

Abstract
Importance of the field
Head and neck squamous cell carcinoma (HNSCC) is the eighth leading cause of cancer death worldwide. Despite advances in surgery and chemoradiation therapy, there has been little improvement in survival rates over the past 4 decades. Additionally, surgery and chemoradiotherapy have serious side effects. The development of agents with greater efficacy and tolerability is needed.
Areas covered in this review
EGFR is the only proven molecular target for HNSCC therapy. Cetuximab, the sole FDA-approved molecular targeted HNSCC therapy, and other potential targeted therapies are being evaluated in preclinical, clinical and post-marketing studies. Here, we review the emerging targets for biological agents in HNSCC and the rationale for their selection.
What the reader will gain
Key information in the development of new drug targets and the emergence of new biomarkers are discussed. Readers will gain insight regarding the limitations of current therapies, the impact of recently approved targeted therapies and the influence that predictive biomarkers will have on drug development.
Take home message
The head and neck cancer drug market is rapidly evolving. Coordination between drug and biomarker development efforts may soon yield targeted therapies that can achieve the promise of personalized cancer medicine.
Keywords: carcinoma, cetuximab, EGFR, erlotinib, gefitinib, HNSCC, HPV, panitumumab, SCCHN, squamous
1. Background
Head and neck cancer encompasses a broad category of neoplasms that primarily develop in the oral cavity, pharynx and larynx. Due to the > 90% squamous cell histology [1] of these cancers, they are generally referred to as head and neck squamous cell carcinoma (HNSCC). HNSCC is the eighth leading cause of cancer death worldwide and, each year, ~ 600,000 new cases are reported and > 300,000 people die as a result of HNSCC [2-4]. While total incidence in both males and females is slowly decreasing worldwide, this decrease in incidence has not been seen in all countries [5]. Generally, the worldwide rate of laryngeal cancer has been decreasing while the trend in the rates of oral cavity and pharyngeal cancers has been variable [5]. For example, the incidence of cancers of the oral cavity and pharynx for both sexes is increasing in northern and eastern Europe as well as Japan; decreasing for both sexes in the US, India and China; and increasing for women while decreasing for men in some west European countries [5]. Moreover, when the data are further analyzed by anatomical subsite, the trend in incidence is altered. For example, in the US, the incidence of tongue and tonsillar cancers has been steadily increasing in younger populations [6]. The reason for this heterogeneity remains unclear but it may be linked to rapidly evolving standards in surveillance, changes in alcohol and tobacco use rates and other contributing etiological factors.
While no single etiological factor has been linked to the development of HNSCC, alcohol and tobacco use have been identified as two important risk factors for developing HNSCC [7]. Indeed, a recent pooled analysis of > 11,000 cases and 16,000 controls found that 72% of HNSCC cases could be attributed to the use of tobacco or alcohol or both [8]. Importantly, as part of the International Head and Neck Epidemiology Consortium (INHANCE), the data set used for this study was derived from multiple case-control studies encompassing a diverse set of patient populations and geographic sites. Other recent pooled analyses using all or part of the INHANCE data set (> 25,000 cases and 33,000 controls) reported that the increased risk of HNSCC attributed to both tobacco and alcohol is dose-dependent [9], the type of alcoholic beverage was not consequential [10], the risk of developing HNSCC was only weakly associated with family history of tobacco related cancer [11], and the risk of developing HNSCC is significantly decreased by cessation of alcohol and tobacco use [12]. Despite the identification of alcohol and tobacco as risk factors, a quarter of HNSCC cases cannot be attributed to either. Moreover, this minority of overall cases becomes a majority in younger patients (66.5% not attributed to tobacco or alcohol in patients < 45 years old) [8] and a significantly larger minority in women (42.6% not attributed to tobacco or alcohol in females) [8]. Thus, additional etiological factors seem likely.
HPV has emerged as an increasingly significant risk factor in HNSCC. A meta-analysis of > 5000 HNSCC tumors from 60 studies found HPV prevalence to be 25.9% across all HNSCC sites with the highest prevalence in oropharynx (35.6%) followed by the larynx (24%) and oral cavity (23.5%) [13]. As a sexually transmitted infection, HPV infection has been definitively linked to squamous cell carcinomas of the cervix [14] and anal canal [15]. In a recent pooled analysis of > 5000 cases and 6000 controls in the INHANCE data set, an increased risk of developing oropharyngeal cancer was found in patients with six or more lifetime sexual partners or four or more oral sex partners [16]. Interestingly, this study did not demonstrate a significant relationship between sexual practices and HNSCC of the oral cavity and larynx.
