

Abstract
Poly(ω-pentadecalactone-co-p-dioxanone) [poly(PDL-co-DO)] copolyesters are copolymers of an isodimorphic system, which remain semicrystalline over the whole range of compositions. Here, we evaluated enzymatically synthesized poly(PDL-co-DO) copolymers as new materials for biomedical applications. In vivo experiments using mice showed that the copolyesters are well tolerated, with tissue responses that are comparable to poly(p-dioxanone). In addition, the copolymers were found to degrade hydrolytically at controlled rates over a period of several months under physiological conditions. The poly(PDL-co-DO) copolymers with up to 69 mol% DO units were successfully transformed to free-standing nanoparticles that are capable of encapsulating an anticancer drug, doxorubicin, or a polynucleotide, siRNA. Drug- or siRNA-loaded nanoparticles exhibited controlled and continuous release of agent over many weeks. In addition, siLUC-encapsulated poly(PDL-co-DO) nanoparticles were active in inhibiting luciferase gene expression in LUC-RKO cells. Because of substantial differences in structure and hydrophobicity between PDL and DO units, poly(PDL-co-DO) biodegradation rate and physical properties can be tuned over a wide range depending on the copolymer composition. Our results demonstrate that the semicrystalline and biodegradable poly(PDL-co-DO) copolyesters are promising biomaterials to serve as drug carriers, as well as potential raw materials for constructing bioabsorbable sutures and other medical devices.
Keywords: Poly(ω-pentadecalactone-co-p-dioxanone), doxorubicin, siRNA, biodegradable nanoparticles, drug delivery
1. Introduction
Biodegradable aliphatic polyesters, including poly(glycolide), poly(lactide), poly(glycolide-co-lactide), and poly(p-dioxanone), have been extensively used for fabricating bioabsorbable sutures, orthopedic devices, and controlled release drug carriers [1–7]. These commercial polyesters are currently produced via metal-catalyzed polymerization reactions and thus contain a small amount of residual metal catalysts that are extremely difficult to remove during the polymer purification processes [8–10].
The diverse nature of medical devices demands that the raw materials for constructing such devices possess versatile biodegradability and physical/mechanical properties. Copolyesters containing both highly hydrophobic and hydrophilic repeat units, such as poly(ω-pentadecalactone-co-p-dioxanone) [poly(PDL-co-DO)] [11], are of great interest because of the potential to tune their biodegradability and physical properties over a wide range by adjusting the comonomer ratio of the polymers. In addition, upon complete degradation of poly(PDL-co-DO), the PDL units in the copolymer are converted to ω–hydroxypentadecanoic acid, a hydroxyl-substituted fatty acid that is expected to pose minimal adverse health effects.
Synthesis of poly(PDL-co-DO) copolyesters (Fig. 1) via ring-opening copolymerization of ω-pentadecalactone (PDL) with p-dioxanone (DO) using Candida antarctica lipase B as the catalyst were recently reported [11]. The poly(PDL-co-DO) copolymers were found to contain nearly random sequences of PDL and DO units in the polymer chains. Solid state characterization of the copolyesters with various compositions revealed that the copolymers belong to an isodimorphic system and remain highly crystalline over the whole range of compositions. The poly(PDL-co-DO) copolyesters crystallize in either poly(PDL) or poly(DO) phase except at the pseudoeutectic composition (70 mol% DO) where poly(PDL) and poly(DO) crystals coexist. The potential utility of these copolymers for biomedical applications have not yet been explored. Since the enzymatically synthesized poly(PDL-co-DO) copolymers are metal-free and high purity aliphatic polyesters, they are appropriate for medical uses [11, 12]. Herein, we describe the biocompatibility and biodegradability of poly(PDL-co-DO) copolyesters, the feasibility of transforming the bulk copolymers to free-standing nanoparticles, and use of the nanoparticles to encapsulate therapeutic drugs (e.g., anticancer drug and oligonucleotides) for controlled release and delivery.
Figure 1.

Chemical structure of poly(PDL-co-DO).
