

Abstract
Amyloid formation occurs when a precursor protein misfolds and aggregates, forming a fibril nucleus that serves as a template for fibril growth. Glycosaminoglycans are highly charged polymers known to associate with tissue amyloid deposits that have been shown to accelerate amyloidogenesis in vitro. We studied two immunoglobulin light chain variable domains from light chain amyloidosis patients with 90% sequence identity, analyzing their fibril formation kinetics and binding properties with different glycosaminoglycan molecules. We find that the less amyloidogenic of the proteins shows a weak dependence on glycosaminoglycan size and charge, while the more amyloidogenic protein responds only minimally to changes in the glycosaminoglycan. These glycosaminoglycan effects on fibril formation do not depend on a stable interaction between the two species but still show characteristic traits of an interaction-dependent mechanism. We propose that transient, predominantly electrostatic interactions between glycosaminoglycans and the precursor proteins mediate the acceleration of fibril formation in vitro.
Keywords: amyloid, light chain amyloidosis, kinetics, binding, mechanism
Introduction
Protein misfolding disorders, including the amyloidoses, are a major contributor to human disease. It is also increasingly apparent that amyloid or amyloid-like structures play an important physiological role in bacteria [1], fungi [2, 3], and even mammals [4]. Furthermore, amyloid fibrils have shown promise in the development of stable, generic protein scaffolds for use in nanomaterials and synthetic biology [5]. Each of these areas would benefit greatly from a greater understanding of the nature of amyloid formation and the different factors that might enhance or diminish the tendency to form fibrils in living systems. Glycosaminoglycans may be one such factor.
Glycosaminoglycans, or GAGs have been observed in association with tissue deposits of amyloid for many years [6–9]. However, it remains an open question what role they play, if any, in the pathogenesis of protein misfolding diseases. Towards that end, numerous studies have been performed in vitro looking at GAG effects on fibril formation rates and toxicity [10–13]. Different GAGs have been demonstrated to accelerate fibril formation by immunoglobulin light chains, gelsolin, α-synuclein, acylphosphatase, and β2-microglobulin [14–21]. In the specific case of light chain amyloidosis, this acceleration has been shown to be highly dependent on the type of GAG used [14, 21].
GAGs are highly polymorphic molecules composed of repeating disaccharide units of either an uronic acid or galactose paired with an N-acetylated or N-sulfated hexosamine. The type of disaccharide, the number of disaccharide repeats, and the number of sulfate molecules per disaccharide can all be varied in vivo to increase the diversity of the GAG population. Recent studies have suggested that glycosaminoglycans size and charge can vary considerably from one organ to another within the body, and it has been shown that the modification of the GAG portion of proteoglycans can significantly alter cell signaling and adhesion [22–24]. All of these studies point to a possible privileged role for GAGs in the pathogenesis of amyloid diseases. However, studies showing that nucleic acids and other polyanions can dramatically accelerate amyloid fibril formation cast doubt on this interpretation, suggesting instead that the observed GAG effect is mainly the result of the size and charge of the GAG molecule [25, 26].
We approach the study of amyloid fibril formation through the disease light chain amyloidosis, or AL. AL is a hematological disorder in which a clonal population of plasma cells expands and secretes large amounts of free immunoglobulin light chain. The light chain protein circulates in the bloodstream until it misfolds and aggregates in target organs and tissues [27]. The proteins AL-09 and AL-103 are both derived from the κI O18:O8 germline sequence; we have previously shown that in spite of their high sequence similarity, these proteins diverge in terms of structure and their fibril formation behavior. These two proteins share greater than 90% sequence identity, yet AL-09 forms amyloid fibrils rapidly without regard to the solution conditions while AL-103 forms fibrils much more slowly and shows a high sensitivity to the solution conditions [28]. Structurally, AL-09 crystallizes with the dimer interface rotated 90º from that of the canonical Bence-Jones interface seen with AL-103 and other light chain proteins [29, 30]. We hypothesize that the altered dimer interface allows AL-09 to populate misfolded intermediate conformation more easily than the canonical dimer interface structures found in other light chains allow them to do.
Amyloid fibril formation characteristically follows nucleated polymerization kinetics. There is a variable lag phase followed by a much more rapid elongation phase. By adding a small amount of preformed fibrils you can seed the fibril formation reaction, reducing or eliminating the lag phase. The reaction plateaus at equilibrium, leaving a small amount of soluble protein still in solution [31]. Changes in the kinetics reflect differences in the amyloid formation pathway, allowing us to study the effect of different perturbations on the system even without detailed molecular knowledge of fibril structure.
In this paper we will look at the in vitro fibril formation properties of these closely related light chains in order to better understand the role of glycosaminoglycans in amyloid fibril formation. We specifically sought to illuminate the role of GAG size and charge in fibril nucleation and elongation by using highly purified heparin derivatives and comparing them to other preparations of GAGs and GAG-like molecules. These kinetic experiments have shown that the acceleration of fibril formation of one of our proteins, AL-103, depends weakly on heparin size and charge. Through the study of fibril formation kinetics as well as the use of isothermal titration calorimetry, we have shown that while heparan sulfate binds to AL fibrils and precursor proteins, there is no correlation between GAG binding and the acceleration of in vitro fibril formation. Thus, stable interactions between GAGs and amyloid fibrils or precursor proteins are not necessary for GAG-dependent acceleration of fibril formation. However, we also see that the GAG-dependent acceleration of fibril formation depends on the concentration of the GAG species and that the presence of moderate amounts of salt in the reaction eliminates the effects observed with GAGs. We also observe enhanced acceleration of fibril formation in the presence of GAGs at low pH values where the protein is highly charged. Based on this evidence we propose that the GAG-dependent acceleration of fibril formation for these proteins is the result of a transient electrostatic interaction that acts to stabilize the fibril nucleus.
