B). The TMRM signal represented in B was normalized to the MitoTraker Green (MTG) signal to confirm that the increased TMRM signal was not dependent upon increased mitochondrial number and further show it was AHR dependent. The values were normalized to DMSO treated Hepa1c1c7 signal (C).
To evaluate the effect of the inner membrane hyperpolarization on cellular energetics, ATP levels were also examined. In the CH12.LX cells, only a statistically insignificant moderate decrease in ATP levels was detected upon TCDD (30 nM, 6h) treatment (Fig 8). The same decrease was also observed in the vehicle treated cells. Given that TCDD induces a dissociation of the AHR from ATP5α1 and a hyperpolarization of the inner membrane in the absence of a measurable change in ATP, it is hypothesized that the change in membrane potential serves to maintain ATP levels following a decrease in ATP synthase efficiency upon loss of AHR interaction.
Figure 8.
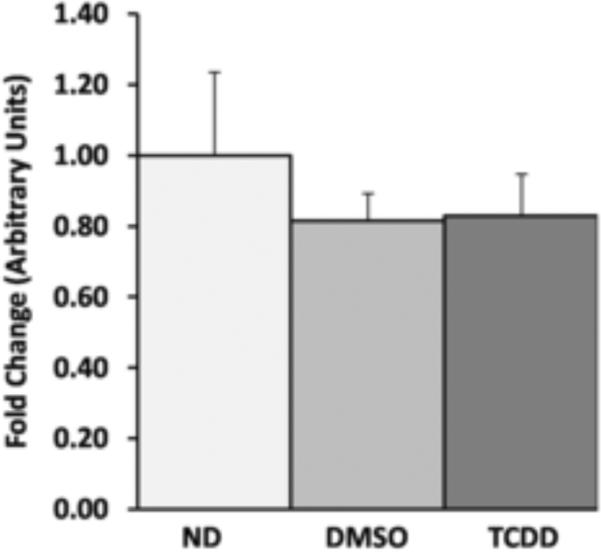
To determine the influence of the mitochondrial inner membrane hyperpolarization on cellular energy, the levels of ATP were examined in CH12.LX cells. Cellular ATP was measured in naïve, DMSO (0.1% vehicle control), and TCDD (30nM) dosed CH12.Lx cells using a luciferase reporter assay. The data was normalized to cell number and the ATP levels of the no dosed (ND) were set to 1. No significant differences were observed between the groups (D).
Discussion
The xenobiotic-induced toxicities mediated through the AHR are complex in their species- and tissues-specific effects. The underlying mechanisms for these toxicities remain poorly defined and may be due, in part, to a lack of a global understanding of the role the AHR plays in different cellular processes, such as gene regulation, protein interactions, and cellular energetics. To date, investigations of the AHR have described numerous aspects of the receptor's function in both xenobiotic response and development. Moreover, it has been shown, using nuclear localization signal (NLS) and DNA binding domain (DBD) mutant mice that normal cellular trafficking is necessary but not sufficient for TCDD-induced biological responses (Bunger et al., 2003; Bunger et al., 2008). These results support the classic view that most TCDD-induced toxicities involve AHR nuclear translocation and transcription; however, they do not rule out the possibility that other AHR-interacting proteins play important roles in the process (Blankenship and Matsumura, 1997; Ge and Elferink, 1998; Chan et al., 1999; Puga et al., 2000). Introduction of mutations to the AHR have provided important new tools to begin to define and dissect the endogenous role for this receptor. For example, cellular localization of the DBD mutant AHR is constitutively nuclear, while mutations in the NLS disrupt normal compartmental trafficking of the AHR (Bunger et al., 2003; Bunger et al., 2008). Similarly, the triple cytochrome p450 knockout mouse displays aberrant bioavailability of the xenobiotics that influence the AHR's normal cellular signaling (Dragin et al., 2008). These recent findings are important but do not directly address, nor rule out, the possibility of secondary effects mediated by AHR protein interaction in xenobiotic-induced toxicity. Therefore, the present investigation sought to identify novel AHR interacting proteins and correlate their function to changes in TCDD-induced toxicity.