2. Materials and methods
2.01. Materials
ω-Pentadecalactone (PDL, ≥98%), toluene (anhydrous, 99.8%), and diphenyl ether (99%) were obtained from Aldrich Chemical Co. and were used as received. p-Dioxanone (DO, ≥99%) was purchased from Leap Labchem Scientific Co. in China. It was dried over calcium hydride and then was vacuum distilled under nitrogen. Immobilized CALB (Candida antarctica lipase B supported on acrylic resin) or Novozym 435, doxorubicin (DOX), poly(vinyl alcohol) (PVA, Mw= 30,000–70,000, 87–89% hydrolyzed), chloroform (HPLC grade), chloroform-d, and methanol (99.9%) were also obtained from Aldrich Chemical Co. The lipase catalyst was dried at 50 °C under 2.0 mmHg for 20 h prior to use. Stably expression luciferase RKO (LUC-RKO) cells were maintained in DMEM (Gibco) containing 10% (v/v) fetal bovine serum (FBS), 1% (w/v) penicillin-streptomycin, and 0.3 mg/ml Hygromycin B at 37 °C under a 5% CO2 humidified atmosphere. siRNA targeted against the luciferase gene (siLUC): sense sequence 5'-G.C.U.A.U.G.A.A.G.C.G.C.U.A.U.G.G.G.C.dT.dT-3'; antisense sequence 5'-P.G.C.C.C.A.U.A.G.C.G.C.U.U.C.A.U.A.G.C. dT.dT-3'. siRNA mimic (m-siRNA): sense sequence 5'-G.G.C.T.A.C.G.T.C.C.A.G.G.A.G.C.G.C.A.C.C-3'; antisense sequence 5'-T.G.C.G.C.T.C.C.T.G.G.A.C.G.T.A.G.C.C.T.T-3'. siLUC were custom synthesized by Dharmacon (Lafeyette, CO), and the m-siRNA was custom synthesized by W.M. Keck Facility (Yale University).
2.02. Synthesis of poly(PDL-co-DO) copolyesters
The poly(PDL-co-DO) copolymers with various compositions and reference poly(p-dioxanone) (PDO) were synthesized according to procedures reported previously [11]. The composition, molecular weight, and polydispersity of the purified polymers are shown in Table 1. As described previously [11], the poly(PDL-co-DO) copolyesters contain near random arrangements of PDL and DO units in the polymer chains.
Table 1.
Composition, Molecular Weight, and Polydispersity of The Synthesized Poly(PDL-co-DO) Copolyesters
| Sample | P58D42 | P31D69 | P24D76 | PDO |
|---|---|---|---|---|
| PDL/DO feed ratio (mol/mol) | 30:70 | 25:75 | 15:85 | 0:100 |
| PDL/DO unit ratio (mol/mol)a | 58:42 | 31:69 | 24:76 | 0:100 |
| Mn (Da)b | 26900 | 18000 | 14300 | 6000 |
| Mw (Da)b | 52800 | 37000 | 26000 | 22000 |
| Polydispersity (Mw/Mn) | 2.0 | 2.1 | 1.8 | 3.7 |
Determined by the proton NMR spectra,
Measured by GPC using narrow polydispersity polystyrene standards.
2.03. Preparation of free-standing films from poly(PDL-co-DO) copolymers and PDO reference homopolymer
Solutions of poly(PDL-co-DO) copolymers in chloroform and PDO in 1,1,1,3,3,3-hexafluoro-2-propanol (0.5 g of polymer dissolved in 3 ml of solvent) were each evenly spreaded on a micro glass slide placed on a flat surface. The polymer solutions were left at ambient temperature for 12 h to allow evaporation of the solvents to form polymer films. The glass slides carrying the polymer films were then soaked in de-ionized water for 24 h to release the films from glass surface. The free-standing polyester films were dried in air at ambient temperature for 24 h and were subsequently baked in an oven at 45 °C for additional 24 h. The polymer films had approximate thickness of 0.3–0.5 mm.
2.04. In vivo testing of biocompatibility of poly(PDL-co-DO) copolymers
All procedures were performed in accordance with the regulations adopted by the National Institutes of Health and approved by the Animal Care and Use Committee of Yale University. Disk-shaped implants were punched out from polymer films using a sterile 6 mm diameter biopsy punch (Acuderm Inc.). Discs were implanted SC as described previously [13]. At 2 and 4 wk, implants were excised en bloc, fixed in formalin and embedded in paraffin, sectioned and stained with hematoxylin and eosin, and analyzed by histomorphometry as described previously [13].
2.05. Fabrication of blank and DOX-loaded poly(PDL-co-DO) nanoparticles
Blank and doxorubicin (DOX)-loaded nanoparticles were fabricated using a modified oil-in-water single emulsion technique. Specifically, 100 mg of poly(PDL-co-DO), and optionally 5 mg of DOX, were dissolved in 2 mL of methylene chloride. The resultant solution was added to 4 mL of 5 wt% poly(vinyl alcohol) (PVA) aqueous solution. The emulsion mixture was sonicated three times with each sonication lasting for 10 seconds. Subsequently, the sonicated mixture was poured into 100 mL of 0.3 wt% PVA aqueous solution and the whole mixture was magnetically stirred in an open beaker at room temperature for 3 h. This process allows nanoparticles to form via gradual evaporation of the CH2Cl2 solvent. The nanoparticles were isolated by centrifugation, washed with de-ionized water three times, and then frozen and lyophilized.
2.06. Nanoparticle characterization and measurement of DOX contents in nanoparticles
The surface morphology and particle sizes of poly(PDL-co-DO) nanoparticles were analyzed using a XL30 ESEM scanning electron microscope (FEI Company). Particle samples were mounted on an aluminum stub using carbon adhesive tape and sputter-coated with a mixture of gold and palladium (60:40) in an argon atmosphere under low pressure using a Dynavac Mini Coater. The image-analysis application program, ImageJ (developed by Wayne Rasband, NIH), was used to measure particle diameters, calculate average particle sizes, and determine particle size distributions.