Results
Figure S1 shows unseeded fibril formation (or fibril nucleation) in the presence of 1mg/mL GAGs and GAG-like molecules for both AL-09 and AL-103. The rate of fibril formation is represented as the t50, or time at which the fibril formation reaction is 50% complete (Figure 1). We do not see any enhancement of the AL-09 fibril formation rate in the presence of GAGs and in fact see a decreased rate of fibril formation in the presence of heparan sulfate. Figures 1b and 1c show rapid fibril formation occurring across a range of heparin molecules of different lengths (figure 1b) and charge levels (figure 1c). Only the reaction with de-O-sulfated heparin showed any difference, slowing slightly from the reaction with protein alone. In figure 1d, AL-103 shows that under physiological solution conditions, AL-103 alone does not form fibrils. However, AL-103 shows a much stronger response to the presence of GAGs and dextran sulfate than that seen for AL-09. Fibril formation occurs in less than 100 hours in the presence of heparan and dextran sulfate. We also observe acceleration in the presence of polysaccharide IV heparin, with fibril formation occurring after approximately 300 hours. Figure 1e shows a gradual increase in the AL-103 fibril formation t50 values in the presence of heparin oligosaccharides of varying size. Only disaccharide heparin does not promote AL-103 fibril formation over the time course of this experiment (>1500 hours, figure 1e, shown with a *). Because the concentrations of the GAGs were based on weight, the molar concentration of disaccharide in this experiment was 20-fold higher than that of the polysaccharide heparin. The lack of fibril formation at such a high relative concentration strongly illustrates the importance of molecule length, but the lack of statistical significance among t50 values with the other heparin molecules of varying sizes makes a more quantitative assessment of the length dependence impossible. We observe in figure 1f that AL-103 fibril formation depends on the sulfation level of the GAGs. In solution with de-N-sulfated heparin, AL-103 shows a slower rate of fibril formation than seen with normo- or over-sulfated heparin, while we see no fibril formation on this time scale (>1000 hours) with de-O-sulfated heparin. GAGs alone did not show any ThT fluorescence (not shown). All fibril formation assays were confirmed by transmission electron microscopy (figure S2). Briefly, AL amyloid fibrils formed in vitro tend to form dense networks of short, rod-like structures as has been previously reported with other AL amyloid fibrils [14, 32]. Some individual amyloid fibril segments are observed within the dense network of fibrils. These images confirm that the increase in Thioflavin T fluorescence is due to the formation of mature amyloid fibrils and not an oligomeric or prefibrillar species.
Figure 1.

t50 values for fibril nucleation in the presence of different GAGs and GAG-like molecules.
a) AL-09 fibril nucleation in the presence of different GAGs. b) GAG length dependence of AL-09 fibril nucleation. c) GAG charge dependence of AL-09 fibril nucleation. d) AL-103 fibril nucleation in the presence of different GAGs. e) GAG length dependence of AL-103 fibril nucleation. f) GAG charge dependence of AL-103 fibril nucleation. Error bars correspond to SEM of triplicate samples. * represents a reaction where no fibril formation occurred.
In figure 2 we analyze the effect of GAGs and GAG-like molecules on fibril elongation (seeded) reactions. The presence of seeds accelerates the rate of AL-103 fibril formation, while AL-09 is not affected by the presence of seeds. The reactions show several small changes but no trends or large deviations. For AL-09, heparan sulfate and dextran sulfate decrease the lag time (or increase the seeding efficiency) of the reactions slightly (figure 2a), but there is no significant difference in seeding efficiency or the rate of fibril elongation due to heparin length (figure 2b) or charge (figure 2c). AL-103 shows the same small increase in seeding efficiency in the elongation reactions containing heparan and dextran sulfate (figure 2d) and a slight decrease in the t50 value in the presence of decasaccharide (figure 2e), but no difference in the elongation rate (as observed in figure S3). The seeding efficiency for AL-103 shows a significant increase in the presence of over-O-sulfated heparin and a decrease when de-O-sulfated heparin is present in the reaction. Overall, the only effect we see from the presence of GAGs is perturbation of the seeding efficiency; we do not see measurable differences in the elongation rate due to the presence of GAGs in solution.
Figure 2.

The presence of GAGs and GAG-like molecules has a minimal effect in the seeding efficiency as shown with the t50 values of the elongation reactions.
a) AL-09 fibril elongation (in the presence of preformed seeds) in the presence of different GAGs. b) GAG length dependence of AL-09 fibril elongation. c) GAG charge dependence of AL-09 fibril elongation. d) AL-103 fibril elongation (in the presence of preformed seeds) in the presence of different GAGs. e) GAG length dependence of AL-103 fibril elongation. f) GAG charge dependence of AL-103 fibril elongation. Error bars correspond to SEM of triplicate samples.
Having confirmed that GAGs can accelerate fibril formation, we sought to determine whether or not this effect was caused by a specific interaction between the two species. Isothermal titration calorimetry (ITC) experiments (figure 3) show that heparan sulfate binds to both the AL-09 and AL-103 precursor proteins (figures 3a and 3c) and to AL-09 and AL-103 fibrils (figures 3b and 3d), with an estimated dissociation constant in the low micromolar range. However, neither dextran sulfate nor any heparin derivative tested (disaccharide, tetrasaccharide, hexasaccharide, polysaccharide, de-O-sulfated, or over-O sulfated) bound to AL-09 and AL-103 fibrils or soluble protein with any measurable affinity over a range of concentrations and temperatures (data not shown), in spite of their clear acceleration of AL-103 fibril formation (figure 1d–1f). These results suggest that enhancement of fibril formation does not necessarily require a binding event that will be detected using ITC.
Figure 3.