In the ~ 2 decades since HNSCC incidence peaked [1], advances in the basic biological sciences have led to an exponentially increasing understanding of the microscopic mechanisms involved in cancer. Specifically, the relationship among cancer, the processes of cell proliferation, and the molecular signals that govern the balance between cancer cell life and death has been increasingly elucidated. While much remains to be explored, the medical and scientific communities have identified a plethora of potential molecular targets important in the progression of HNSCC. The most important of these targets so far has been the EGFR pathway. This critical signaling pathway has been the focus of basic and clinical research for decades which, in the form of cetuximab, yielded the first molecular targeted therapy for the treatment of HNSCC. Additionally, other biological pathways that modulate angio-genesis, inflammation, cell survival, cell death and many other important aspects of HNSCC tumor biology have been identified. Potential drug targets include elements of the VEGF pathway, signal transducer and activator of transcription-3 (STAT3) pathway, Akt pathway, Src-family kinases and HPV associated proteins. This review focuses on both the continued development of EGFR targeted therapies and the progress of newly emerging targets as they relate to the treatment of head and neck cancer.
2. Medical need
In the past 3 decades, survival rates for HNSCC have remained relatively unchanged. Over this time, there has been a 3% decrease in 5-year overall survival to 66% for cancers of the larynx and a 7% increase to 61% for cancers of the oral cavity and pharynx while the survival rate for all malignancies has improved from 50 to 66% [1,17]. Additionally, two-thirds of HNSCC patients continue to present with locally advanced disease [1]. For these patients, 5-year survival rates are < 50% and post-treatment quality of life is severely reduced [18].
Paradoxically, while overall survival rates have remained relatively stagnant, numerous advances in treatment have occurred. Extensive surgical resection, for decades the mainstay of primary treatment for advanced HNSCC, has given way to a reduced role as part of an integrative management strategy combining surgery, chemoradiotherapy and more recently biological therapy. This shift in the treatment paradigm has generally yielded equivalent or slightly better efficacy with significantly better organ preservation and cosmetic results [19,20]. Also, advances including new endoscopic, reconstructive and robotic techniques have continued to reduce postoperative complications [21]. Improvements in chemoradiotherapy, such as the establishment of concurrent platinum based chemotherapy with external beam radiotherapy as the gold standard of primary treatment, as well as the addition of induction chemotherapy and altered fractionation regimens, have increased both survival rates and organ preservation [19,22]. Moreover, advances in imaging have allowed for the development of techniques, such as intensity-modulated radiation therapy, which deliver high doses of radiation precisely to tumors while sparing surrounding tissue [23].
Overall, despite improvements in each component of modern integrated management, only a marginal increase in overall survival has occurred while both acute and chronic toxicities persist [19,22]. Specifically, the aggressive treatment necessitated by advanced HNSCC disease often results in severe side effects (primarily xerostomia and dysphagia) that can have a dramatic impact on post-treatment quality of life [23]. Moreover, the recurrence rate of patients treated for HNSCC also remains unacceptably high at up to 50% within the first 2 years [19]. Thus, there is an urgent need for drugs that can be safely integrated into current treatment regimens that improve both the tolerability and the efficacy of primary and secondary treatment.
3. Existing treatment
3.1 Early stage and locally advanced disease
Accurate staging is critical in the treatment of HNSCC. The approximately one-third of patients who present with early stage, locally contained disease [1] are generally treated with either surgery or radiotherapy both of which can yield excellent results and cure rates of better than 90% for stage I and 70% for stage II patients [24,25]. The selection of either modality has traditionally been based on a wide array of factors including accessibility of the lesion, organ preservation, patient and institutional preference, and risk of second primary tumor or recurrence. Treatment options for patients with locally advanced HNSCC (stage III and stage IV) are more complicated and treatment choice is dependent on a multitude of factors but primarily the site of the lesion and its accessibility for excision [26]. Broadly, tumors of the oral cavity are generally treated with primary surgical excision and tumors of the pharynx and larynx are treated with chemoradiation as the primary treatment [26]. The survival and locoregional control benefits of concurrent chemoradiation have been demonstrated in several large studies [27-31] including a pivotal multi-center Phase III trial of ~ 300 patients that demonstrated the benefit (14% projected 3-year overall survival benefit) of adding cisplatin to conventional radiotherapy [27]. Moreover, a meta-analysis of > 17,000 patients in 93 clinical trials found an absolute 5-year survival benefit of 6.5% (over radiotherapy alone) for concomitant chemotherapy and radiotherapy compared to only 2.4% for induction chemotherapy followed by radiotherapy [22]. Additional adjuvant treatment, which is indicated for high risk patients (extracapsular nodal spread, positive margins, perineural invasion, etc.) and those with residual tumor, generally consists of aggressive postoperative chemoradiotherapy or salvage surgery with neck dissection after chemoradiation [26].