The amount of DOX encapsulated in poly(PDL-co-DO) nanoparticles was determined by measuring the intrinsic fluorescence of DOX using a SpectraMax spectrofluorometer (Molecular Devices). DOX-loaded nanoparticles (5 mg) were dissolved in dimethyl sulfoxide (DMSO, 1 ml). The fluorescence intensity of DOX in the solution was measured at 590 nm emission wavelength (with 470 nm excitation wavelength).
2.07. In vitro drug release profiles of DOX-loaded poly(PDL-co-DO) nanoparticles
To determine drug release rates, a pre-determined amount of DOX-loaded nanoparticles was incubated in Dulbecco's phosphate buffered saline (PBS) solution at 37 °C on a rotary shaker set at 100 rpm. Aliquots of the supernatant were taken at various time intervals and analyzed by fluorescence spectroscopy to determine amount of released DOX.
2.08. Hydrolytic degradation of poly(PDL-co-DO) nanoparticles
Blank poly(PDL-co-DO) nanoparticles were suspended in a 10 mM PBS solution with pH value of 7.4. The resultant mixture in a flask was placed in a rotary shaker, and was shaked at 100 rpm speed, 37 °C constant temperature. At different time intervals, a small fraction of the mixture was removed from the flask and was centrifuged to separate the nanoparticles from the buffer solution. The sediment formed upon centrifugation was lyophilized and subsequently analyzed by gel permeation chromatography (GPC) using polystyrene standards to determine the molecular weight of the degraded polymer particles.
2.09. Studies on siRNA-loaded poly(PDL-co-DO) nanoparticles
Nanoparticle fabrication
Poly(PDL-co-DO) nanoparticles encapsulating siRNA were prepared by a double-emulsion solvent evaporation technique. In brief, siRNA was reconstituted in deionized water, and was then combined with spermidine at different N/P ratios (molar ratios of the triamine nitrogen to the polynucleotide phosphate). The resultant mixture was stirred at room temperature for 15 min to allow spermidine-siRNA complexes to form. This aqueous solution (0.1 ml) was then added dropwise to a poly(PDL-co-DO) solution (50 mg/ml, 2 ml) in methylene chloride to form the first emulsion. Addition of this emulsion to 4 ml of 5% PVA aqueous solution and subsequent sonication (three times, 10 seconds each time) of the whole mixture formed a double emulsion. This double emulsion was poured into 0.3% PVA aqueous solution (100 ml) and the final emulsion was stirred in an open beaker for 3 h to allow the methylene chloride solvent to evaporate. The particles, which form as a result of solvent evaporation, were collected by centrifugation, washed with de-ionized water three times, and then lyophilized.
Encapsulation efficiency measurement
To determine loading of siRNA in poly(PDL-co-DO) nanoparticles, 5–10 mg of siRNA-loaded nanoparticles were vortexed in 0.5 ml of methylene chloride and the copolymer was allowed to dissolve at room temperature for 30 min. The released siRNA was extracted twice from the organic phase with each time using 0.5 ml TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH = 7.4). The TE buffer was added to the organic solution, and the resultant mixture was vortexed vigorously for 1 min and then centrifuged at 12,000 rpm for 5 min at 4 °C. The two portions of the extracting aqueous solution were combined and the total solution was analyzed for double-stranded RNA content using the QuantIT™ PicoGreen™ assay according to the procedures recommended by the manufacturer (Invitrogen). A standard curve correlating fluorescence and siRNA concentration was used to determine the amount of siRNA in the aqueous solution.
In vitro siRNA release profile
siRNA-loaded nanoparticles (5–10 mg) were suspended in 1.0 ml PBS solution (pH = 7.4) and incubated at 37 °C with gentle shaking (100 rpm). The amount of released siRNA from the nanoparticles was measured at various time intervals over 60 days. During each measurement, the particle-solution mixture was centrifuged at 13,000 rpm for 5 min. The supernatant was removed and its siRNA content was analyzed using the same method as described above in “Encapsulation Efficiency Measurement” section. The precipitated particles were re-suspended in an equal volume of fresh PBS solution and incubated under the same conditions for continuous monitoring of siRNA release.
2.10. In vitro luciferase gene silencing using siRNA-loaded poly(PDL-co-DO) nanoparticles
LUC-RKO cells were seeded in 24-well plates (1.5×105 cells per well of 0.5 ml size) and were allowed to grow overnight. The cells were treated with different doses of siRNA-loaded P31D69 [poly(PDL-co-69%DO), see Table 1 for characteristics] nanoparticles in culture medium at 37 °C. After 24 h incubation of the cells, the culture medium was replaced with the fresh medium. The cells were washed twice with PBS solution after additional 24–48 h growth and were subsequently treated with 200 μl Report Lysis Buffer at room temperature for 15 min. The mixture of cell and buffer in each well was transferred into a microcentrifuge tube and was vortexed for 20 seconds. The cell lysates were collected by centrifugation at 12,000 rpm for 2 min at 4 °C. Twenty microliters of the supernatant was mixed with 100 μl Luciferase Assay Reagent (Luciferase Assay System, Promega Co., Madison, WI) and the luminescence of the mixture was measured by a luminometer. The protein concentrations of the samples were determined by using a protein assay kit (Micro BCA™ Protein assay kit, Pierce). Luciferase activity of a sample was normalized on the basis of its protein content.