Interaction of heparan sulfate with fibrils and fibril precursor proteins
ITC shows the binding of heparan sulfate glycosaminoglycans to both AL-09 and AL-103 at 35ºC. 5.4mg/mL heparan sulfate was titrated into a solution of a) soluble 40μM AL-09, b) AL-09 amyloid fibrils (approx. 20μM), c) soluble 40μM AL-103, and d) AL-103 fibrils (approx. 20μM).
In an attempt to resolve this apparent contradiction, we sought to determine whether or not the enhancing effect we observed with fibril formation depended on the concentration of the GAGs. Such a concentration dependence should arise if there is an interaction between the protein and GAG molecules. Figure 4 shows the concentration dependence of the GAG effect on fibril formation, comparing fibril formation t50 values of AL-09 and AL-103 with either 1.0 or 0.1 mg/mL GAGs or GAG-like molecules. AL-09 shows robust fibril formation at both concentrations (figure 4a and 4b), but AL-103 shows no fibril formation at the lower concentration (figure 4d). Only dextran sulfate shows enhancement of ThT fluorescence at the low concentration, peaking at about 100 hours before falling off, likely due to inner filter effects. (figure S4). This concentration dependence of the GAG effect on AL-103 fibril formation supports the idea that the GAG molecules interact with the amyloidogenic precursor proteins in order to produce the observed acceleration of fibril formation.
Figure 4.

GAG concentration dependence of fibril formation t50 values
a) AL-09 fibril nucleation in the presence of 1.0mg/mL GAGs or GAG-like molecules. b) AL-09 fibril nucleation in the presence of 0.1mg/mL GAGs or GAG-like molecules. c) AL-103 fibril nucleation in the presence of 1.0mg/mL GAGs or GAG-like molecules. d) AL-103 fibril nucleation in the presence of 0.1mg/mL GAGs or GAG-like molecules. Error bars correspond to SEM of triplicate samples. * represents a reaction where no fibril formation occurred.
To further test the potential interaction between the two species, we added moderate concentrations of salt to the fibril formation reactions in an attempt to screen any electrostatic contribution to binding. We chose the sulfate and perchlorate anions for this experiment because they fall on different ends of the Hofmeister series and it has previously been demonstrated that there is a relationship between the Hofmeister anion effect on protein stability and amyloid formation [33]. Figure 5 and figure S5 shows a comparison of AL-09 and AL-103 fibril formation in the presence of 0.5M sodium sulfate or sodium perchlorate and 0.1mg/mL heparin, heparan sulfate, and dextran sulfate. We selected lower GAG concentrations for this experiment to accentuate the difference between the two anions studied. AL-09 presents high fibril formation t50 values in the presence of sodium sulfate and in the presence of both dextran sulfate and sodium sulfate (figure 5a), but the rest of the reactions in sulfate and perchlorate do not differ significantly, restricting our ability to interpret these results. However, AL-103 fibril formation is clearly dominated by the choice of salt present. The t50 values in the presence of sodium sulfate are higher than in the presence of sodium perchlorate, consistent with what we observed for AL-09. In figure 4 we observe that AL-103 fibril formation occurred after 67±5 hours at this concentration of dextran sulfate. In the presence of sodium sulfate and dextran sulfate, AL-103 forms fibrils after 249±39 hours (figure 5b). In the presence of sodium perchlorate and dextran sulfate, AL-103 forms fibrils after 38±1 hours. In both the perchlorate and sulfate reactions, the fibril formation is indistinguishable in the presence or absence of dextran sulfate, indicating that the presence of either salt disrupts dextran sulfate’s effect on fibril formation. Moreover, AL-103 was unable to form fibrils in the presence of 0.1 mg/mL polysaccharide IV heparin or heparan sulfate, yet when we incubate these GAGs in the presence of the salts, AL-103 forms fibrils at comparable t50 values of the reactions with salt alone. This signals an electrostatic contribution to the GAG-mediated acceleration of fibril formation. In the presence of moderate amounts of salt, the GAG-protein interaction is disrupted and the salt’s effect on fibril formation dominates.
Figure 5.

Moderate concentrations of salt dominate the fibril formation kinetics
GAGs and GAG-like molecules are present at a concentration of 0.1 mg/mL; salts are present at a concentration of 0.5M. a) AL-09 fibril nucleation in the presence of the sulfate, perchlorate anions. b) d) AL-103 fibril nucleation in the presence of the sulfate and perchlorate anions. * represents a reaction where no fibril formation occurred.
To further illustrate the importance of electrostatic contributions to the GAG effect, figure 6 shows the pH dependence of heparan sulfate mediated acceleration of AL fibril formation. The previously discussed acceleration of fibril formation occurs even though our AL proteins have a net negative charge at pH 7.4. As shown in figure 6a (AL-09) and figure 6b (AL-103), by lowering the solution pH and increasing the net positive charge on the protein we can further enhance the rate of fibril formation. At pH 5 both AL-09 (theoretical pI=4.83) and AL-103 (theoretical pI=5.62) are near their isoelectric point, while the reactions at pH 3 are significantly below the isoelectric point and thus have a positive charge (AL-09 has a theoretical charge of +7; AL-103 has +9). In both instances, the rates of fibril formation at low pH values increased over that found at pH 7, where the proteins have a net negative charge (AL-09 has a theoretical charge of -2; AL-103 has -1). We have previously shown that low pH alone can enhance AL-103 fibril formation (t50 for that reaction is equal to 295 hours) [28], but in the case of AL-103 (figure 6b) there is unambiguously a further acceleration at low pH in the presence of heparan sulfate.
Figure 6.
