

Abstract
Methamphetamine (METH) is a drug of abuse with neurotoxic and vascular effects that may be mediated by reactive oxygen species (ROS). However, potential sources of METH-induced generation of ROS are not fully understood. This study is focused on the role of NAD(P)H oxidase (NOX) in METH-induced dysfunction of brain endothelial cells. Treatment with METH induced a time-dependent increase in phosphorylation of NOX subunit p47, followed by its binding with gp91 and p22, and the formation of an active NOX complex. An increase in NOX activity was associated with elevated production of ROS, alterations of occludin levels and increased transendothelial migration of monocytes. Inhibition of NOX by NSC 23766 attenuated METH-induced ROS generation, changes in occludin protein levels and monocyte migration. Because an active NOX complex is localized to caveolae, we next evaluated the role of caveolae in METH-mediated toxicity to brain endothelial cells. Treatment with METH induced phosphorylation of ERK1/2 and caveolin-1 protein. Inhibition of ERK1/2 activity or caveolin-1 silencing protected against METH-induced alterations of occludin levels. These findings indicate an important role of NOX and functional caveolae in METH-induced oxidative stress in brain endothelial cells that contribute to the subsequent alterations of occludin levels and transendothelial migration of inflammatory cells.
Keywords: blood–brain barrier, brain endothelial cells, drug abuse, methamphetamine, oxidative stress, tight junctions
Introduction
Methamphetamine (METH) is a highly addictive synthetic psychostimulant causing neurotoxicity in humans. METH neurotoxicity has been characterized by enhanced release and reduced synaptic reuptake of major monoamine neurotransmitters, including dopamine (DA) and serotonin (5-HT), and is accompanied by a decrease in the number of dopamine transporter (DAT) binding sites. METH enters neurons via the DAT or 5-HT transporter and displaces both vesicular and intracellular DA and 5-HT. The displaced amines can then be oxidized producing reactive oxygen species (ROS). Indeed, increased tissue oxidative stress is known to be one of the major causes of neurotoxicity following METH administration [1, 2]. However, cultured brain endothelial cells have also been shown to be a target for METH toxicity despite the lack of dopaminergic or serotonergic innervations. For example, exposure to METH can alter endothelial cell redox status by depleting cellular glutathione levels [3]. METH exposure leads to intracellular ROS generation in brain endothelial cells and disruption of blood–brain barrier (BBB) functions [4–6].
The BBB is a specialized system of capillary endothelial cells interconnected by intercellular tight junctions (TJs) that form a selectively barrier, which controls the internal environment of the central nervous system (CNS). TJs play a role in regulating the exchanges of substances between the brain and blood resulting in maintaining the homeostatic environment of the brain. TJs are constituted by transmembrane proteins such as occludin, claudins and junctional associated molecules (JAMs). In addition, cytoplasmic zonula occludens (ZO) proteins interact with transmembrane proteins and link them to the actin cytoskeleton [7].
Occludin, a 60–65 kD phosphoprotein, is highly expressed in cerebral endothelium, whereas it is much more sparsely distributed in peripheral endothelia [8]. It consists of four transmembrane domains, which bind to the two extracellular loops of claudin forming the paracellular component of the TJ. Mutation or overexpression of occludin in cultured cells affected both electrical resistance and flux of non-charged solutes [9]. Occludin is anchored to actin cytoskeleton via its binding to ZO-1 protein. Phosphorylation and expression of occludin are regulated by cellular redox status [10].
NADPH oxidase (NOX) is the major enzyme responsible for superoxide production and an important source of ROS in phagocytic cells, which protect the host from bacteria and fungi [11, 12]. Recently, NOX has also been identified in other cell types, including adventitial fibroblasts [13], vascular smooth muscle [14], renal mesangial cells [15] and endothelial cells [16–18]. The NOX complex consists of two membrane-bound components, gp91 (also known as Nox2) and p22, as well as several cytosolic regulatory subunits, including p40, p47, p67 and the small GTPase Rac. Once activated, p47 is phosphorylated and relocated to the cell membrane where it is assembled with gp91/p22 to form an active NOX complex. Although expressed at a lower level, the gp91 containing NOX has also been observed to contribute to ROS formation by vascular endothelial cells [19].
Taking under consideration that ROS may disrupt the BBB and that NOX containing gp91 is an important source of ROS, we hypothesize that NOX is involved in METH-induced alteration of the BBB in brain microvasculature. The present study indicates that exposure to METH results in NOX activation, leading to ROS generation, altered occludin levels and increased transendothelial migration of monocytes.
Materials and methods
Cell culture and treatment factors
Immortalized human brain microvascular endothelial cells (hCMEC/D3 cells) [20] were cultured in EBM-2 medium, supplemented with EGM-2 Bullet-kit, which contains insulin-like growth factor-I (IGF-I), epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), hydrocortisone, ascorbate, gentamycin and 2.5% foetal bovine serum (FBS) (Lonza, Walkersville, MD, USA). All cell culture plates were coated with rat tail collagen type I (BD Biosciences, San Jose, CA, USA) for 1 hr. Methamphetamine hydrochloride (US Pharmacopeia, Rockville, MD, USA) was dissolved in water and added directly into serum-depleted hCMEC/D3 cells with EBM medium (Lonza). Treatment with METH at the concentrations of up to 200 μM for up to 24 hrs did not induce cell death in our model system. NSC 23766 (a selective inhibitor of Rac1 and NOX) was added to the cell culture media 16 hrs before METH exposure and U0126 (a selective inhibitor of MEK1/2) was added 30 min. before treatment with METH.