2.11. Statistical analysis
Statistical tests were performed with a two-sided Student's T-test. A P-value of 0.05 or less was considered to be statistically significant.
3. Results and discussion
3.1. Biocompatibility of poly(PDL-co-DO) copolymers
The host response to films made from poly(PDL-co-DO) was evaluated at 2 and 4 wk after subcutaneous implantation in 3 month-old C57Bl6 mice. All materials studied—PDO, P58D42 and P24D76—elicited a typical foreign body response (FBR) characterized by the presence of foreign body giant cells at the material surface and the formation of collagenous capsules largely devoid of blood vessels (Fig. 2). PDO implants displayed accelerated degradation and could not be detected in 4 wk samples (Fig. 2B). Despite the complete degradation of PDO, remnants of the FBR could be observed in the form of enlarged inflammatory cells and collagenous capsule. In contrast to PDO, P58D42 and P24D76 did not undergo degradation at 4 wk and both copolymers were encapsulated (Fig. 2 D, F). Overall, capsule thickness appeared to increase between 2 and 4 wk in all samples and this was confirmed by evaluation of capsule thickness via histomorphometry (Fig. 3). P24D76 elicited the most robust encapsulation at 2 and 4 wks with capsule formation significantly larger than that elicited by PDO. Capsule formation elicited by P58D42 did not differ from either PDO or P24D76.
Figure 2.

Foreign body response to poly(PDL-co-DO) copolyesters. Representative images of copolyesters implanted SC in mice for 2 (A, C, E) and 4 wk (B, D, F) and stained with H&E are shown. PDO (A, B), P58D42 (C, D), and P24D76 (E, F) elicited normal recruitment of inflammatory cells and foreign body giant cell formation (arrows). PDO implants displayed accelerated degradation and could not be detected at 4 wk. Remnants of a persistent inflammatory response and encapsulation (arrowhead in B) were evident. Bar = 50 micron (A–F).
Figure 3.
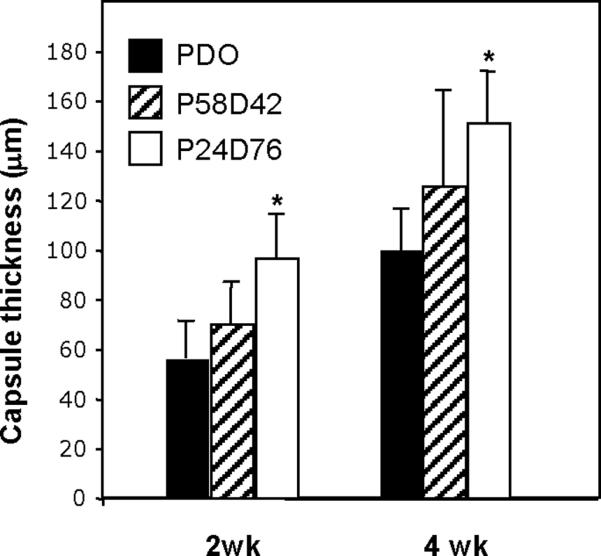
Variation in capsule thickness elicited by copolyesters. Polymers were implanted s.c. for 2 and 4 wk, explanted, and analyzed by histomorphometry. P24D76 elicited the formation of thicker capsules. A total of 25 sections from three mice/group were analyzed. * p ≤ 0.05; n=3.
3.2. Characterization of blank and DOX-loaded poly(PDL-co-DO) nanoparticles
Free-standing, blank nanoparticles were successfully fabricated with P58D42 [poly(PDL-co-42%DO), Table 1] and P31D69 [poly(PDL-co-69%DO)] by using a single emulsion evaporation technique (Fig. 4, A–D). Following the same procedures, P24D76 [poly(PDL-co-76%DO), Table 1] formed nanoparticle agglomerates (Fig. 4, E and F) due presumably to the higher hydrophilic DO unit content and lower molecular weight of the copolymer. As we reported earlier, adding trehalose prior to lyophilization alleviates aggregation of copolyester nanoparticles [14]. The SEM images of A, C, and E in Fig. 4 correspond to the nanoparticles without trehalose treatment, whereas the micrographs B, D, and F represent the trehalose-treated particles. As the result of its low solubility in common organic solvents, reference polymer PDO could not be transformed to nanoparticles. Because of their ability to form singly suspended nanoparticles, P58D42 and P31D69 nanoparticles were employed for subsequent biodegradation and drug delivery studies.
Figure 4.