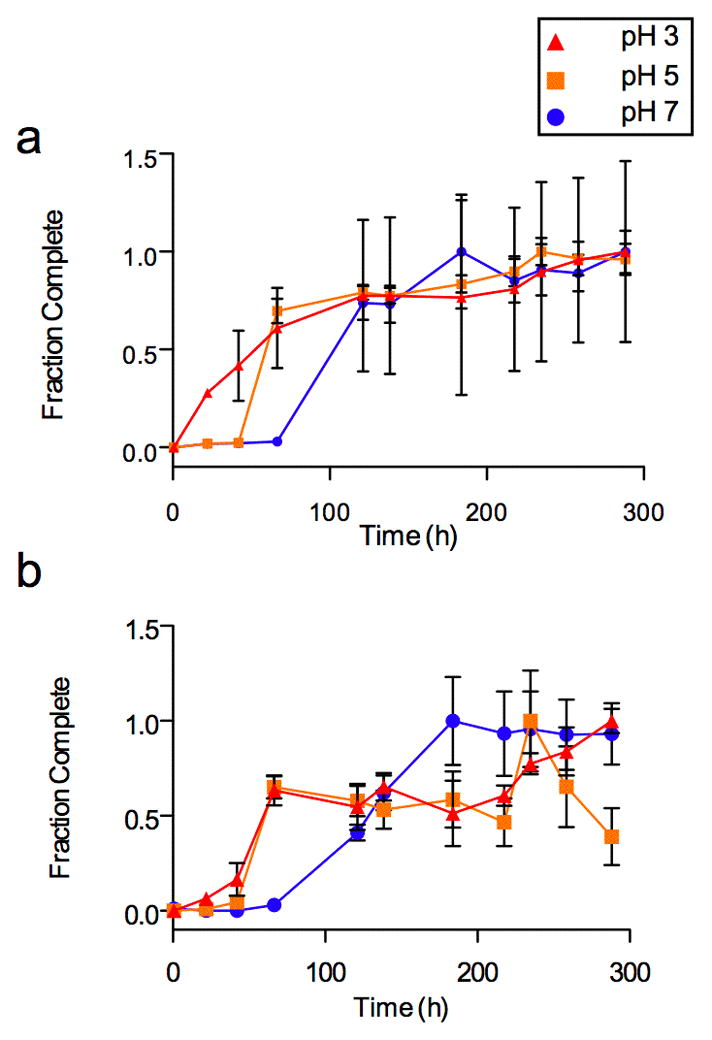
pH dependence of the GAG effect on fibril formation
Fibril formation is shown by the increase in ThT fluorescence (normalized) versus time in hours. Heparan sulfate was present at a concentration of 0.1mg/mL. a) AL-09 fibril nucleation in the presence of heparan sulfate, pH 3–7. b) AL-103 fibril nucleation in the presence of heparan sulfate, pH 3–7.
Our data with AL-103 show a clear acceleration of fibril formation due to GAGs and GAG-like molecules, consistent with previous studies. AL-09 fibril formation in the presence of GAGs is more ambiguous, but this befits the difficulty of accelerating AL-09 fibril formation that we have observed previously [28].We observe charge and size dependence on the acceleration of AL-103 amyloid formation. The evidence of binding based on ITC creates a contradiction; we can measure interactions with only heparan sulfate, indicating that while binding may occur it is not necessary for the acceleration of fibril formation observed with heparin and dextran sulfate. AL-103 amyloid formation reactions show several other hallmarks of an electrostatic interaction, namely concentration dependence and disruption through the addition of salt. To account for these apparent contradictions, we propose that the acceleration of fibril formation due to GAGs is the result of a transient electrostatic interaction that acts to stabilize and accelerate the formation of a fibril nucleus (Figure 7). This model explains the unique features of the fibril formation kinetics that point to an interaction between the two species while reconciling with the fact that no such interaction can be detected.
Figure 7.
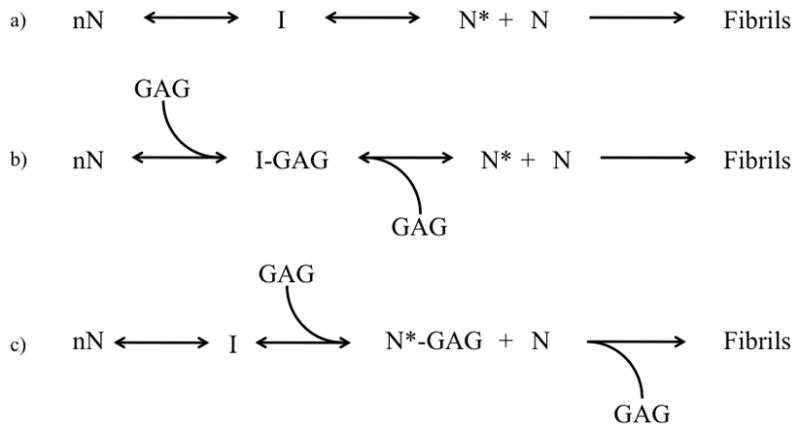
Proposed model of glycosaminoglycan effect on AL fibril formation
Under normal conditions, shown in (a), an unknown number of natively folded AL-103 monomers (nN) undergo a conformational change. This altered conformation is prone to aggregate, forming an intermediate I en route to the formation of a nucleus N*. Additional monomers interact with N* to form amyloid fibrils. AL-09 and AL-103 differ in the efficiency of the first two steps; AL-09 appears to populate the nucleus in a very favorable way without the enhancement of cofactors such as GAGs. Based on our fibril formation and calorimetry data, we hypothesize that glycosaminoglycans (GAGs) accelerate AL-103 fibril formation by interacting with and stabilizing a prefibrillar conformation. This is depicted in (b) and (c). In (b), the interaction occurs between the GAG molecule and an intermediate, while in (c) the interaction occurs with the nucleus itself. At this point our data do not give any indication as to which of (b) or (c) are more likely.