The findings reported here demonstrate a novel and intriguing link between the AHR and the mitochondrial machinery responsible for cellular energy production. The mitochondrial ATP synthase is a multi-subunit complex, localized to the inner mitochondrial membrane. ATP5α1 is one of the proteins that comprise the soluble catalytic F1 subunit of this complex. Changes in ATP5α1 levels have been linked to various disease processes, including cancer, and to play a role in tumor progression and the Warburg effect (Baran et al., 2007; Seth et al., 2009; Pennington et al., 2010; Yusenko et al., 2010). The characterization of the interaction between the AHR and ATP5α1 and subsequent changes in mitochondrial function, might link these observations with TCDD-induced tumor promotion (Ray and Swanson, 2009). More importantly, the AHR:ATP5α1 interaction might explain previous observations linking TCDD exposure with changes in mitochondrial function and gene expression (Senft et al., 2002; Paajarvi et al., 2005; Aly and Domenech, 2009; Shertzer et al., 2009; Forgacs et al., 2010). For example, Senft and colleagues observed an AHR-dependent increase in TCDD-induced H2O2 and superoxide production that was independent of Cyp1a1 and Cyp1a2 (Senft et al., 2002). In contrast to the observed TCDD-mediated hyperpolarization in CH12.LX and Hepa1c1c7 cells, several reports have described a TCDD-induced decrease in mitochondrial membrane polarization (Paajarvi et al., 2005; Aly and Domenech, 2009). Presently the reasons for these disparate results are unclear but might be attributable to cell type specific and methodological reasons. Finally, it is tempting to speculate that AHR:ATP5α1 interactions play a broader role in TCDD-induced metabolic syndrome (Uemura et al., 2009). For example, the TCDD-induced dissociation of the AHR from the ATP synthase complex might negatively impact its enzymatic efficiency and lead to mitochondrial stress. The ensuing mitochondrial stress could play a role in the metabolic dysfunction observed following TCDD exposure. Shertzer, et. al. also observed that hepatocytes from TCDD-treated mice displayed a mitochondrial membrane hyperpolarization and suggested that this was due to a defect in ATP synthase (Shertzer et al., 2006). The mitochondrial-localized AHR might play an important role in cellular energetic by maintaining ATP synthase efficiency and TCDD exposure disrupts this function, leading to mitochondrial stress.
Localization of the AHR to the mitochondria is a novel finding and worthy of examination. Though the receptor itself has not previously been associated with the mitochondria, its cytosolic partners have been. The ARA9 has been reported to aid in mitochondrial transport through interaction with TOM20 and Hsc70 (Yano et al., 2003) (MacKenzie and Payne, 2007). In support of this we have identified Hsp8, the murine homolog of Hsc70, in both AHR-TAP and ARA9-TAP MS data sets (data not shown). Hsp90 and Hsc70 have also been reported to facilitate mitochondrial transport through interaction with TOM70 (Young et al., 2003). Hsp90, itself, localizes to the mitochondria in tumor cells and influence the organelle's function (Kang et al., 2007). Finally, Hsp90 has been demonstrated to facilitate the mitochondrial import of proteins lacking traditional mitochondrial localization sequences (MLS), (Budas et al.; Young et al., 2003; Halestrap, 2006). Therefore, even though the AHR lacks a recognized MLS, its cytoplasmic partners (i.e. Hsp90 and ARA9) provide a possible mechanism by which the receptor is capable of translocating to the mitochondria. Finally, the possibility that the AHR:ATP5α1 interaction is mediated through an interaction with the Hsp90 and/or ARA9 must also be considered. Though this possibility has not been tested, directly, ATP5α1 was only observed in a single TAP-ARA9 experiment (data not shown). In contrast, it was observed in multiple TAP/MS runs for the AHR.
Taken together, the data presented here suggest a direct role for the AHR in mitochondrial function and cellular energy production. However, it is important to emphasize that this role played by the AHR within the mitochondrial compartment must not be essential for the organelle's function, at least in mice, since targeted deletion of AHR is not lethal. Here we also observed that treatment with TCDD concordantly produced a decrease in the amount of AHR in the mitochondria and a hyperpolarization of the inner membrane, which occurred in a transcriptionally-independent manner. In light of these observations, we propose that the hyperpolarization of the inner membrane occurs in response to decreased efficiency of the ATP synthase complex and that this inefficiency is a result of the reduced interaction between the AHR and the ATP synthase complex. This model is supported by the observation that the AHR deficient BCL1 cells have a resting membrane potential that is measurably higher than the AHR expressing CH12LX cells. Interestingly, and perhaps only coincidental, CH12.LX cells express remarkably high levels of AHR and exhibit a very rapid doubling rate, approximately every 12–14 h. In contrast, the BCL-1 cells exhibit a remarkably slow doubling rate, approximately every 24–28 h. The HepaC12 cells did not exhibit the same higher resting membrane potential as the BCL-1 cell line; however, the doubling time for the Hepa C12 cell line is comparable to the BCL-1 cell line. Though the results presented here do not rule out a role for the AHR in cell cycle control, it is interesting to speculate that these doubling times are influenced by the putative decrease in ATP synthase efficiency following loss of the receptor. Finally, the discovery of a role for the AHR in cellular energy production establishes a possible mechanism by which dioxin-induced toxic endpoints such as wasting syndrome or immune system suppression may be occurring.