Western blotting
Treated hCMEC/D3 cells were washed twice with phosphate-buffered saline (PBS) and lysed in a NP40-lysis buffer containing 25 mM Hepes (pH 7.4), 150 mM NaCl, 4 mM EDTA, 1% Nonidet P-40 (NP-40) (Roche, Indianapolis, IN, USA), 1 mM Na3VO4 and 25 mM NaF for 30 min. on ice. The lysates were centrifuged at 14,000 ×g for 10 min. and the supernatants were used as NP40-soluble fraction. The pellet was further homogenized using a conical glass homogenizer with a fitted Teflon pestle (Kontes Glass Go., Vineland, NJ, USA) in SDS-RIPA buffer containing 25 mM Hepes (pH 7.4), 4 mM EDTA, 25 mM Na3VO4 and 1% SDS, and used as NP40-insoluble fraction. In addition, Mem-PER® Eukaryotic Membrane Protein Extraction Reagent Kit was used according to the manufacturer’s instructions to fractionate membrane proteins (Pierce Rockford, IL, USA). Protein concentrations were determined using BCA™ Protein Assay Kit (Thermo Scientific, Rockford, IL, USA). Samples were then separated on a 4–15% sodium dodecyl sulphate-polyacrylamide gel (SDS-PAGE), transferred onto PVDF membrane (Bio-Rad Laboratories, Hercules, CA, USA), blocked with 3% bovine serum albumin (BSA) dissolved with Tris-buffered saline containing 0.1% Tween-20 (TBS-T), and incubated with the respective antibodies overnight at 4°C. Antibody against occludin was obtained from Zymed Laboratories (Carlsbad, CA, USA). Antibodies against p47, p22 and gp91 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal antibody against serine-phosphorylated p47 was from Abcam (Cambridge, MA, USA). Antibody against phosphorylated caveolin-1 (pY14) was from BD Transduction Laboratories (Franklin Lakes, NJ, USA) and anti-caveolin-1 (Cav-1) antibody was from Affinity BioReagents (Golden, CO, USA). Rabbit polyclonal antibody against p-p42/p44 MAPK or p42/p44 MAPK was purchased from Cell Signaling Laboratories (Danvers, MA, USA). Monoclonal anti-β-actin antibody (recognizing both F-actin and G-actin) conjugated with peroxidase was purchased from Sigma-Aldrich (St. Louis, MO, USA). Polyclonal horseradish peroxidase (HRP)-conjugated glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was from Santa Cruz Biotechnology and rabbit polyclonal anti-β-tubulin antibody was obtained from Abcam. All antibodies were diluted with 3% BSA in TBS-T buffer. Anti-Cav-1 and anti–β-actin antibodies were diluted at 1:5000 and all other antibodies were diluted at 1:2000. Immunoblots were analysed by the ECL Western blot detection system (Amersham Biosciences, Piscataway, NJ, USA) and quantified using ImageJ (http://rsb.info.nih.gov/ij/). The area under the curve (AUC) of the specific signal was corrected for the AUC of the loading control. The mean value for the control group was set as 1 or 100% and the values in the treated groups were calculated accordingly to allow for ratio comparisons.
Immunoprecipitation
hCMEC/D3 cells were lysed in NP-40 buffer containing freshly added protease and phosphatase inhibitor cocktail solution (Roche). Total protein lysates (500 μg) were incubated with 1 μg of anti-p47 antibody for 16 hrs with gentle rotation. Twenty microlitres of 50% slurry of rProtein A/G Agarose (Santa Cruz Biotechnology) were added; the samples were incubated for an additional 1 hr and centrifuged at 5000 rpm for 3 min. to collect immune complexes. The supernatants were also collected and analysed for GAPDH levels as an internal control. The immunoprecipitates were washed three times in lysis buffer, resolved by 4–15% SDS-PAGE, transferred unto a PVDF membrane and analysed as described in the Western blotting section.
Immunofluorescence
Monolayers of hCMEC/D3 cells grown on type I collagen-coated glass cover slips were fixed by a 15 min. immersion in 4% paraformaldehyde and permeabilized for 10 min. in PBS containing 0.1% Triton X-100. Non-specific binding was blocked with 3% BSA in TBS-T for 1 hr. Cover slips were incubated overnight at 4°C in a humidified atmosphere with anti-occludin antibody diluted at 1:100 in 3% BSA in TBS-T buffer. After three washes with PBS, the cover slips were incubated with secondary Alexafluor 488-conjugated donkey anti-mouse antibody diluted at 1:400 in 3% BSA in TBS-T for 1 hr, washed repeatedly with PBS, and mounted with UltraCruz™ Mounting Medium with DAPI (Santa Cruz Biotechnology). In negative controls, all staining procedures were the same; however, no primary antibodies were applied. The images were captured by a confocal microscope (Olympus Fluoview V5; Olympus America, Melville, NY, USA) or fluorescence microscope (Nikon Eclipse E600; Nikon Americas, Melville, NY, USA) at 400× using the same setting for all samples.
Pixel intensity was measured using software provided by the manufacturer (Olympus America) to evaluate the relative changes in immunoreactivity in at least 30 cells randomly selected in each visual field. The results represent the analysis of at least five independent cultures under each experimental condition. Occludin immunoreactivity in the membrane fraction of hCMEC/D3 cells was quantified by measuring the pixel intensity corresponding to the cell–cell borders.
Measurement of ROS
ROS generation was measured in hCMEC/D3 cells with the fluorescence probe, dihydroethium (DHE; Invitrogen, Carlsbad, CA). Cells were treated with METH for the indicated time and further incubated with 10 μM DHE for 20 min. at 37°C. Cells were washed twice with PBS, trypsinized and harvested by centrifugation at 1300 rpm for 3 min. The supernatants were discarded, the cells were resuspended in PBS and analysed by flow cytometry (FACSCalibur; Becton-Dickinson Corp., Franklin Lakes, NJ, USA). Fluorescence was also visualized under a confocal microscope (Olympus Fluoview V5.0).
NOX activity assay
Treated hCMEC/D3 cells were washed with ice-cold PBS and harvested in 500 μl Tris-sucrose buffer (pH 7.1) containing 10 mM Tris, 340 mM sucrose, 1 mM EDTA and protease inhibitors mixture. Cell suspension was sonicated and centrifuged at 1475 ×g at 4°C for 15 min. to remove nuclei and unbroken cells. The supernatant was then centrifuged at 60,000 ×g at 4°C for 30 min. The pellet was suspended in 100 μl of Tris-sucrose buffer, and NOX activity was evaluated by a lucigenin-enhanced chemiluminescence method as described by Sekhar et al.[21]. Intensity of chemiluminescence was measured using MikroWin2000 software and CentroXS3 LB 960 Microplate Luminometer (Berthold Technologies, Oak Ridge, TN, USA).
Monocyte transmigration assay
hCMEC/D3 cells were seeded at the density of 2 × 105 cells per insert on rat tail collagen type I collagen-coated Transwell® Permeable Supports (1.12 cm2 diameter, 3.0 μm pores; Corning incorporated, NY, USA) and cultured until reaching confluency, typically for 3 days. The cells were serum-depleted for 24 hrs and 100 μM of NSC 23766 was added for the last 16 hrs. U937 cells (a human leukaemic monocyte lymphoma cell line) were labelled with the cell tracker, calcein-AM (Invitrogen), according to the manufacturer’s instructions and added into the endothelial monolayers in the amount of 1 × 105 cells per insert. Transendothelial migration of fluorescently labelled monocytes was measured for 5 hrs in the presence or absence of 10 μM of METH. Measurement of relative fluorescence was acquired from the lower chamber using the SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Caveolin-1 silencing
Caveolin-1 (Cav-1) silencing was performed as described previously [22] using small interfering RNA (siRNA) targeting human Cav-1 from Santa Cruz Biotechnology. Silencer Negative Control #1 siRNA (Applied Biosystems, Austin, TX, USA) was used as non-specific control siRNA. hCMEC/D3 cells were transfected overnight with 100 nM of Cav-1 or control siRNA using GeneSilencer siRNA Transfection Reagent (Genlantis, San Diego, CA, USA). Then, the cells were washed and allowed to recover for 2 days in normal medium before METH exposure.