Discussion
As immunoglobulins, AL-09 and AL-103 necessarily differ at several points in their amino acid sequence (figure S6). The two proteins have a similar isoelectric point (theoretical pI=4.83 for AL-09, 5.62 for AL-103) and only the K42Q mutation of AL-09 alters the balance of cationic residues. The changes in sequence between the two proteins should not significantly perturb their ability to interact with a highly anionic polymer like the GAGs. This is reflected in figure 3, where the two proteins bind to heparan sulfate with similar affinity. However, this study has shown that AL-09 and AL-103 do differ in their response to GAGs in solution. How can their behavior differ so significantly if the interaction is so similar? This behavior is entirely consistent with their response to other co-solutes. Though the sequences of the two proteins are highly similar, we have previously found that the rate of fibril formation for AL-09 is relatively insensitive to the solution conditions even after rigorous purification of any preformed oligomeric species from solution [28]. This is in direct contrast to AL-103, which shows strong differences in the rate of fibril formation under different solution conditions. Our hypothesis is that the differences we have observed in the dimerization of AL-09 strongly predispose this particular AL protein to fibril formation [30]. Thus, the difference in fibril formation between the two proteins in the presence of GAGs can best be attributed to AL-09’s relative ease of fibril formation rather than to any property of the GAGs. We also note that in spite of the huge acceleration in AL-103 fibril formation due to the presence of GAGs, its fibril formation rate at best matches that of AL-09. These observations support a dominant role for protein sequence differences in the fibril formation pathway and the pathogenesis of AL. It is worth noting that we have observed some variability in the t50 values of the AL-09 fibril formation reaction in physiological solution conditions. In our previous study [28], the t50 was ~250 h, while the t50 for AL-09 alone in this study is ~40 h. We have recently observed well-to-well variability that resembles these differences in the kinetics for AL-09, suggesting that the reaction, while stochastic, may follow at least two divergent pathways.
The effect of GAGs on amyloid fibril formation has been studied intensely due to their association with amyloid deposits in vivo [6–9]. The central question is, does this association have any importance for the pathogenesis of AL or any amyloid disorder? It was recently proposed that GAGs act as a scaffold for the growth of amyloid fibrils based on interactions between the sulfate groups of the GAGs and the amyloidogenic proteins [34]. This mechanism provides a plausible role for GAGs in the formation of amyloid fibrils in living tissue, but based on our results it must be modified to apply to light chain amyloidosis. The central importance of an electrostatic interaction is consistent with the fibril formation kinetics we observed, both for the possible length dependence of the GAG effect and for the disruption of the GAG effect by salt. However, our ITC data clearly show that equilibrium binding of the type described in the scaffolding mechanism is not a requirement for observing acceleration of fibril formation in these AL proteins. While it is certainly possible for an interaction to occur without a detectable change in enthalpy, we think that is unlikely in this case based on previous studies of GAG-protein interactions [35] and the large electrostatic component of this interaction demonstrated by the salt titration in figure 5. We propose that, in vitro, the GAG mediated acceleration of AL fibril formation occurs as a result of an electrostatic interaction between the sulfate groups and a short-lived intermediate during the formation of the fibril nucleus (Figure 7). This interaction would necessarily stabilize the fibril nucleus or its precursor, accelerating the rate of fibril formation dramatically. At a molecular level, positively charged lysine or arginine residues will interact with the negatively charged sulfate moieties of the GAGs and GAG-like molecules. Further studies are needed to identify the specific interaction or interactions necessary for this mechanism.
We showed in figure 2 that there is no difference in the fibril elongation rate in the seeded reactions-the only differences occur in the efficiency of the seeding process as shown in the lag time before the exponential phase of the reaction. These changes in the seeding efficiency and the lack of change in the elongation rate further supports the idea that any GAG effect in the reaction is predominantly occurring at the nucleation stage of fibril formation rather than continually along the length of the growing fibril.
This nucleation dominant mechanism could provide a role for GAGs in the pathogenesis of AL. By stabilizing the nucleus, GAGs and other biological polyanions could reduce the lag time of fibril formation to biologically relevant timescales. Furthermore, through this mechanism the anionic charge density could also influence the location of fibril formation by encouraging local fibril nucleation. However, we have also clearly documented binding between mature fibrils and heparan sulfate (figure 3), again raising the question of whether or not this association occurs incidentally. This notion is especially relevant in light of the suggestion that soluble oligomers rather than fibrils are the disease-bearing agents in the amyloid disorders [36]. AL-09 responds to the presence of heparan sulfate more than any other GAG studied, slowing its fibril nucleation (figure 1a) and enhancing its elongation (figure 2a). While these are minor differences, it is possible that there is an additional effect due to heparan sulfate that is too subtle for our assays.
It is also curious that our immunoglobulin light chains show an affinity for heparan sulfate without showing any affinity for either heparin or dextran sulfate. Particularly from an electrostatic viewpoint these are all highly similar molecules. However, it is known that heparan sulfate proteoglycans have a special role in cell signaling and in association with amyloid fibrils [22, 37]. It is possible that these molecules have a native affinity for proteins in general or for proteins containing the Ig fold (a ubiquitous structural element). We should also note that the heparin used in these assays was highly purified and fractionated while the heparan sulfate was much more polydisperse. Heparin is commercially available in a highly purified form because of its biomedical applications, whereas heparan sulfate is still purified from tissue extracts. It is possible that this difference influenced the ability of heparan sulfate to bind our light chains.
One interesting feature of this study and of our previous work is the ability of dextran sulfate to mimic the effect of GAGs on amyloid fibril formation. The interest in the role of GAGs is based strongly on the association of GAGs with amyloid deposits in vivo, but dextran sulfate has no such association. The influence of a ‘generic’ sulfated polysaccharide (and other polyanions [25]) indicates that any proposed mechanism by which these molecules accelerate fibril formation must not depend on unique interactions. Rather, it appears that the quasi-linear arrangement of negative charges on the associated polymer is the determining factor in the acceleration of fibril formation rather than a specific association between GAG molecules and the amyloidogenic proteins. In some ways this may be analogous to the role of lipid membranes in the formation of fibrils by α-synuclein among others [38].