Highlights
-
-
The AHR interacts with the mitochondrial protein, ATP5α1.
-
-
Cell fractionation experiments show that the AHR can be found in the mitochondria.
-
-
TCDD-exposure induces a hyperpolarization of the mitochondrial inner membrane.
-
-
The hyperpolarization is AHR-dependent
-
-
The hyperpolarization occurs without altering ATP levels within the cell
Acknowledgement
We would like to thank Drs Christopher Bradfield and Garry Nolan for their willingness to share cell lines and reagents. This work was supported by NIH grant P42 ES04911.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest The authors declare that there are no conflicts of interest.
References
- Aly HA, Domenech O. Cytotoxicity and mitochondrial dysfunction of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in isolated rat hepatocytes. Toxicol Lett. 2009;191:79–87. doi: 10.1016/j.toxlet.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Arnold LW, LoCascio NJ, Lutz PM, Pennell CA, Klapper D, Haughton G. Antigen-induced lymphomagenesis: identification of a murine B cell lymphoma with known antigen specificity. J Immunol. 1983;131:2064–2068. [PubMed] [Google Scholar]
- Baran AA, Silverman KA, Zeskand J, Koratkar R, Palmer A, McCullen K, Curran WJ, Jr., Edmonston TB, Siracusa LD, Buchberg AM. The modifier of Min 2 (Mom2) locus: embryonic lethality of a mutation in the Atp5a1 gene suggests a novel mechanism of polyp suppression. Genome Res. 2007;17:566–576. doi: 10.1101/gr.6089707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhoover MA, Hall JM, Greenlee WF, Thomas RS. Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol Pharmacol. 2009;77:195–201. doi: 10.1124/mol.109.059675. [DOI] [PubMed] [Google Scholar]
- Bishop GA, Haughton G. Induced differentiation of a transformed clone of Ly-1+ B cells by clonal T cells and antigen. P NATL ACAD SCI USA. 1986;83:7410–7414. doi: 10.1073/pnas.83.19.7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship A, Matsumura F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced activation of a protein tyrosine kinase, pp60src, in murine hepatic cytosol using a cell-free system. Mol Pharmacol. 1997;52:667–675. doi: 10.1124/mol.52.4.667. [DOI] [PubMed] [Google Scholar]
- Bradfield CA, Poland A. A competitive binding assay for 2,3,7,8-tetrachlorodibenzo-p-dioxin and related ligands of the Ah receptor. Mol Pharmacol. 1988;34:682–688. [PubMed] [Google Scholar]
- Budas GR, Churchill EN, Disatnik MH, Sun L, Mochly-Rosen D. Mitochondrial import of PKCepsilon is mediated by HSP90: a role in cardioprotection from ischaemia and reperfusion injury. Cardiovasc Res. 88:83–92. doi: 10.1093/cvr/cvq154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Glover E, Moran SM, Walisser JA, Lahvis GP, Hsu EL, Bradfield CA. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol Sci. 2008;106:83–92. doi: 10.1093/toxsci/kfn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC, Bradfield CA. Resistance to 2,3,7,8-Tetrachlorodibenzo-p-dioxin Toxicity and Abnormal Liver Development in Mice Carrying a Mutation in the Nuclear Localization Sequence of the Aryl Hydrocarbon Receptor. J Biol Chem. 2003;278:17767–17774. doi: 10.1074/jbc.M209594200. 10.1074/jbc.M209594200. [DOI] [PubMed] [Google Scholar]
- Carver LA, LaPres JJ, Jain S, Dunham EE, Bradfield CA. Characterization of the Ah receptor-associated protein, ARA9. J Biol Chem. 1998;273:33580–33587. doi: 10.1074/jbc.273.50.33580. [DOI] [PubMed] [Google Scholar]
- Chan WK, Yao G, Gu YZ, Bradfield CA. Cross-talk between the aryl hydrocarbon receptor and hypoxia inducible factor signaling pathways. Demonstration of competition and compensation. J Biol Chem. 1999;274:12115–12123. doi: 10.1074/jbc.274.17.12115. [DOI] [PubMed] [Google Scholar]
- Daum G, Gasser SM, Schatz G. Import of proteins into mitochondria. Energy-dependent, two-step processing of the intermembrane space enzyme cytochrome b2 by isolated yeast mitochondria. J Biol Chem. 1982;257:13075–13080. [PubMed] [Google Scholar]