SEM images of the blank nanoparticles fabricated from various poly(PDL-co-DO) copolymers. A, B: P58D42; C, D: P31D69; E, F: P24D76. B, D, F represent the trehalose-treated particles that were resuspended in distilled water, and then mounted on an aluminum stub with glass cover slip.
To test their characteristics as drug delivery vehicles, P58D42 and P31D69 nanoparticles were prepared with encapsulated DOX (Fig. 5). The nanoparticles exhibited a smooth surface and near spherical shape (Fig 5A,B). Nanoparticles formed with P58D42 and P31D69 had similar size distributions, varying from 150 to 450 nm (with most particles having a diameter between 175 and 300 nm) and 125 to 300 nm, respectively (Fig. 5C,D). The average particle size, particle yield, DOX content, and DOX encapsulation efficiency were determined for nanoparticles made from each polymer (Table 2). Drug loading and encapsulation efficiency were higher for the particles of DO-rich copolymer P31D69 vs. those of P58D42, which is probably due to the increased hydrophilicity of the former polymer and resultant stronger interactions of the polymer chains with DOX during nanoparticle preparation.
Figure 5.

SEM images and particle size distributions of DOX-loaded nanoparticles prepared from poly(PDL-co-DO) copolymers P58D42 (A,C) and P31D69 (B,D).
Table 2.
Characterization of DOX-loaded Poly(PDL-c-DO) Nanoparticles
| Polymer | Mean Size (nm) | Particle Yield (wt%) | Drug Loading (wt%) | Encapsulation Efficiency (%) |
|---|---|---|---|---|
| P58D42 | 235 ± 47 | 60 | 1.3 | 26 |
| P31D69 | 207 ± 44 | 54 | 2.1 | 42 |
3.3. Hydrolytic degradation of blank poly(PDL-co-DO) nanoparticles
The kinetics of hydrolytic degradation of poly(PDL-co-DO) nanoparticles was investigated by measuring the polymer molecular weight (Mn and Mw) as a function of the incubation time in PBS solution for P58D42 and P31D69 nanoparticles (Fig. 6). Over the period of 70 days, the number-average molecular weight (Mn) decreased from 22,000 to 6,900 for P31D69 nanoparticles and from 17,000 to 7,000 for P58D42 nanoparticles (Fig. 6A). The changes in the weight-average molecular weight (Mw) of the polymers followed a similar trend (Fig. 6B). Clearly, both P58D42 and P31D69 copolyesters possess significant degradability under physiological conditions. The degradation profiles of the copolymers were biphasic with an initial rapid degradation occurring over the first 3 days followed by a gradual molecular weight decrease over the subsequent 60–70 days. As reported previously, poly(PDL-co-DO) copolyesters are copolymers of an isodimorphic system and remain semicrystalline over the whole range of the copolymer compositions. Thus, the observed initial fast degradation of P58D42 and P31D69 could result mainly from hydrolytic cleavage of the polymer chains in the amorphous regions of the particle samples since these regions are more accessible to water. The subsequent slower degradation rates are likely due to the polymer chain cleavage associated with less-accessible crystalline phases of the copolymers.
Figure 6.

Changes of number-average molecular weight (A) and weight-average molecular weight (B) as a function of degradation time for blank P58D42 (■) and P31D69 (○) nanoparticles incubated in PBS solution (pH = 7.4) at 37 °C.
3.4. In vitro drug release profile of DOX-loaded poly(PDL-co-DO) nanoparticles
DOX was slowly released from DOX-loaded P58D42 and P31D69 nanoparticles (Fig. 7). Nanoparticles exhibited a biphasic release of the drug, consisting of an initial burst followed by a sustained release. Approximately 30–40% of the total encapsulated DOX was released within the first day, with an additional 30–40% of the drug continually released over the next 20 days. The rapid release of DOX during the first day is consistent with an initial period of rapid polymer degradation of the poly(PDL-co-DO) particles (Fig. 6). The DOX release rate is higher for P31D69 nanoparticles than for P58D42 nanoparticles, suggesting that the increase in DO unit content and, thus hydrophilicity of the poly(PDL-co-DO) copolymer, accelerated release for DOX.
Figure 7.

Cumulative DOX release from DOX-encapsulated P58D42 and P31D69 nanoparticles incubated in PBS at 37 °C. Data are given as mean ± SD (n = 3).
3.5. Preparation and drug release of m-siRNA-loaded poly(PDL-co-DO) nanoparticles
Targeted post-transcriptional silencing of genes using short interfering RNA (siRNA) is a promising strategy to treat various diseases [15–17]. The major limitations of siRNA as a therapeutic are rapid degradation of unprotected siRNA, its non-specific distribution, and poor cellular uptake [18, 19]. Thus, delivery systems capable of protecting and transporting siRNA across the extra- and intracellular barriers to the tumor site are essential [20]: nanoparticles of degradable polymers have a great potential in this role [21–24]. However, it is a challenge to encapsulate siRNA in sufficient density into polymeric nanoparticles because of its high solubility in aqueous medium [25–27].