Cetuximab, a mAb against EGFR, was approved by the FDA in 2006 as the first molecular targeted therapy for HNSCC. Regulatory approval for cetuximab in conjunction with radiotherapy was based on a Phase III trial that demonstrated significant improvements in locoregional control and survival when cetuximab was compared to radiotherapy alone [32]. Recently, results from this landmark study were updated to include 5-year survival data demonstrating that cetuximab plus radiotherapy provided a sustained survival benefit of ~ 9% (5-year overall survival of 45.6 vs 36.4%) [33]. Importantly, the 9% increase in survival provided by adding cetuximab to radiotherapy compared favorably to the benefit (6.5%) provided by adding platinum based chemotherapy to radiotherapy [22]. While these results are indeed promising, the results of cetuximab addition to chemoradiotherapy in a large, multi-center Phase III study of locally advanced HNSCC have not yet been reported.
3.2 Metastatic or recurrent disease
In metastatic or recurrent disease, which comprises 10% of initial presentations [1] and occurs in 50% of patients who were treated for locally advanced HNSCC [4,34], the goals of therapy are both palliation of symptoms and extension of survival. In general, treatment options are diverse and can consist of single or multi-agent chemotherapy as well as salvage surgery, when the lesion is accessible, and/or re-irradiation [19]. The standard of care for most patients with recurrent or metastatic HNSCC is multiagent chemotherapy, with the combination of a platinum compound and 5-fluorouracil (5-FU) as the most widely used regimen [35]. Several large studies have compared single chemotherapeutic agents with two agent combination therapy and, in general, have found that combination treatment is superior in response rates and as palliative therapy but does not improve overall survival [36,37]. More recently, promising results from studies of taxane compounds (paclitaxel and docetaxel) in combination with cisplatin [38-40] have prompted a direct comparison with 5-FU and cisplatin (FC) for recurrent and metastatic disease. However, the results from this study indicated that paclitaxel and cisplatin (PC) did not provide any improvement in response rate (29.8% FC vs 26% PC) or survival (median survival 8.7 months FC vs 8.1 months PC) when compared to FC [41].
While the addition of a third cytotoxic agent to the standard two agent therapy has been effective but limited by toxicity [42-44], the addition of cetuximab to FC was recently approved by the FDA. Approval was based on the results of a Phase III study (EXTREME) that demonstrated that cetuximab, when added to a 5-FU and platinum based chemotherapy improved median overall survival by 2.7 months (10.1 vs 7.4 months for chemotherapy alone) [45]. Moreover, cetuximab as a monotherapy has been demonstrated to be effective in second-line treatment of metastatic and recurrent HNSCC patients [46-48]. Importantly, it is generally accepted that the toxic effects of cetuximab (primarily an acneiform skin rash) are far less severe than those that arise from conventional chemotherapy and that the addition to cetuximab to chemotherapy, radiotherapy or both has no effect on compliance [32,33,45]. Recently, the quality of life assessment of patients from the EXTREME trial has been reported [49]. Quality of life was assessed based on cancer specific and head and neck specific questionnaire data. Importantly, addition of cetuximab to 5-FU and a platinum compound significantly improved patients’ overall quality of life and did not adversely affect social functioning. Surprisingly, the addition of cetuximab was found to significantly improve symptoms of pain, dysphagia, speech and social eating. These tolerability advantages become especially important as the use of cetuximab expands.
4. Market review
Cetuximab sales led to a worldwide revenue in 2009 of > $2 billion [50-52] of which, ~ 30 – 40% is attributable to HNSCC [53]. As the only current regulatory agency approved biological agent for HNSCC, cetuximab is expected to sustain continuing sales growth for the foreseeable future. Moreover, considerable momentum is building to replace cisplatin with cetuximab or future biological agents as the standard of care in HNSCC [54] which may increase sales of cetuximab or newer agents substantially. However, this potential change in the standard of care and its associated increase in cost remain to be justified by a direct comparison of cetuximab to platinum based chemotherapy. As cancer therapy continues its shift towards biological agents and increasingly individually tailored regimens, the cost of these drugs is expected to increase considerably due to the high cost of biological agents relative to conventional chemotherapeutics. Accordingly, the overall size of the head and neck cancer drug market is also expected to increase.