We have also considered the possibility that the GAGs and GAG-like molecules accelerate fibril formation through macromolecular crowding, but the low GAG concentrations (relative to those seen in typical crowding assays [39–41]) used in these assays and the acceleration of AL-103 fibril formation even in the presence of low molecular weight heparin derivatives speak against this possibility.
At this point, further study is needed before we will be able to describe the exact mechanism through which GAGs might influence the pathogenesis of AL at the molecular level or to determine the precise role of GAGs in the pathophysiology of these diseases.
Materials and Methods
Cloning, Expression, Extraction, and Purification
Recombinant AL-09 and AL-103 proteins were expressed in E. coli and purified as described previously [14, 29]. All proteins were purified by HiLoad 16/60 Superdex 75 column on an AKTA FPLC (GE Healthcare) system. Pure protein was verified by SDS polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis. If necessary, additional purification was performed using a Biorad Uno Q1 anion exchange column.
Reagents
Purified heparin derivatives (disaccharide, tetrasaccharide, hexasaccharide, octasaccharide, decasaccharide, polysaccharide (MW 12000 Da), de-N-sulfated, de-O-sulfated, and over-O-sulfated heparin) were obtained from Neoparin, Inc. (Alameda, CA); all other reagents were obtained from Sigma-Aldrich and were used without further purification. Over-O-sulfated heparin contains at least 3.5 sulfate molecules per disaccharide unit, while the de-O- and de-N-sulfated heparin each have more than 90% of their respective sulfate groups removed. GAG samples were dissolved in 10mM pH 7.4 Tris-HCl, filtered, and either used immediately or aliquoted and stored at −20ºC. Samples were not refrozen after thawing.
Fibril Seeds
AL-09 seeds were prepared as described previously [14]. Briefly, 20 μM AL-09 in the presence of 0.5 M Na2SO4 in 10mM Tris-HCl (pH 7.4) was incubated at 50ºC. AL-103 seeds were prepared from a solution of 20 μM AL-103 incubated in 10mM sodium acetate, boric acid, and sodium citrate (ABC) buffer at pH 2. Samples were incubated at 37ºC and shaken at 250 rpm. Fibril formation was verified by Thioflavin T (ThT) fluorescence and electron microscopy.
Fibril Formation
Fibril formation kinetics were followed by measuring ThT fluorescence on a plate reader (Analyst AD, Molecular Devices) with an excitation wavelength of 440nm and an emission wavelength of 480 nm. All experiments were performed in triplicate using black 96-well polystyrene plates. Plates were incubated at 37ºC and shaken continuously on a Lab-Line titer plate shaker. Each well contained 20μM protein, 150mM NaCl, 0.02% NaN3, 1mg/mL GAGs, and 10μM ThT in 10mM Tris-HCl buffer (pH 7.4) unless otherwise indicated. For pH dependent reactions, a combination of acetate, borate, and citrate (ABC) buffers were used as described in our previous studies [28]. Salt concentrations were chosen based on estimates of GAG sulfate concentration from the literature [14–21, 33, 42, 43]. Any preformed aggregates were removed from protein stocks prior to fibril formation assays by ultracentrifugation to the sedimentation time of a 0.5S particle [44]. Elongation (seeded) experiments included 1.0μL of fibril seeds in each well. The seeds were prepared as described above, washed twice in 10mM Tris-HCl (pH 7.4), and sonicated for 5 minutes in a Branson model 8510 bath sonicator prior to the start of the reaction. Only interior wells of the 96-well plates were used for experiments; the exterior wells were filled with buffer to delay evaporation over time. Total volume for each reaction was 260μL. The average of the triplicate wells was range scaled. t50 values represent the midpoint of the exponential phase of the reaction, the time where fibril formation is 50% complete. Further calculations for the fibril formation assays were not done, as they required the assumption of too many as yet unknown mechanistic details.
Isothermal Calorimetry (ITC)
ITC was performed with a VP-ITC titration calorimeter (Microcal, Northampton, MA). Soluble protein was prepared in 10mM Tris-HCl (pH 7.4) with 150mM NaCl for a final concentration of 40μM. Fibrils were prepared as described for fibril seeds and washed twice with 10mM Tris-HCl (pH 7.4) containing 150mM NaCl. For calculation purposes, fibril concentration was estimated to be 20μM. GAGs and GAG-like molecules were prepared in the same buffer at concentrations of up to 8mg/mL for polysaccharide heparin, 5.4mg/mL for heparan sulfate, and 10mg/mL for dextran sulfate. A 298μL syringe was used to deliver the GAGs in 58 injections (1 at 3 μL followed by 57 at 5μL) into the calorimetric cell containing 1.41mL of either the precursor protein or fibrils. ITC experiments were performed at both 25 and 35ºC and at multiple concentrations of protein and ligand. Control titrations were done to measure the heat arising from the injection of GAGs into the solution.
Electron Microscopy (EM)
A 3μl fibril sample was placed on a 300 mesh copper formvar/carbon grid and air-dried. The sample was negatively stained with 4% uranyl acetate, washed, air-dried and inspected on a Philips Tecnai T12 transmission electron microscope at 80kV.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. William Kirk for technical assistance with ITC. Funding provided by NIH GM071514 (MRA), AHA SDG0630077N (MRA), and NIH F30DK082169 (DJM). We would also like to acknowledge members of the Ramirez-Alvarado lab for review of this manuscript.