P31D69 nanoparticles were chosen as the carrier for siRNA delivery because relatively high loading and encapsulation efficiency was achieved with DOX (Table 2). To facilitate identification of optimal process conditions for preparing siRNA-encapsulated nanoparticles, a more stable siRNA mimic was used in preliminary studies. Two complementary DNA oligonucleotides were annealed to generate a double-stranded product (which we call mimic siRNA, or m-siRNA), which has the same molecular weight and charge density of siRNA, but contains deoxynucleotides for increased stability. P31D69 nanoparticles loaded with m-siRNA had a smooth surface and near spherical shape with an average diameter of approximately 200 nm (Fig. 8).
Figure 8.

SEM image of m-siRNA-encapsulated P31D69 nanoparticles.
The m-siRNA:polymer ratio influenced m-siRNA loading and encapsulation efficiency (Table 3). With increasing m-siRNA to polymer ratio, the actual m-siRNA loading increased significantly, but the particle size and zeta potential of the nanoparticles did not change significantly. To improve the loading and encapsulation efficiency of nucleic acids, spermidine (a low molecular weight natural triamine) was added to condense and stabilize nucleic acids prior to its encapsulation [28]. Cationic molecules are capable of condensing polynucleotides (such as RNA and DNA) via electrostatic interactions [21, 29, 30], thus effecting the polynucleotide solubility in aqueous medium. Upon increasing the N/P ratio (molar ratio of spermidine nitrogen to the nucleic acid phosphate) from 4:1 to 20:1, the zeta potential, m-siRNA loading, and encapsulation efficiency of the particles decreased moderately (Table 4). The observed lower m-siRNA loading and encapsulation efficiency at a high N/P ratio could result from increased aqueous solubility of m-siRNA-spermidine complexes containing excessive amount of the amine molecules. Use of spermidine in large excess might increase its presence on the surface of the particles, which could partially neutralize and reduce the surface negative charges. However, changes in the N/P ratio had minimal effects on the average size of the nanoparticles (Table 4).
Table 3.
Effects of m-siRNA to Polymer Ratio on The Properties and Encapsulation Efficiency of m-siRNA/P31D69 Nanoparticles
| m-siRN A/Polymer Ratio (pmoles/mg) | Particle Size (nm) | Zeta Potential (mV) | m-siRNA Loading (pmoles/mg polymer) | Encapsulation Efficiency (%) |
|---|---|---|---|---|
| 125 | 203 ± 48 | −25.2 ± 1.4 | 37.5 ± 0.8 | 30.0 ± 0.6 |
| 250 | 196 ± 40 | −28.7 ± 1.9 | 55.2 ± 3.9 | 22.1 ± 1.5 |
| 500 | 225 ± 51 | −33.1 ± 2.4 | 130.7 ± 7.7 | 26.1 ± 1.5 |
Table 4.
Effects of Spermidine to m-siRNA Ratio on The Properties and Encapsulation Efficiency of m-siRNA/P31D69 Nanoparticles
| N/P Ratioa | Particle Size (nm) | Zeta Potential (mV) | m-siRNA Loading (pmoles/mg polymer) | Encapsulation Efficiency (%) |
|---|---|---|---|---|
| 4 : 1 | 193 ± 43 | −23.6 ± 1.2 | 83.0 ± 0.9 | 33.2 ± 0.3 |
| 8 : 1 | 211 ± 49 | −22.1 ± 0.8 | 75.8 ± 2.5 | 30.3 ± 1.0 |
| 16 : 1 | 201 ± 35 | −18.7 ± 1.3 | 76.7 ± 7.5 | 30.7 ± 3.0 |
| 20 : 1 | 221 ± 54 | −16.5 ± 2.2 | 64.6 ± 1.5 | 25.9 ± 0.6 |
Molar ratio of spermidine nitrogen to the nucleic acid phosphate.
The in vitro rate of release of m-siRNA from nanoparticles with different loadings was studied (Fig. 9). All particle formulations exhibit a biphasic release profile, comparable to that of the DOX-encapsulated nanoparticles, with a small burst release over the first 3 days followed by a gradual, sustained release over subsequent 60 days. The highest cumulative percentage of m-siRNA released from the nanoparticles was observed for the particle sample with m-siRNA loading of 55 pmoles/mg polymer. Specifically, the nanoparticles released ~20% m-siRNA in the first day and additional 25% m-siRNA gradually for the next 60 days. We speculate that the sustained release of polynucleotides from these nanoparticles might be crucial for using such particles to continuously inhibit expression of genes in living cells.
Figure 9.

m-siRNA release profiles of P31D69 nanoparticles with various m-siRNA loadings upon incubation in PBS at 37 °C. Data are given as mean ± SD (n = 3).