- Denison MS, Fisher JM, Whitlock JP., Jr. The DNA recognition site for the dioxin-Ah receptor complex. Nucleotide sequence and functional analysis. J Biol Chem. 1988;263:17221–17224. [PubMed] [Google Scholar]
- Dragin N, Shi Z, Madan R, Karp CL, Sartor MA, Chen C, Gonzalez FJ, Nebert DW. Phenotype of the Cyp1a1/1a2/1b1−/− triple-knockout mouse. Mol Pharmacol. 2008;73:1844–1856. doi: 10.1124/mol.108.045658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgacs AL, Burgoon LD, Lynn SG, LaPres JJ, Zacharewski T. Effects of TCDD on the expression of nuclear encoded mitochondrial genes. Toxicol Appl Pharmacol. 2010;246:58–65. doi: 10.1016/j.taap.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa-Sehara A, Sogawa K, Yamane M, Fujii-Kuriyama Y. Characterization of xenobiotic responsive elements upstream from the drug-metabolizing cytochrome P-450c gene: a similarity to glucocorticoid regulatory elements. Nucleic Acids Res. 1987;15:4179–4191. doi: 10.1093/nar/15.10.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge N-L, Elferink CJ. A Direct Interaction between the Aryl Hydrocarbon Receptor and Retinoblastoma Protein. Linking Dioxin Signaling to the Cell Cycle. J Biol Chem. 1998;273:22708–22713. doi: 10.1074/jbc.273.35.22708. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. Mitochondria and preconditioning: a connexin connection? Circ Res. 2006;99:10–12. doi: 10.1161/01.RES.0000233145.94073.b8. [DOI] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Kearney PC, Woolson EA, Isensee AR, Helling CS. Tetrachlorodibenzodioxin in the environment: sources, fate, and decontamination. Environ Health Persp. 1973;5:273–277. doi: 10.1289/ehp.7305273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Burgoon LD, Gier LE, Dere E, Zacharewski TR, Hogenesch JB, LaPres JJ. Identification and characterization of genes susceptible to transcriptional cross-talk between the hypoxia and dioxin signaling cascades. Chem Res Toxicol. 2006:1284–1293. doi: 10.1021/tx060068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MacKenzie JA, Payne RM. Mitochondrial protein import and human health and disease. Biochim Biophys Acta. 2007;1772:509–523. doi: 10.1016/j.bbadis.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J COMP PHYSIOL B. 2005;175:221–230. doi: 10.1007/s00360-005-0483-3. [DOI] [PubMed] [Google Scholar]
- Paajarvi G, Viluksela M, Pohjanvirta R, Stenius U, Hogberg J. TCDD activates Mdm2 and attenuates the p53 response to DNA damaging agents. Carcinogenesis. 2005;26:201–208. doi: 10.1093/carcin/bgh289. [DOI] [PubMed] [Google Scholar]
- Pennington K, Peng J, Hung CC, Banks RE, Robinson PA. Differential effects of wild-type and A53T mutant isoform of alpha-synuclein on the mitochondrial proteome of differentiated SH-SY5Y cells. J Proteome Res. 2010;9:2390–2401. doi: 10.1021/pr901102d. [DOI] [PubMed] [Google Scholar]
- Perdew GH. Association of the Ah receptor with the 90-kDa heat shock protein. J Biol Chem. 1988;263:13802–13805. [PubMed] [Google Scholar]
- Poland A, Glover E. Chlorinated biphenyl induction of aryl hydrocarbon hydroxylase activity: A study of the structure activity relationship. Mol. Pharmacol. 1977;13:924. [PubMed] [Google Scholar]
- Poland A, Glover E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: Segregation of toxicity with the Ah locus. Mol. Pharm. 1980;17:86–94. [PubMed] [Google Scholar]
- Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem. 1976;251:4936–4946. [PubMed] [Google Scholar]
- Poland A, Greenlee WF, Kende AS. Studies on the mechanism of action of the chlorinated dibenzo-p-dioxins and related compounds. Ann N Y Acad Sci. 1979;320:214–230. doi: 10.1111/j.1749-6632.1979.tb56603.x. [DOI] [PubMed] [Google Scholar]
- Poland A, Kende A. The genetic expression of aryl hydrocarbon hydroxylase activity: Evidence for a receptor mutation in nonresponsive mice. Cold Spring Harbor Laboratory. 1977:847–867. [Google Scholar]
- Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- Prokipcak RD, Denison MS, Okey AB. Nuclear Ah receptor from mouse hepatoma cells: effect of partial proteolysis on relative molecular mass and DNA-binding properties. Arch Biochem Biophys. 1990;283:476–483. doi: 10.1016/0003-9861(90)90670-t. [DOI] [PubMed] [Google Scholar]
- Puga A, Barnes SJ, Dalton TP, Chang C, Knudsen ES, Maier MA. Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J Biol Chem. 2000;275:2943–2950. doi: 10.1074/jbc.275.4.2943. [DOI] [PubMed] [Google Scholar]
- Ray S, Swanson HI. Activation of the aryl hydrocarbon receptor by TCDD inhibits senescence: a tumor promoting event? Biochem Pharmacol. 2009;77:681–688. doi: 10.1016/j.bcp.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JV, Bradfield CA. AH Receptor Signaling Pathways. ANNU REV CELL DEV BI. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- Senft AP, Dalton TP, Nebert DW, Genter MB, Puga A, Hutchinson RJ, Kerzee JK, Uno S, Shertzer HG. Mitochondrial reactive oxygen production is dependent on the aromatic hydrocarbon receptor. Free Radic Biol Med. 2002;33:1268–1278. doi: 10.1016/s0891-5849(02)01014-6. [DOI] [PubMed] [Google Scholar]
- Seth R, Keeley J, Abu-Ali G, Crook S, Jackson D, Ilyas M. The putative tumour modifier gene ATP5A1 is not mutated in human colorectal cancer cell lines but expression levels correlate with TP53 mutations and chromosomal instability. J Clin Pathol. 2009;62:598–603. doi: 10.1136/jcp.2009.064436. [DOI] [PubMed] [Google Scholar]
- Shertzer HG, Genter MB, Shen D, Nebert DW, Chen Y, Dalton TP. TCDD decreases ATP levels and increases reactive oxygen production through changes in mitochondrial F(0)F(1)-ATP synthase and ubiquinone. Toxicol Appl Pharmacol. 2006;217:363–374. doi: 10.1016/j.taap.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shertzer HG, Schneider SN, Kendig EL, Clegg DJ, D'Alessio DA, Johansson E, Genter MB. Tetrahydroindenoindole inhibits the progression of diabetes in mice. Chem Biol Interact. 2009;177:71–80. doi: 10.1016/j.cbi.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Slavin S, Strober S. Spontaneous murine B-cell leukaemia. Nature. 1978;272:624–626. doi: 10.1038/272624a0. [DOI] [PubMed] [Google Scholar]
- Tian Y, Rabson AB, Gallo MA. Ah receptor and NF-[kappa]B interactions: mechanisms and physiological implications. Chem Biol Interact. 2002;141:97–115. doi: 10.1016/s0009-2797(02)00068-6. [DOI] [PubMed] [Google Scholar]
- Uemura H, Arisawa K, Hiyoshi M, Kitayama A, Takami H, Sawachika F, Dakeshita S, Nii K, Satoh H, Sumiyoshi Y, Morinaga K, Kodama K, Suzuki T, Nagai M. Prevalence of metabolic syndrome associated with body burden levels of dioxin and related compounds among Japan's general population. Environ Health Persp. 2009;117:568–573. doi: 10.1289/ehp.0800012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gurp JR, Hankinson O. Isolation and characterization of revertants from four different classes of aryl hydrocarbon hydroxylase-deficient hepa-1 mutants. Mol Cell Biol. 1984;4:1597–1604. doi: 10.1128/mcb.4.8.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengellur A, LaPres JJ. The Role of Hypoxia Inducible Factor 1{alpha} in Cobalt Chloride Induced Cell Death in Mouse Embryonic Fibroblasts. Toxicol Sci. 2004;82:638–646. doi: 10.1093/toxsci/kfh278. [DOI] [PubMed] [Google Scholar]
- Yano M, Terada K, Mori M. AIP is a mitochondrial import mediator that binds to both import receptor Tom20 and preproteins. J Cell Biol. 2003;163:45–56. doi: 10.1083/jcb.200305051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- Yusenko MV, Ruppert T, Kovacs G. Analysis of differentially expressed mitochondrial proteins in chromophobe renal cell carcinomas and renal oncocytomas by 2-D gel electrophoresis. Int J Biol Sci. 2010;6:213–224. doi: 10.7150/ijbs.6.213. [DOI] [PMC free article] [PubMed] [Google Scholar]