Abbreviations
- GAG
glycosaminoglycans
- AL
light chain amyloidosis
- ThT
Thioflavin T
- ITC
isothermal titration calorimetry
- SEM
standard error of the mean
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang X, Hammer ND, Chapman MR. The molecular basis of functional bacterial amyloid polymerization and nucleation. J Biol Chem. 2008;283:21530–21539. doi: 10.1074/jbc.M800466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wickner R, Edskes H, Shewmaker F, Nakayashiki T. Prions of fungi: inherited structures and biological roles. Nat Rev Micro. 2007;5:611–618. doi: 10.1038/nrmicro1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tyedmers J, Madariaga ML, Lindquist S. Prion switching in response to environmental stress. Plos Biol. 2008;6:e294. doi: 10.1371/journal.pbio.0060294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maji S, Perrin M, Sawaya M, Jessberger S, Vadodaria K, Rissman R, Singru P, Nilsson K, Simon R, Schubert D, Eisenberg D, Rivier J, Sawchenko P, Vale W, Riek R. Functional Amyloids As Natural Storage of Peptide Hormones in Pituitary Secretory Granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamada D, Yanagihara I, Tsumoto K. Engineering amyloidogenicity towards the development of nanofibrillar materials. Trends Biotechnol. 2004;22:93–97. doi: 10.1016/j.tibtech.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Magnus JH, Stenstad T, Kolset SO, Husby G. Glycosaminoglycans in extracts of cardiac amyloid fibrils from familial amyloid cardiomyopathy of Danish origin related to variant transthyretin Met 111. Scand J Immunol. 1991;34:63–69. doi: 10.1111/j.1365-3083.1991.tb01521.x. [DOI] [PubMed] [Google Scholar]
- 7.Ohishi H, Skinner M, Sato-Araki N, Okuyama T, Gejyo F, Kimura A, Cohen AS, Schmid K. Glycosaminoglycans of the hemodialysis-associated carpal synovial amyloid and of amyloid-rich tissues and fibrils of heart, liver, and spleen. Clin Chem. 1990;36:88–91. [PubMed] [Google Scholar]
- 8.Alexandrescu A. Amyloid accomplices and enforcers. Protein Science. 2004;14:1–12. doi: 10.1110/ps.04887005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snow AD, Kisilevsky R. Temporal relationship between glycosaminoglycan accumulation and amyloid deposition during experimental amyloidosis. A histochemical study. Lab Invest. 1985;53:37–44. [PubMed] [Google Scholar]
- 10.Pollack SJ, Sadler, Hawtin SR, Tailor VJ, Shearman MS. Sulfated glycosaminoglycans and dyes attenuate the neurotoxic effects of beta-amyloid in rat PC12 cells. Neurosci Lett. 1995;184:113–116. doi: 10.1016/0304-3940(94)11182-i. [DOI] [PubMed] [Google Scholar]
- 11.Timmer NM, Schirris TJ, Bruinsma IB, Otte-Holler I, van Kuppevelt TH, de Waal RM, Verbeek MM. Aggregation and cytotoxic properties towards cultured cerebrovascular cells of Dutch-mutated Abeta40 (DAbeta(1–40)) are modulated by sulfate moieties of heparin. Neurosci Res. 2010;66:380–389. doi: 10.1016/j.neures.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Valle-Delgado JJ, Alfonso-Prieto M, de Groot NS, Ventura S, Samitier J, Rovira C, Fernandez-Busquets X. Modulation of A{beta}42 fibrillogenesis by glycosaminoglycan structure. FASEB J. 2010 doi: 10.1096/fj.09-153551. Article in press. [DOI] [PubMed] [Google Scholar]
- 13.Woods AG, Cribbs DH, Whittemore ER, Cotman CW. Heparan sulfate and chondroitin sulfate glycosaminoglycan attenuate beta-amyloid(25–35) induced neurodegeneration in cultured hippocampal neurons. Brain Res. 1995;697:53–62. doi: 10.1016/0006-8993(95)00775-l. [DOI] [PubMed] [Google Scholar]
- 14.McLaughlin RW, De Stigter JK, Sikkink LA, Baden EM, Ramirez-Alvarado M. The effects of sodium sulfate, glycosaminoglycans, and Congo red on the structure, stability, and amyloid formation of an immunoglobulin light-chain protein. Protein Sci. 2006;15:1710–1722. doi: 10.1110/ps.051997606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suk JY, Zhang F, Balch WE, Linhardt RJ, Kelly JW. Heparin accelerates gelsolin amyloidogenesis. Biochemistry. 2006;45:2234–2242. doi: 10.1021/bi0519295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohlberg JA, Li J, Uversky VN, Fink AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry. 2002;41:1502–1511. doi: 10.1021/bi011711s. [DOI] [PubMed] [Google Scholar]
- 17.Motamedi-Shad N, Monsellier E, Torrassa S, Relini A, Chiti F. Kinetic analysis of amyloid formation in the presence of heparan sulfate. Faster unfolding and change of pathway. Journal of Biological Chemistry. 2009:1–24. doi: 10.1074/jbc.M109.018747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borysik A, Morten I, Radford S, Hewitt E. Specific glycosaminoglycans promote unseeded amyloid formation from β2-microglobulin under physiological conditions. Kidney Int. 2007;72:174–181. doi: 10.1038/sj.ki.5002270. [DOI] [PubMed] [Google Scholar]
- 19.Relini A, De Stefano S, Torrassa S, Cavalleri O, Rolandi R, Gliozzi A, Giorgetti S, Raimondi S, Marchese L, Verga L, Rossi A, Stoppini M, Bellotti V. Heparin Strongly Enhances the Formation of β2-Microglobulin Amyloid Fibrils in the Presence of Type I Collagen. Journal of Biological Chemistry. 2007;283:4912–4920. doi: 10.1074/jbc.M702712200. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto S, Yamaguchi I, Hasegawa K, Tsutsumi S, Goto Y, Gejyo F, Naiki H. Glycosaminoglycans enhance the trifluoroethanol-induced extension of beta 2-microglobulin-related amyloid fibrils at a neutral pH. J Am Soc Nephrol. 2004;15:126–133. doi: 10.1097/01.asn.0000103228.81623.c7. [DOI] [PubMed] [Google Scholar]