Statistical analysis
Every experiment was independently performed at least three times. Data were expressed as the mean ± S.E.M. One or two-way ANOVA was used to analyse the significance of the differences between the control and experimental groups. Value of P < 0.05 was considered significant.
Results
METH alters occludin protein levels in cerebral endothelial cells
Disruption of BBB integrity is considered an important element of METH-induced neurotoxicity. Therefore, we first evaluated the effects of METH exposure (5–200 μM) on occludin protein levels in hCMEC/D3 cells (Fig. 1A). hCMEC/D3 cells were treated for 6 hrs in these experiments. The basal occludin level was distributed equally in NP40-soluble and insoluble fractions of hCMEC/D3 cells. METH treatment decreased occludin levels in NP-40 soluble fraction in a dose-dependent manner. These effects were significant in cells exposed to METH at 10 μM or higher concentrations. Based on these results, we selected 10 μM as the METH concentration used in the remaining experiments. The effects of METH on occludin levels in the NP-40 insoluble fraction were less pronounced and reached statistical significance only at the highest concentration (200 μM).
Fig 1.

METH treatment induces alterations of occludin protein levels in hCMEC/D3 cells. (A) hCMEC/D3 cells were treated with the indicated doses of METH for 6 hrs, lysed with NP40-lysis buffer, followed by analysis of occludin levels by Western blotting in NP40-soluble (S) and NP40-insoluble (In) fractions. β-Actin levels were assessed as a loading control. The blots (left) reflect representative Western blot data and the bar graph (right) represents densitometry analysis of the band intensity ratio of occludin to actin from these experiments. (B) hCMEC/D3 cells were exposed to 10 μM METH for the indicated time periods, lysed and occludin levels were analysed by Western blotting as in (A). (C) Representative images of immunoreactivity of occludin as determined by immunofluorescence (green staining) in hCMEC/D3 cells treated with METH (10 μM) for 6 hrs. Nuclear staining with DAPI was used to visualize the nuclei (blue staining). METH treatment resulted in diminished and discontinued immunoreactivity of occludin (arrows). Scale bar, 20 μm. (D) METH-induced changes in occludin level in the membrane fraction of hCMEC/D3 cells. Cells were treated with METH as in (B), followed by isolation of the membrane fraction. Occludin levels were then assessed by Western blotting as in (A). β-Actin and β-tubulin were determined as loading controls. The data in (A), (B) and (D) are mean ± S.E.M. All experiments were repeated at least three times. *Statistically significant as compared to the respective control.
Time-dependent experiments (Fig. 1B) indicated that exposure to 10 μM METH resulted in a relatively rapid decrease in occludin levels in NP40-soluble fraction. Indeed, decreased occludin levels were observed after 1 hr of METH exposure and remained reduced for the duration of treatment in this cellular fraction. Treatment with 10 μM METH also altered occludin levels in NP-40 insoluble fraction; however, these changes were statistically significant only in cells exposed to METH for 6 hr.
To confirm and further characterize METH-induced changes, hCMEC/D3 cells were immunostained for occludin levels (Fig. 1C). Compared to control cells, cultures exposed to 10 μM METH revealed a weaker occludin immunoreactivity that was markedly disrupted and discontinued at the areas corresponding to the cell–cell borders (arrows). We next determined the time-dependent effects of METH (10 μM) exposure on occludin levels in the isolated membrane fractions of hCMEC/D3 cells. As indicated in Figure 1D, diminished occludin levels were evident in membranes of cells exposed to METH between 1 and 6 hrs, reaching the lowest levels following a 3 hrs exposure and partially recovering 24 hrs post-METH treatment.
Another important transmembrane TJ protein is claudin-5 [7]. As determined by Western blotting, protein levels of claudin-5 were not affected by treatment with 10 μM METH for up to 24 (data not shown).
Exposure to METH stimulates formation of an active NOX complex
Induction of oxidative stress is widely believed to be an important factor in METH-induced toxicity [1, 2]. Because NOX is the major source of ROS in vasculature, we evaluated whether exposure to METH can stimulate NOX in hCMEC/D3 cells. Our initial studies concentrated on the phosphorylation of p47, which is essential for the activation of NOX. Western blot analysis indicated that p47 was rapidly phosphorylated upon METH treatment (Fig. 2A). As determined by the densitometric analysis, the ratio of phosphorylated p47 (p-p47) to total p47 was significantly elevated from 30 min. to 3 hrs of METH exposure, that is, at the similar exposure time as METH-induced alterations of occludin levels.
Fig 2.

METH exposure stimulates the formation of an active NOX complex. (A) Phosphorylation of p47 (p-p47) NOX subunit as compared to total p47 protein levels in cells exposed to 10 μM METH for the indicated time as determined by Western blotting. (B) Formation of an active NOX complex as determined by co-immunoprecipitation of p47 with the membrane-associated NOX subunits gp91 and p22 in hCMEC/D3 cells treated with 10 μM METH for the indicated times. The blots in A and B (left) are representative images of three experiments and the bar graphs depict the mean ± S.E.M. of the densitometric analyses of target proteins normalized to actin (A) or p47 (B) levels from these experiments. GAPDH levels were measured in the supernatants obtained after precipitation of immune complexes as an internal control. (C) NOX activity in hCMEC/D3 cells treated with 10 μM METH for the indicated period of time. (D) hCMEC/D3 cells were pre-treated with 100 μM NSC 23766 (NSC) for 16 hrs followed by exposure to 10 μM METH for 30 min. Values in C and D represent mean ± S.E.M., n= 3. *Statistically significant as compared to the respective control. †Values in the METH+NSC group are significantly different as compared to those in the METH group.
Phosphorylation of cytoplasmic p47 on multiple serine residues induces its translocation to the membrane fraction and its interaction with membrane subunits of NOX, as well as phosphoinosistide lipid products [23]. To evaluate whether exposure to METH can result in the formation of an active NOX complex in hCMEC/D3 cells, p47 was immunoprecipitated with anti-p47 antibody followed by Western blot analysis of gp91 or p22. As indicated in Figure 2B, binding of p47 to gp91 and p22 was significantly increased by METH treatment. Enhanced immunoprecipitation of p47 with gp91 and p22 was induced in cells treated with METH for as short as 10 min. To ensure that the same protein amount was used for immunoprecipitation, GAPDH protein levels were measured as internal control in the supernatants obtained after precipitation of immune complexes.