5. Current research goals
As in the development of all cancer therapies, the principal goals of new HNSCC drugs are to increase overall patient survival and reduce treatment toxicity. While improvements continue to be made by optimizing conventional radio and chemotherapies, both approaches have nearly reached their limits of efficiently balancing efficacy and toxicity. As a result, development of new drugs for HNSCC has increasingly focused on molecular targeted therapies specific to the biology of head and neck cancers. These research efforts can broadly be divided into three categories: the continuing optimization of EGFR targeted therapies, the clinical development of established therapeutic targets and the preclinical investigation of new mechanisms of action.
Within the realm of EGFR targeted therapies, several important questions remain unanswered. First, in order to expand the use of cetuximab, a direct comparison of its efficacy in combination with radiotherapy to that of platinum based chemoradiotherapy must be studied. Currently, there are three Phase III trials underway (Table 1) either comparing cetuximab to cisplatin or adding cetuximab to cisplatin. Other EGFR targeted therapies, including EGFR targeted tyrosine kinase inhibitors (TKIs), have not yet demonstrated efficacy in a Phase III study. In addition to expanding the use of cetuximab for more HNSCC patients, major research efforts are underway to improve its efficacy for existing indications. Due to the fact that only a subset of patients will respond to EGFR targeting despite ubiquitous EGFR overexpression in HNSCC [55-57], identification of biomarkers for efficacy and toxicity is an important area of investigation. Likewise, studies investigating adjuvant therapies that make EGFR targeting more effective are also needed.
Table 1.
Currently active Phase III trials of cetuximab in HNSCC.
| ClinicalTrials.gov identifier | Study location | No of patients | Study parameters |
|---|---|---|---|
| NCT00716391 | Spain | 458 | Cetuximab and RT vs cisplatin and RT following TPF induction chemotherapy in locally advanced HNSCC |
| NCT00265941 | USA | 942 | Addition of cetuximab to cisplatin and RT in locally advanced HNSCC |
| NCT00956007 | USA | 700 | Addition of cetuximab to intensity-modulated RT in previously resected HNSCC |
| NCT01012258 | China | 65 | Evaluation of efficacy and tolerability of cetuximab in addition to RT in a Chinese patient population |
| NCT00999700 | Italy | 278 | TPF induction chemotherapy followed by cetuximab and RT vs concurrent cisplatin and RT |
HNSCC: Head and neck squamous cell carcinoma; RT: Radiotherapy; TPF: Docetaxel, cisplatin and 5-fluorouracil.
Drug candidates targeting biological pathways other than EGFR remain investigational but are under active development. The commercialization of agents targeting other pathways such as angiogenesis and intracellular signal transduction networks has followed a model based on the development of EGFR targeted therapies. Generally, these emerging therapies are being evaluated first in the setting of recurrent or metastatic disease and then as an adjuvant to chemoradiotherapy in locally advanced disease. Moreover, the combination of emerging molecular targeted therapies with EGFR targeted therapies or with each other is also actively being studied. Briefly, the goals of clinical studies investigating non-EGFR targeted therapies are: i) establishing efficacy as an adjuvant to chemoradiotherapy or cetuximab; ii) determining toxicities and iii) recording data on factors influencing prognosis and/or response to therapy. Research efforts for preclinical HNSCC therapeutic targets are focused on collecting a supporting base of evidence in cell culture and animal models as well as in other malignancies.
6. Scientific rationale
6.1 EGFR pathway
EGFR is a transmembrane glycoprotein expressed at the plasma membrane that plays a critical role in mediating the relationship between extracellular signals and cellular homeostasis. As a member of the human EGFR (HER) family, its structure consists of an extracellular ligand binding domain, transmembrane domain and an intracellular tyrosine kinase domain [58]. EGFR, which is also known as Her1 and ErbB1, can bind a variety of protein ligands including EGF, amphiregulin and TGF-α [58]. Signaling via EGFR begins with ligand binding to the extracellular domain followed by dimerization with other EGFR and HER family molecules. Dimerization allows for auto- and trans-phosphorylation of the tyrosine kinase domain which leads to the activation of several well-characterized cell survival and proliferation pathways including the MAPK, Jak/STAT and AKT pathways [58]. In the case of HNSCC, heterodimerization with HER2 has been shown to be of prognostic value and may be particularly important [59-62]. Thus, aberrations of EGFR signal activation, duration or intensity can result in the dysregulation of cellular homeostasis that is a hallmark of cancer.