- 21.Ren R, Hong Z, Gong H, Laporte K, Skinner M, Seldin DC, Costello CE, Connors LH, Trinkaus-Randall V. The role of glycosaminoglycan sulfation in the formation of immunoglobulin light chain amyloid oligomers and fibrils. J Biol Chem. 2010;285:37672–37682. doi: 10.1074/jbc.M110.149575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 23.Varki A. Nothing in Glycobiology Makes Sense, except in the Light of Evolution. Cell. 2006;126:841–845. doi: 10.1016/j.cell.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 24.Shi X, Zaia J. Organ-specific Heparan Sulfate Structural Phenotypes. Journal of Biological Chemistry. 2009;284:11806–11814. doi: 10.1074/jbc.M809637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calamai M, Kumita JR, Mifsud J, Parrini C, Ramazzotti M, Ramponi G, Taddei N, Chiti F, Dobson CM. Nature and significance of the interactions between amyloid fibrils and biological polyelectrolytes. Biochemistry. 2006;45:12806–12815. doi: 10.1021/bi0610653. [DOI] [PubMed] [Google Scholar]
- 26.Gomes MP, Cordeiro Y, Silva JL. The peculiar interaction between mammalian prion protein and RNA. Prion. 2008;2:64–66. doi: 10.4161/pri.2.2.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baden EM, Sikkink LA, Ramirez-Alvarado M. Light chain amyloidosis - current findings and future prospects. Curr Protein Pept Sci. 2009;10:500–508. doi: 10.2174/138920309789351949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin DJ, Ramirez-Alvarado M. Comparison of amyloid fibril formation by two closely related immunoglobulin light chain variable domains. Amyloid. 2010;17:129–136. doi: 10.3109/13506129.2010.530081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Randles EG, Thompson JR, Martin DJ, Ramirez-Alvarado M. Structural alterations within native amyloidogenic immunoglobulin light chains. J Mol Biol. 2009;389:199–210. doi: 10.1016/j.jmb.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baden EM, Owen BA, Peterson FC, Volkman BF, Ramirez-Alvarado M, Thompson JR. Altered dimer interface decreases stability in an amyloidogenic protein. J Biol Chem. 2008;283:15853–15860. doi: 10.1074/jbc.M705347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wetzel R. Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res. 2006;39:671–679. doi: 10.1021/ar050069h. [DOI] [PubMed] [Google Scholar]
- 32.Qin Z, Hu D, Zhu M, Fink AL. Structural characterization of the partially folded intermediates of an immunoglobulin light chain leading to amyloid fibrillation and amorphous aggregation. Biochemistry. 2007;46:3521–3531. doi: 10.1021/bi061716v. [DOI] [PubMed] [Google Scholar]
- 33.Sikkink LA, Ramirez-Alvarado M. Salts enhance both protein stability and amyloid formation of an immunoglobulin light chain. Biophys Chem. 2008;135:25–31. doi: 10.1016/j.bpc.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Motamedi-Shad N, Monsellier E, Chiti F. Amyloid formation by the model protein muscle acylphosphatase is accelerated by heparin and heparan sulphate through a scaffolding-based mechanism. J Biochem. 2009;146:805–814. doi: 10.1093/jb/mvp128. [DOI] [PubMed] [Google Scholar]
- 35.Ancsin JB, Kisilevsky R. The heparin/heparan sulfate-binding site on apo-serum amyloid A. Implications for the therapeutic intervention of amyloidosis. J Biol Chem. 1999;274:7172–7181. doi: 10.1074/jbc.274.11.7172. [DOI] [PubMed] [Google Scholar]
- 36.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elimova E, Kisilevsky R, Szarek WA, Ancsin JB. Amyloidogenesis recapitulated in cell culture: a peptide inhibitor provides direct evidence for the role of heparan sulfate and suggests a new treatment strategy. FASEB J. 2004;18:1749–1751. doi: 10.1096/fj.03-1436fje. [DOI] [PubMed] [Google Scholar]
- 38.Bodner CR, Maltsev AS, Dobson CM, Bax A. Differential phospholipid binding of alpha-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry. 2010;49:862–871. doi: 10.1021/bi901723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatters D. Macromolecular Crowding Accelerates Amyloid Formation by Human Apolipoprotein C-II. Journal of Biological Chemistry. 2002;277:7824–7830. doi: 10.1074/jbc.M110429200. [DOI] [PubMed] [Google Scholar]
- 40.Minton A. Models for Excluded Volume Interaction between an Unfolded Protein and Rigid Macromolecular Cosolutes: Macromolecular Crowding and Protein Stability Revisited. Biophysical Journal. 2004;88:971–985. doi: 10.1529/biophysj.104.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munishkina LA, Cooper EM, Uversky VN, Fink AL. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J Mol Recognit. 2004;17:456–464. doi: 10.1002/jmr.699. [DOI] [PubMed] [Google Scholar]
- 42.Cacace MG, Landau EM, Ramsden JJ. The Hofmeister series: salt and solvent effects on interfacial phenomena. Q Rev Biophys. 1997;30:241–277. doi: 10.1017/s0033583597003363. [DOI] [PubMed] [Google Scholar]
- 43.Broering JM, Bommarius AS. Evaluation of Hofmeister effects on the kinetic stability of proteins. J Phys Chem B. 2005;109:20612–20619. doi: 10.1021/jp053618+. [DOI] [PubMed] [Google Scholar]
- 44.Zagorski MG, Yang J, Shao H, Ma K, Zeng H, Hong A. Methodological and chemical factors affecting amyloid beta peptide amyloidogenicity. Methods Enzymol. 1999;309:189–204. doi: 10.1016/s0076-6879(99)09015-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.