To confirm the assembly of an active enzyme, we measured the NOX complex activity upon treatment with 10 μM METH. As shown in Figure 2C, NOX activity was markedly increased after METH treatment with the maximal stimulation at 30 min., followed by the gradual return to the basal level at later time points. Additional treatment with NSC 23766, a specific inhibitor of NOX confirmed the specificity of the responses (Fig. 2D). Similar effects were also observed using other NOX inhibitors, diphenyleneiodonium chloride (DPI) and apocynin (data not shown).
NOX complex mediates METH-induced ROS generation
In the next series of experiments, we evaluated whether METH-induced activation of NOX can result in stimulation of oxidative stress in hCMEC/D3 cells using DHE fluorescence as the specific marker of superoxide production. DHE upon reaction with superoxide anions forms a red fluorescent product (ethidium), which intercalates with DNA [24, 25]. In cells exposed to 10 μM METH for 30 min., DHE fluorescence markedly increased (Fig. 3A, left). In FACS analysis, this effect corresponded to a shift of the histogram to the right (Fig. 3A, right, arrow). METH-induced cellular oxidative stress was confirmed using chloromethy-2’, 7’-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), a marker of a wide spectrum of ROS (data not shown).
Fig 3.

METH-induced superoxide production is mediated by an active NOX complex in hCMEC/D3 cells. (A) hCMEC/D3 cells were treated with 10 μM METH for the indicated time period and the production of superoxide was measured by the oxidation of DHE as determined by flow cytometry. Relative superoxide levels (left) were expressed as percentage of control. (Right) Reflects an example of FACS analysis in hCMEC/D3 cells exposed to METH for 30 min. The arrow indicates a shift of histogram in the METH group indicating increased production of superoxide. (B) Inhibition of NOX protects against METH-induced production of superoxide. hCMEC/D3 cells were pre-treated with 100 μM NSC 23766 (NSC), followed by exposure to 10 μM METH for 30 min. Production of superoxide (left) was quantified by DHE staining and FACS analysis as in (A) (left) and visualized under the confocal microscope (right). Scale bar, 50 μm. The values in (A) and (B) represent the mean ± S.E.M., n= 3; *Statistically significant as compared to the respective control. †Values in the METH+NSC group are significantly different as compared to those in the METH group.
To address the question of whether NOX activation is involved in METH-induced ROS generation, we pharmacologically inhibited NOX with NSC 23766 (100 μM), followed by a 30 min. exposure to METH (10 μM) and the assessment of DHE fluorescence by FACS analysis and confocal microscopy. As indicated in Figure 3B (left), inhibition of NOX significantly protected against the METH-induced DHE fluorescence. These effects were also confirmed under confocal microscopy as shown in Figure 3B (right), where a marked increase in red fluorescence corresponded to METH-induced formation of superoxide. Taken together, these observations provide evidence that ROS generated in hCMEC/D3 cells in response to METH treatment are mediated by an active NOX complex.
Inhibition of NOX protects against METH-induced alterations of occludin levels and transendothelial migration of monocytes
Our next series of experiments was devoted to investigating whether activation of NOX plays a role in METH-induced changes in occludin levels. hCMEC/D3 cells were pre-treated with 100 μM NSC 23766, followed by 3 hrs exposure to 10 μM METH. Consistent with the results presented in Figure 1, occludin levels were decreased by METH treatment in the NP40-soluble fraction with minimal changes in the NP-40-insoluble fraction. Cells exposed to METH in the presence of NSC 23766 exhibited significant protection against alterations of occludin levels in the NP-40 soluble fraction. However, exposure to NSC 23766 also resulted in redistribution of occludin as shown by decreased levels of this TJ protein in the NP-40 insoluble fraction of hCMEC/D3 cells (Fig. 4A).
Fig 4.

Inhibition of NOX protects against METH-induced occludin changes and transendothelial migration of monocytes. (A) hCMEC/D3 cells were exposed to 10 μM METH for 3 hrs in the presence or absence of NOX inhibitor NSC 23766 (NSC, 100 μM). Occludin levels were evaluated in NP-40 soluble and insoluble fractions by Western blotting as in Figure 1. The blots reflect representative data from three different experiments, and the bar graphs represent quantified results (mean ± S.E.M.) from these experiments as analysed by densitometry. (B) hCMEC/D3 cells were treated with METH as in (A), followed by evaluation of occludin by immunofluorescence labelling. The representative images (left) were obtained by confocal microscope. Fluorescence intensity at the cell–cell borders (right) was calculated by determination of pixel intensity per area (μm2) in at least 30 cells out of approximately 140 cells in each visual field. Scale bar, 20 μm. (C) Inhibition of NOX protects against METH-induced transendothelial migration of monocytes. Confluent hCMEC/D3 cells were pre-treated with NSC 23766 as in (A), followed by exposure to 10 μM METH for 5 hrs. Calcein-AM–labelled monocytes were added to treated or control hCMEC/D3 cultures and cocultured for 5 hrs. Values are mean ± S.E.M. *Statistically significant as compared to the respective control. †Values in the NSC or METH + NSC group are significantly different as compared to those in the Vehicle or METH group, respectively.
We also determined the effects of NSC 23766 on METH-induced alterations of protein levels of occludin using immunofluorescence labelling (Fig. 4B, left). Analysing the obtained data, we specifically focused on occludin immunoreactivity in the areas corresponding to the cell–cell borders, which was then quantified and plotted in a form of a bar graph (Fig. 4B, right). Exposure to NSC 23766 alone did not affect occludin immunoreactivity. Notably, METH treatment significantly reduced occludin-positive staining; however, these effects were fully reversed by NSC 23766 pre-treatment.
Integrity of occludin may regulate the barrier functions of brain endothelial cells and paracellular migration of inflammatory cells. Therefore, we also determined the involvement of NOX in METH-induced monocyte migration across monolayers of brain endothelial cells. Treatment of hCMEC/D3 cells with 10 μM of METH for 5 hrs resulted in a 33% increase in transendothelial monocyte passage. Pre-treatment with NSC 23766 protected against these effects (Fig. 4C), demonstrating the role of NOX in METH-induced barrier dysfunction of brain endothelial cells. In addition, treatment with NSC 23766 alone significantly decreased transendothelial migration of monocytes.
Functional caveolae and ERK1/2 signalling are detrimental for METH-induced ROS production and alterations of occludin levels
An active NOX complex was shown to be associated with caveolae [26]. In addition, signalling pathways localized to caveolae may be involved in the regulation of TJ proteins [22]. Therefore, in the last series of experiments, we determined the involvement of functional caveolae in METH-induced changes in signalling pathway activation and occludin protein levels. Treatment with METH induced a rapid (within 10 min.) phosphorylation of ERK1/2 without changing the total level of this kinase. In addition, a 30 min. exposure to METH increased phosphorylation of Cav-1 protein, the structural and regulatory protein in caveolae. The antibody used in these experiments specifically recognized phosphorylation at Tyr14. Interestingly, inhibition of ERK1/2 activity by a specific inhibitor U0126 completely protected against METH-induced phosphorylation of Cav-1, indicating that this process is dependent on active ERK1/2 (Fig. 5A).