3.6. In vitro luciferase gene silencing study
Using the optimal process conditions developed for encapsulation of polynucleotides in poly(PDL-co-DO) nanoparticles as described above, P31D69 nanoparticles carrying siLUC were prepared at 27% encapsulation efficiency, which contained 67 pmoles of siLUC per mg of the polymer and had the N/P ratio of 4:1. This siLUC-loaded, P31D69 nanoparticles were used to determine the effectiveness of this approach in inhibition of luciferase gene expression in the LUC-RKO cells. The LUC-RKO cells were treated with different doses (from 10 to 50 nM) of the siRNA in the nanoparticles for 48 or 72 h and compared to a commercial transfection agent, Lipofectamine 2000 (LF2K), which was applied according to the manufacturer's recommended procedures. At 48 h post-transfection and 10 nM siLUC dosage, approximately 30% reduction of luciferase gene expression in LUC-RKO cells was achieved with siLUC/P31D69 nanoparticles (Fig. 10). In comparison, about 76% of the gene expression was inhibited by siLUC/LF2K under the same conditions. Upon increasing the transfection time from 48 to 72 h, the luciferase expression level decreased further by additional 14% for the cells treated with siLUC/P31D69 nanoparticles while no significant change was observed for those cells transfected with siLUC/LF2K. Interestingly, siLUC/LF2K gene inhibition was independent of dose; in contrast, siLUC/P31D69 nanoparticles exhibit dose-dependent inhibition. Thus, when treated with siLUC/P31D69 nanoparticles at 50 nM siLUC dosage for 72 h, the luciferase gene expression in LUC-RKO cells was reduced by 62%. These results indicate that controlled and continuous release of siRNA from nanoparticles could allow such siRNA delivery systems to control expression of target genes in cells over time.
Figure 10.

Inhibition of luciferase gene expression in LUC-RKO cells upon treatment with siLUC/P31D69 nanoparticles or siLUC/LF2K using various siLUC doses and incubation time. Data are given as mean ± SD (n = 3).
4. Conclusion
Poly(PDL-co-DO) copolymers were evaluated as new materials for biomedical applications. In vivo experiments using mice showed that the copolyesters are well tolerated when implanted subcutaneously. In addition, the copolymers were found to degrade hydrolytically over a period of several months under physiological conditions (e.g., 37 °C, pH 7.4). The poly(PDL-co-DO) copolymers with up to 69 mol% DO units were successfully transformed to free-standing nanoparticles that are capable of encapsulating a typical anticancer drug or a polynucleotide (e.g., siRNA). The drug-loaded poly(PDL-co-DO) nanoparticles exhibited controlled and continuous release of the drugs over an extensive period of time (20–60 days). In particular, siLUC-encapsulated poly(PDL-co-DO) nanoparticles were active in inhibiting luciferase gene expression in LUC-RKO cells. Because of substantial differences in structure and hydrophobicity between PDL and DO units, poly(PDL-co-DO) degradation rate and physical properties can vary over a wide range depending on the copolymer composition. The experimental results reported in this paper demonstrate that the semicrystalline and biodegradable poly(PDL-co-DO) copolyesters are promising biomaterials to serve as drug carriers, as well as potential raw materials for constructing bioabsorbable sutures and orthopedic devices.
Acknowledgement
This work was supported by grants from the National Institutes of Health (EB000487 and HL085416) and the China Scholarship Council (CSC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–47. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- [2].Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70:1–20. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- [3].Alexis F, Pridgen EM, Langer R, Farokhzad OC. Nanoparticle technologies for cancer therapy. Drug Deliv. 2010;197:55–86. doi: 10.1007/978-3-642-00477-3_2. [DOI] [PubMed] [Google Scholar]
- [4].Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–51. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- [5].Chu CC. Chapter 42. In: Bronzino JD, editor. The Biomedical Engineering Handbook. Third edition CRC Press; Boca Raton, Florida: 2006. [Google Scholar]
- [6].Kohn J, Langer R. In: Biomaterials Science: an Introduction to Materials in Medicine. Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, editors. Academic Press; New York: 1996. pp. 64–72. [Google Scholar]
- [7].Barrows TH. Degradable implant materials: A review of synthetic absorbable polymers and their applications. Clin Mater. 1986;1:233–57. [Google Scholar]
- [8].Odian GG. Principles of Polymerization. Wiley-Interscience; New York: 2004. [Google Scholar]