Fig 5.
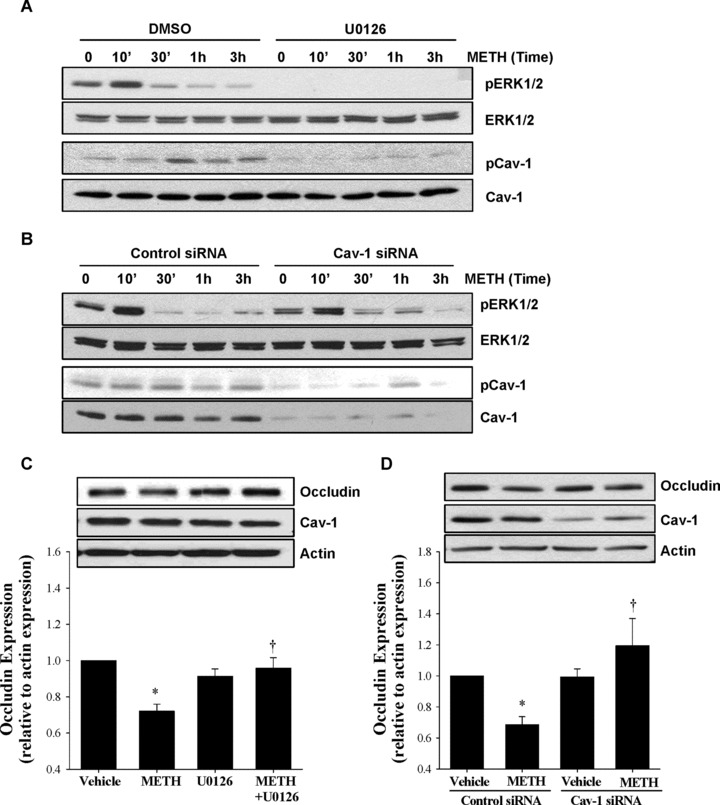
Functional caveolae and ERK1/2 signalling are detrimental for METH-induced alterations of TJ proteins. (A) hCMEC/D3 cells were exposed to 10 μM METH for the indicated time period. In addition, selected cultures were pre-treated with U0126 (1 μM; for 30 min.), followed by METH exposure. Phosphorylation of ERK1/2 (pERK1/2) and caveolin-1 (pCav-1), as well as total levels of these proteins (ERK1/2 and Cav-1, respectively), were determined by Western blotting. (B) hCMEC/D3 cells were transfected with control or Cav-1 siRNA 3 days before METH treatment as described in the Materials and Methods section, followed by METH (10 μM) treatment for the indicated time period. pERK1/2, pCav-1 and the total levels of these proteins were determined as in (A). (C) hCMEC/D3 cells were pre-treated with U0126 as in (A) and exposed to 10 μM METH for 3 hrs. Occludin and total Cav-1 levels were evaluated by Western blotting. Vehicle used was DMSO. (D) Caveolae were disrupted in hCMEC/D3 cells by transfecting Cav-1 siRNA as in (B) and the cells were exposed to 10 μM METH for 3 hrs. Controls were transfected with control siRNA. The blots reflect representative Western blot data and the bar graphs represent quantified results (mean ± S.E.M.) as analysed by densitometry. All experiments were repeated three times. *Statistically significant as compared to the respective control. †Values in the METH + U0126 or METH + Cav-1 siRNA group are significantly different as compared to those in the METH group.
To address the role of caveolae in these events, hCMEC/D3 cells were transfected with Cav-1 siRNA. This procedure resulted in a decrease of Cav-1 protein to negligible levels and prevented phosphorylation of Cav-1 (Fig. 5B). In contrast, Cav-1 silencing did not affect ERK1/2 phosphorylation, indicating that ERK1/2 activation is upstream from Cav-1 phosphorylation. The role of ERK1/2 signalling in METH-induced alterations of TJ proteins was confirmed by the observation that pre-treatment with U0126 protected against altered occludin levels (Fig. 5C). Interestingly, Cav-1 silencing also prevented METH-induced changes in occludin levels (Fig. 5D), indicating the role of functional caveolae in the regulation of TJ protein levels.
Discussion
METH is a frequently abused drug via the intranasal, intravenous, smoked and, less commonly, the oral route [27]. Although the onset of psychostimulatory effects of METH depends on the route of exposure, METH plasma concentrations continuously increase for 4 hrs post a single drug administration with the mean plasma METH values ∼0.3 μM after administration of 15 mg METH [28]. Higher mean plasma METH values were reported following METH vapour inhalation, reflecting faster absorption via smoking [29–31]. However, METH is commonly abused in multiple dose cycles, with an interdose interval of 0.5–3 hrs, which may continue for several days [31, 32]. Indeed, METH abusers typically use 20–40 mg METH more than once a day for approximately 15–20 days per month [33]. METH body burden in abusers is estimated at ∼50 mg, with the blood concentrations in the range of 0.1–11.1 μM [34].
Although METH toxicity has recently been reported using different cell systems, several of these studies employed very high concentrations of METH that markedly exceeded the plasma levels of METH abusers. For example, treatment with 1.68 mM METH induced intracellular oxidative stress and mitochondrial alterations in a human dopaminergic neuroblastoma SH-SY5Y cell line [35] and 4 mM METH induced similar effects in cultured mouse astrocytes [36]. Similarly, generation of ROS and toxicity to human brain endothelial cells were demonstrated using METH at the dose of 2.5 mM [6]. Thus, this study is unique in terms of using an METH concentration of 10 μM that is relevant to human drug abuse. Using sensitive detection methods, we demonstrated that such pathologically relevant doses of METH can induce intracellular oxidative stress and alter TJ integrity in human cerebral microvascular endothelial cells.
A wildly recognized mechanism of METH-induced pathology is related to stimulation of cellular oxidative stress and several studies implicated ROS as an important mediator of BBB disruption and neurotoxicity induced by this drug. The intracellular sources of ROS generated by METH are not fully understood; however, mitochondrial toxicity has been considered [37]. Indeed, exposure to METH decreased mitochondrial membrane potential, increased mitochondrial mass, enhanced protein nitrosylation and diminished protein levels of complexes I, III and IV of the electron transport chain in primary human T cells. These changes were associated with impaired functions of T cells. Importantly, antioxidants attenuated METH-induced mitochondrial damage, further indicating a cross-relationship between METH cytotoxicity, mitochondrial damage and cellular oxidative stress [37]. Another potential source of METH-induced ROS is activation of cytochrome P-450 (CYP450) [38].
Novel results of this study focus on the role of METH-induced activation of NOX, a ROS-producing enzyme that was originally identified and characterized in phagocytes and is also expressed within the cerebral vasculature [39]. Similar to the leukocyte form, NOX expressed in endothelial cells contains both gp91 and p22 [19], that is, the subunits that binding to p47 was increased in hCMEC/D3 cells in response to METH treatment in this study (Fig. 2). Excessive generation of superoxide and its derivatives by NOX within the brain microvasculature may cause lipid peroxidation and membrane disruption, thus damaging BBB integrity [40]. Indeed, our studies revealed that a 30 min. treatment with 10 μM METH can stimulate the formation of NOX complex, associated with increased enzyme activity and generation of ROS in hCMEC/D3 cells. To prove the direct involvement of NOX in METH-induced ROS generation, we inhibited this enzyme activity with NSC 23766. As indicated in Figure 3, this strategy significantly blocked production of superoxide in METH-treated hCMEC/D3 cells. These experiments provide direct evidence that NOX can be activated by low doses of METH (10 μM) in cerebral microvascular endothelial cells and contribute to the induction of oxidative stress.
Disruption of the BBB and alterations of TJ protein levels are associated with neurotoxicity of METH. In line with these observations, one of the early events observed in this study were METH-induced alterations of occludin levels in hCMEC/D3 cells. These changes were analysed both by Western blotting in NP-40 soluble and insoluble cellular fractions and by immunostaining. Surprisingly, the effects of METH on occludin levels in the NP-40 insoluble fraction were less pronounced compared to those in the NP-40 soluble fraction. Although occludin in the NP-40 insoluble fraction is considered to be incorporated into TJ complexes [41], our immunostaining results do not support this notion. In fact, occludin-positive immunoreactivity was markedly diminished and disrupted in the areas corresponding to the cell-cell borders (Figs 1C and 4B). Moreover, occludin protein levels were significantly decreased in the membranes extracted by a commercially available kit (Fig. 1D).
Important results of this study provide evidence that inhibition of NOX by NSC 23766 can protect against METH-induced diminished occludin levels. These results are in agreement with the reports showing the role of cellular oxidative stress in alterations of TJ expression [42, 43]. It was demonstrated that increased oxidative stress activated protein tyrosine kinases [44], RhoA, and PI3 kinase [45], resulting in the disruption of the barrier properties of brain endothelial cells and enhanced permeability across the in vitro model of the BBB. Part of this sequence of events has been linked to the activation of myosin light chain kinase, indicating the role of cytoskeleton in METH-induced vascular toxicity [5]. It should be pointed out that NSC 23766 blocks NOX by inhibition of Rac1, leading to activation of the Rho pathway [46]. Although the Rho signalling is linked to TJ assembly [47], alterations of Rho activity may contribute to a decrease in monocyte migration as observed in hCMEC/D3 cells cocultured with monocytes and exposed to this drug (Fig. 4C). Modulation of the Rho pathway may also be responsible for occludin redistribution in the NP-40 insoluble fraction of hCMEC/D3 cells (Fig. 4A).
Previous results from our laboratory emphasized the role of degradative processes, namely activation of matrix metalloproteinases and proteasome, in degradation of TJ proteins [48]. In addition, an interesting mechanism of caveolae-mediated internalization of occludin was recently described as a process associated with CCL2-induced TJ remodelling in brain endothelial cells [49]. It was observed that within a 1 hr CCL2 treatment, TJ proteins containing occludin internalized via a caveolae-dependent pathway into recycling endosomes. Internalized TJ proteins did not appear to undergo degradation and TJ recycling contributed to TJ complex recovery. This alternative process may also contribute to METH-induced alterations of occludin levels in the cellular membrane fraction observed in this study.
Another possibility of METH-induced alterations of TJ complexes might be related to induction of inflammatory responses and increased levels of pro-inflammatory cytokines, which can activate the ERK1/2 pathway and thus influence phosphorylation of TJ proteins. Indeed, exposure to METH results in increased levels of IL-1β, IL-6 and TNFα, as shown both in whole brain homogenates and in individual cell types [2, 3, 50, 51]. However, an increase in cytokine levels is typically observed a few hours post-METH exposure. In contrast, we observed the maximum of ERK1/2 phosphorylation already 10 min. following METH treatment. This time-dependent sequence suggests that activation of the MAPK pathway might be the effect of METH via NOX activation rather than a secondary effect mediated by cytokine induction.
The ultimate outcome of the disrupted integrity of the BBB can be enhanced endothelial permeability and elevated transendothelial leukocyte migration causing subsequent tissue damage. Therefore, we also evaluated the effects of METH on transendothelial migration of U937 cells. Consistent with alterations of occludin protein levels, exposure to METH induced an increase in migration of human monocytic cells across hCMEC/D3 monolayers. These effects appeared to be mediated by METH-induced activation of NOX, because an inhibition of this enzyme protected against enhanced cell migration. Overall, these results demonstrate a functional significance of ‘leaky’ barrier properties of brain endothelial cells because of altered occludin levels that may stimulate paracellular transendothelial migration of leukocyte and thus contribute to METH-induced neuroinflammatory responses. Nevertheless, exposure to METH is known to induce a variety of redox-responsive transcription factors and pro-inflammatory genes that also can influence adhesion and transendothelial migration of inflammatory cells.
Caveolae are a subgroup of lipid rafts abundant in endothelial cells that play a role in the regulation of various endothelial functions [52]. For example, caveolae and caveolae-associated pathways have emerged as regulators of TJ integrity [22] and the platform necessary for the assembly and activation of NOX [26]. Cav-1 protein was initially identified as a tyrosine-phosphorylated substrate of v-src[53]; however, recent evidence indicates that it can play a regulatory role in caveolae-associated signalling and organize the association of signalling molecules within caveolae [54]. Down-regulation of Cav-1 expression either by gene silencing or knock-out approaches eliminates functional caveolae in endothelial cells.
Novel results of this study demonstrate that treatment with METH stimulates phosphorylation of Cav-1 in tyrosine 14 residue (Tyr14). Although literature data suggest that Cav-1 Tyr14 is a principal target for Src kinase phosphorylation [53], this study clearly indicates that Cav-1 phosphorylation at Tyr14 is regulated by the ERK1/2 pathway in response to METH exposure. Indeed, ERK1/2 inhibition completely protected against METH-induced phosphorylation of Cav-1 at Tyr14 (Fig. 5A). These results are important because phosphorylation of Cav-1 appears to be involved in the regulation of paracellular permeability [55]. For example, an increase in Cav-1 phosphorylation was determined in endothelial cells exposed to hydrogen peroxide and protection against this effect attenuated hydrogen peroxide-induced hyperpermeability of endothelial cell monolayers [56].