- [9].Albertsson AC, Stridsberg KM, Ryner M. Controlled ring-opening polymerization: polymers with designed macromolecular architecture. Adv Polym Sci. 2002;157:41–65. [Google Scholar]
- [10].Bezwada RS, Jamiolkowski DD, Lee IY, Agarwal V, Persivale J, Trenka-Benthin S, et al. Monocryl® suture, a new ultra-pliable absorbable monofilament suture. Biomaterials. 1995;16:1141–8. doi: 10.1016/0142-9612(95)93577-z. [DOI] [PubMed] [Google Scholar]
- [11].Jiang Z, Azim H, Gross RA, Focarete ML, Scandola M. Lipase-catalyzed copolymerization of [omega]-pentadecalactone with p-dioxanone and characterization of copolymer thermal and crystalline properties. Biomacromolecules. 2007;8:2262–9. doi: 10.1021/bm070138a. [DOI] [PubMed] [Google Scholar]
- [12].Akieda H, Shioya Y, Kajita M, Ozu K. Method of producing poly (p-dioxanone), poly (p-dioxanone) monofilaments and method for producing the same. US Patent. 2002:6448367. [Google Scholar]
- [13].Krady MM, Zeng J, Yu J, MacLauchlan S, Skokos EA, Tian W, et al. Thrombospondin-2 modulates extracellular matrix remodeling during physiological angiogenesis. Am J Pathol. 2008;173:879–91. doi: 10.2353/ajpath.2008.080128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu J, Jiang Z, Zhang S, Saltzman WM. Poly ([omega]-pentadecalactone-cobutylene-co-succinate) nanoparticles as biodegradable carriers for camptothecin delivery. Biomaterials. 2009;30:5707–19. doi: 10.1016/j.biomaterials.2009.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cun D, Foged C, Yang M, Frøkjær S, Nielsen HM. Preparation and characterization of poly (dl-lactide-co-glycolide) nanoparticles for siRNA delivery. Int J Pharm. 2010;390:70–5. doi: 10.1016/j.ijpharm.2009.10.023. [DOI] [PubMed] [Google Scholar]
- [16].Huang C, Li M, Chen C, Yao Q. Small interfering RNA therapy in cancer: mechanism, potential targets, and clinical applications. Expert Opin Ther Targets. 2008;12:637–45. doi: 10.1517/14728222.12.5.637. [DOI] [PubMed] [Google Scholar]
- [17].Pirollo KF, Chang EH. Targeted delivery of small interfering RNA: approaching effective cancer therapies. Cancer Res. 2008;68:1247–50. doi: 10.1158/0008-5472.CAN-07-5810. [DOI] [PubMed] [Google Scholar]
- [18].Bartlett DW, Davis ME. Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA-containing nanoparticles. Biotechnol Bioeng. 2007;99:975–85. doi: 10.1002/bit.21668. [DOI] [PubMed] [Google Scholar]
- [19].Patil Y, Panyam J. Polymeric nanoparticles for siRNA delivery and gene silencing. Int J Pharm. 2009;367:195–203. doi: 10.1016/j.ijpharm.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–38. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vijayanathan V, Thomas T, Thomas TJ. DNA Nanoparticles and development of DNA delivery vehicles for gene therapy. Biochemistry. 2002;41:14085–94. doi: 10.1021/bi0203987. [DOI] [PubMed] [Google Scholar]
- [22].Luten J, van Nostrum CF, De Smedt SC, Hennink WE. Biodegradable polymers as non-viral carriers for plasmid DNA delivery. J Control Release. 2008;126:97–110. doi: 10.1016/j.jconrel.2007.10.028. [DOI] [PubMed] [Google Scholar]
- [23].Gary DJ, Puri N, Won YY. Polymer-based siRNA delivery: perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J Control Release. 2007;121:64–73. doi: 10.1016/j.jconrel.2007.05.021. [DOI] [PubMed] [Google Scholar]
- [24].Kim WJ, Kim SW. Efficient siRNA delivery with non-viral polymeric vehicles. Pharm Res. 2009;26:657–66. doi: 10.1007/s11095-008-9774-1. [DOI] [PubMed] [Google Scholar]
- [25].Cohen-Sela E, Chorny M, Koroukhov N, Danenberg HD, Golomb G. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J Control Release. 2009;133:90–5. doi: 10.1016/j.jconrel.2008.09.073. [DOI] [PubMed] [Google Scholar]
- [26].Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J Control Release. 1999;57:171–85. doi: 10.1016/s0168-3659(98)00116-3. [DOI] [PubMed] [Google Scholar]
- [27].Tewes F, Munnier E, Antoon B, Ngaboni Okassa L, Cohen-Jonathan S, Marchais H, et al. Comparative study of doxorubicin-loaded poly (lactide-co-glycolide) nanoparticles prepared by single and double emulsion methods. Eur J Pharm Biopharm. 2007;66:488–92. doi: 10.1016/j.ejpb.2007.02.016. [DOI] [PubMed] [Google Scholar]
- [28].Woodrow KA, Cu Y, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzman WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater. 2009;8:526–33. doi: 10.1038/nmat2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Keller M, Tagawa T, Preuss M, Miller AD. Biophysical characterization of the DNA binding and condensing properties of adenoviral core peptide mu. Biochemistry. 2002;41:652–9. doi: 10.1021/bi0156299. [DOI] [PubMed] [Google Scholar]
- [30].Ghonaim HM, Li S, Blagbrough IS. Very Long Chain N 4, N 9-diacyl spermines: non-viral lipopolyamine vectors for efficient plasmid DNA and siRNA delivery. Pharm Res. 2009;26:19–31. doi: 10.1007/s11095-008-9705-1. [DOI] [PubMed] [Google Scholar]