This study also indicates the role of functional caveolae in the regulation of METH-induced alterations of occludin protein levels. Specifically, Cav-1 silencing completely protected against decreased occludin levels in METH-treated hCMEC/D3 cells (Fig. 5D). Similar protections against alterations of TJ protein levels were observed in brain endothelial cells isolated from Cav-1 deficient mice and exposed to HIV-1 protein Tat [22]. Interestingly, blocking ERK1/2 activation by U0126 also protected against METH-induced alterations of occludin levels (Fig. 5C).
In conclusion, this study demonstrates that METH, at the levels relevant to human abuse, can activate a NOX complex, followed by the subsequent activation of ERK1/2 signalling and phosphorylation of Cav-1 at Tyr14. Next, these events result in the generation of ROS, alterations of occludin protein levels, and compromised barrier function of brain endothelial cells (Fig. 6). Disruption of functional caveolae and/or inhibition of Cav-1 phosphorylation protected against METH-induced production of ROS and disruption of occludin levels. These results suggest that targeting NOX and caveolae-associated pathways may provide an important therapeutic strategy against METH-induced vascular toxicity.
Fig 6.
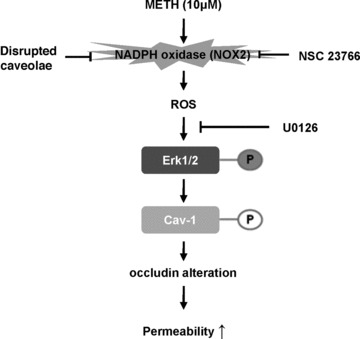
Schematic diagram of METH-induced alteration of occludin protein levels and increased monocyte transendothelial migration. METH in concentrations relevant to drug abuse in humans activates NOX by a caveolae-associated mechanism. Activated NOX results in increased ROS generation with the subsequent phosphorylation of ERK1/2 and Cav-1, followed by alterations of occludin levels. An increase in transendothelial migration of monocytes appears to be an ultimate outcome of these events.
Acknowledgments
This work is supported by grants from the NIH: DA027569, MH63022, MH072567 and NS39254.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Cunha-Oliveira T, Rego AC, Oliveira CR. Cellular and molecular mechanisms involved in the neurotoxicity of opioid and psychostimulant drugs. Brain Res Rev. 2008;58:192–208. doi: 10.1016/j.brainresrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Flora G, Lee Y, Nath A, et al. Methamphetamine-induced TNF-α gene expression and activation of AP-1 in discrete regions of mouse brain. NeuroMolecular Med. 2002;2:71–85. doi: 10.1385/NMM:2:1:71. [DOI] [PubMed] [Google Scholar]
- 3.Lee YW, Hennig B, Yao J, et al. Methamphetamine induces AP-1 and NF-kappaB binding and transactivation in human brain endothelial cells. J Neurosci Res. 2001;66:583–91. doi: 10.1002/jnr.1248. [DOI] [PubMed] [Google Scholar]
- 4.Sharma HS, Kiyatkin EA. Rapid morphological brain abnormalities during acute methamphetamine intoxication in the rat: An experimental study using light and electron microscopy. J Chem Neuroanat. 2009;37:18–32. doi: 10.1016/j.jchemneu.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramirez SH, Potula R, Fan S, et al. Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J Cereb Blood Flow Metab. 2009;29:1933–45. doi: 10.1038/jcbfm.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X, Banerjee A, Banks WA, et al. N-acetylcysteine amide protects against methamphetamine-induced oxidative stress and neurotoxicity in immortalized human brain endothelial cells. Brain Res. 2009;1275:87–95. doi: 10.1016/j.brainres.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24:719–25. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- 8.Hirase T, Staddon J, Saitou M, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110:1603–13. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- 9.Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players. Trends Cell Biol. 1999;9:268–73. doi: 10.1016/s0962-8924(99)01578-0. [DOI] [PubMed] [Google Scholar]
- 10.Haorah J, Knipe B, Leibhart J, et al. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005;78:1223–32. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- 11.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 12.Segal AW. How neutrophills kill microbes. Ann Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pagano PJ, Clark JK, Cifuentes-Pagano ME, et al. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensinI. Proc Natl Acad Sci USA. 1997;94:14483–8. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H Oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 15.Jones S, Hancock J, Jones O, et al. The expression of NADPH oxidase components in human glomerular mesangial cells: detection of protein and mRNA for p47phox, p67phox, and p22phox. J Am Soc Nephrol. 1995;5:1483–91. doi: 10.1681/ASN.V571483. [DOI] [PubMed] [Google Scholar]
- 16.Mohazzab KM, Kaminski PM, Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol Heart Circ Physiol. 1994;266:H2568–72. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- 17.Jones SA, O’Donnell VB, Wood JD, et al. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol Heart Circ Physiol. 1996;271:H1626–34. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 18.Bayraktutan U, Draper N, Lang D, et al. Expression of a functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovasc Res. 1998;38:256–62. doi: 10.1016/s0008-6363(98)00003-0. [DOI] [PubMed] [Google Scholar]
- 19.Gorlach A, Brandes RP, Nguyen K, et al. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res. 2000;87:26–32. doi: 10.1161/01.res.87.1.26. [DOI] [PubMed] [Google Scholar]
- 20.Weksler BB, Subileau EA, Perriere N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–4. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 21.Sekhar KR, Crooks PA, Sonar VN, et al. NADPH Oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–45. [PubMed] [Google Scholar]
- 22.Zhong Y, Smart EJ, Weksler B, et al. Caveolin-1 regulates human immunodeficiency virus-1 Tat-induced alterations of tight junction protein expression via modulation of the Ras signaling. J Neurosci. 2008;28:7788–96. doi: 10.1523/JNEUROSCI.0061-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ago T, Kuribayashi, Hiroaki H, et al. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc Natl Acad Sci USA. 2003;100:4474–9. doi: 10.1073/pnas.0735712100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothe G, Valet G. Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2’,7’-dichlorofluorescin. J Leukoc Biol. 1990;47:440–8. [PubMed] [Google Scholar]
- 25.Carter W, Narayanan P, Robinson J. Intracellular hydrogen peroxide and superoxide anion detection in endothelial cells. J Leukoc Biol. 1994;55:253–8. doi: 10.1002/jlb.55.2.253. [DOI] [PubMed] [Google Scholar]
- 26.Vilhardt F, van Deurs B. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J. 2004;23:739–48. doi: 10.1038/sj.emboj.7600066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domier C, Simon S, Rawson R, et al. A comparison of injecting and noninjecting methamphetamine users. J Psychoactive Drugs. 2000;32:229–32. doi: 10.1080/02791072.2000.10400233. [DOI] [PubMed] [Google Scholar]
- 28.Schepers RJF, Oyler JM, Joseph RE, et al. Methamphetamine and amphetamine pharmacokinetics in oral fluid and plasma after controlled oral methamphetamine administration to human volunteers. Clin Chem. 2003;49:121–32. doi: 10.1373/49.1.121. [DOI] [PubMed] [Google Scholar]
- 29.Cook C, Jeffcoat A, Hill J, et al. Pharmacokinetics of methamphetamine self-administered to human subjects by smoking S-(+)-methamphetamine hydrochloride. Drug Metab Dispos. 1993;21:717–23. [PubMed] [Google Scholar]
- 30.Perez-Reyes M, White W, McDonald S, et al. Clinical effects of methamphetamine vapor inhalation. Life Sci. 1991;49:953–9. doi: 10.1016/0024-3205(91)90078-p. [DOI] [PubMed] [Google Scholar]
- 31.Cho A, Melega W, Kuczenski R, et al. Relevance of pharmacokinetic parameters in animal models of methamphetamine abuse. Synapse. 2001;39:161–6. doi: 10.1002/1098-2396(200102)39:2<161::AID-SYN7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 32.Gawin F, Khalsa-Denison M. Is craving mood-driven or self-propelled? Sensitization and “street” stimulant addiction. NIDA Res Monogr. 1996;163:224–50. [PubMed] [Google Scholar]
- 33.Simon S, Richardson K, Dacey J, et al. A comparison of patterns of methamphetamine and cocaine use. J Addict Dis. 2002;21:35–44. doi: 10.1300/j069v21n01_04. [DOI] [PubMed] [Google Scholar]
- 34.Melega WP, Cho AK, Harvey D, et al. Methamphetamine blood concentrations in human abusers: application to pharmacokinetic modeling. Synapse. 2007;61:216–20. doi: 10.1002/syn.20365. [DOI] [PubMed] [Google Scholar]
- 35.Wu C-W, Ping Y-H, Yen J-C, et al. Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol Appl Pharmacol. 2007;220:243–51. doi: 10.1016/j.taap.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Lau JWS, Senok S, Stadlin A. Methamphetamine-induced oxidative stress in cultured mouse astrocytes. Ann NY Acad Sci. 2000;914:146–56. doi: 10.1111/j.1749-6632.2000.tb05192.x. [DOI] [PubMed] [Google Scholar]
- 37.Potula R, Hawkins BJ, Cenna JM, et al. Methamphetamine causes mitrochondrial oxidative damage in human T lymphocytes leading to functional impairment. J Immunol. 2010;185:2867–76. doi: 10.4049/jimmunol.0903691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumagai Y, Shimojo N. Possible mechanisms for induction of oxidative stress and suppression of systemic nitric oxide production caused by exposure to environmental chemicals. Environ Health Prev Med. 2002;7:141–50. doi: 10.1007/BF02897942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller AA, Drummond GR, Schmidt HHHW, et al. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97:1055–62. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- 40.Gluck MR, Moy LY, Jayatilleke E, et al. Parallel increases in lipid and protein oxidative markers in several mouse brain regions after methamphetamine treatment. J Neurochem. 2001;79:152–60. doi: 10.1046/j.1471-4159.2001.00549.x. [DOI] [PubMed] [Google Scholar]
- 41.Sakakibara A, Furuse M, Saitou M, et al. Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol. 1997;137:1393–401. doi: 10.1083/jcb.137.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Goes A, Wouters D, van der Pol SMA, et al. Reactive oxygen species enhance the migration of monocytes across the blood-brain barrier in vitro. FASEB J. 2001;15:1852–4. doi: 10.1096/fj.00-0881fje. [DOI] [PubMed] [Google Scholar]
- 43.Lochhead JJ, McCaffrey G, Quigley CE, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30:1625–36. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haorah J, Ramirez SH, Schall K, et al. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. 2007;101:566–76. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- 45.Schreibelt G, Kooij G, Reijerkerk A, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007;21:3666–76. doi: 10.1096/fj.07-8329com. [DOI] [PubMed] [Google Scholar]
- 46.Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–41. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 47.Gopalakrishnan S, Raman N, Atkinson SJ, et al. Rho GTPase signaling regulates tight junction assembly and protects tight junctions during ATP depletion. Am J Physiol Cell Physiol. 1998;275:C798–809. doi: 10.1152/ajpcell.1998.275.3.C798. [DOI] [PubMed] [Google Scholar]
- 48.Huang W, Eum SY, Andras IE, et al. PPARα and PPARγ attenuate HIV-induced dysregulation of tight junction proteins by modulations of matrix metalloproteinase and proteasome activities. FASEB J. 2009;23:1596–606. doi: 10.1096/fj.08-121624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamatovic SM, Keep RF, Wang MM, et al. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009;284:19053–66. doi: 10.1074/jbc.M109.000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu Q, Zhang D, Walston M, et al. Chronic methamphetamine exposure alters immune function in normal and retrovirus-infected mice. Int Immunopharmacol. 2002;2:951–62. doi: 10.1016/s1567-5769(02)00047-4. [DOI] [PubMed] [Google Scholar]
- 51.Tocharus J, Khonthun C, Chongthammakun S, et al. Melatonin attenuates methamphetamine-induced overexpression of pro-inflammatory cytokines in microglial cell lines. J Pineal Res. 2010;48:347–52. doi: 10.1111/j.1600-079X.2010.00761.x. [DOI] [PubMed] [Google Scholar]
- 52.Milovanova T, Chatterjee S, Hawkins BJ, et al. Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim Biophys Acta. 2008;1783:1866–75. doi: 10.1016/j.bbamcr.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee H, Volonte’ D, Galbiati F, et al. Constitutive and growth factor-regulated phosphorylation of caveolin-1 occurs at the same site (Tyr-14) in vivo: identification of a c-Src/Cav-1/Grb7 signaling cassette. Mol Endocrinol. 2000;14:1750–75. doi: 10.1210/mend.14.11.0553. [DOI] [PubMed] [Google Scholar]
- 54.Quest AF, Leyton L, Parraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem Cell Biol. 2004;82:129–44. doi: 10.1139/o03-071. [DOI] [PubMed] [Google Scholar]
- 55.Nag S, Manias JL, Stewart DJ. Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol Appl Neurobiol. 2009;35:417–26. doi: 10.1111/j.1365-2990.2008.01009.x. [DOI] [PubMed] [Google Scholar]
- 56.Sun Y, Hu G, Zhang X, et al. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ Res. 2009;105:676–85. doi: 10.1161/CIRCRESAHA.109.201673. [DOI] [PMC free article] [PubMed] [Google Scholar]