Accordingly, aberrant EGFR activity has been well documented in a variety of cancers and squamous cell carcinomas in particular. In HNSCC, data encompassing a diverse patient population have demonstrated that EGFR protein overexpression is present in the majority of tumors [55-57] and EGFR gene amplification occurs in up to 30% of HNSCC tumors [63-65]. EGFR and TGF-α mRNA increases have also been shown to be an early marker for HNSCC carcinogenesis [55,66]. Furthermore, EGFR overexpression has been linked to drug and radiotherapy resistance [67-69]. Despite recent confounding results regarding the link between EGFR gene copy number and clinical variables [70], the majority of evidence suggests that increased EGFR expression and gene copy number are linked to poorer patient outcomes in HNSCC [71-75]. However, clinical trials of EGFR targeted therapies have not consistently demonstrated a correlation between EGFR overexpression and the efficacy of EGFR targeted therapies [70]. Thus, while targeting EGFR is thought to provide both specificity and disruption of signaling pathways vital to HNSCC tumor growth, the best method or combination of methods for exploiting EGFR as a drug target have yet to be elucidated.
Currently, the two primary strategies under investigation as EGFR targeted therapies are inhibition of its tyrosine kinase domain with a small molecule and inhibition of the extracellular ligand binding using mAbs (Figure 1). The TKIs act by blocking the ATP binding domain of EGFR [76]. In contrast to the TKIs, mAbs act by specifically inhibiting ligand binding. Both strategies have been shown to limit tumor growth and angiogenesis as well as increase radiosensitivity [77-79]. Additionally, mAbs against EGFR have been shown to induce immune mediated antitumor processes such as antibody-dependent cellular cytotoxicity (ADCC) [80-83]. In general, EGFR targeted therapies have been shown to inhibit cellular proliferation, survival, invasion and angiogenesis as well as act synergistically with chemoradiation therapies [78,84-89].
Figure 1. Targeted molecular pathways in head and neck cancer.
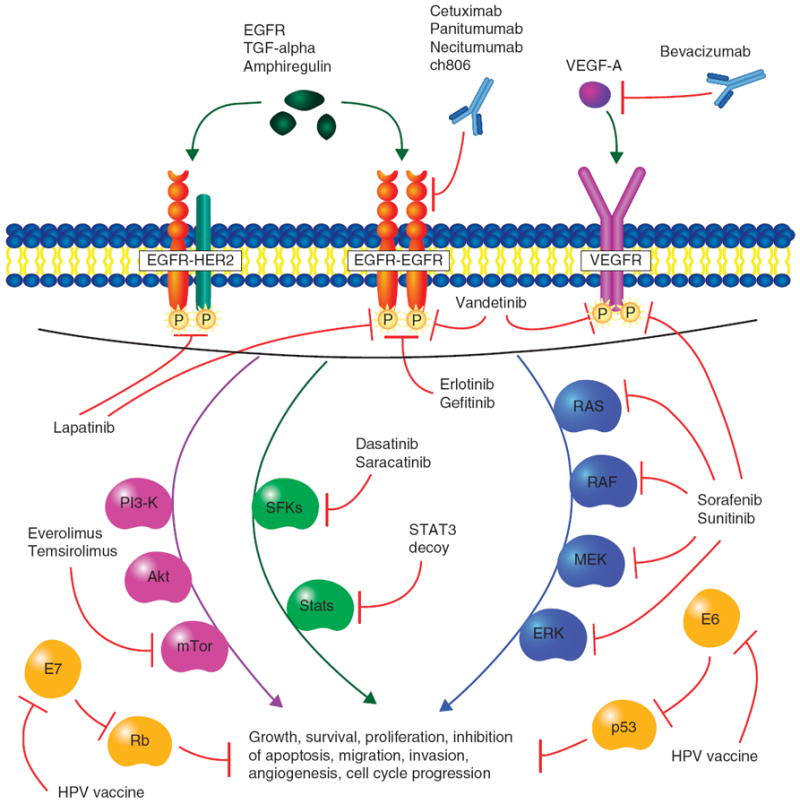
Selected agents that are currently in clinical trials are represented here. Briefly, EGFR and VEGFR signals utilize a variety of downstream molecular pathways including the PI3K-Akt-mTOR, STAT and Ras-MAPK (ERK) pathways. Agents targeting these pathways include mAbs (cetuximab, panitumumab, necitumumab, ch806, bevacizumab), tyrosine kinase inhibitors (erlotinib, gefitinib, vandetanib, lapatinib), multikinase inhibitors (sorafenib, sunitinib), Src family kinase inhibitors (dasatinib, saracatinib), mTOR inhibitors (everolimus, temsirolimus) and nucleic acid decoy (STAT3 decoy). p53 and Rb are tumor suppressors that are inhibited by HPV proteins E6 and E7, respectively. E6 and E7 are constitutively expressed in HPV-16 and -18 infected tumor cells and are targeted by HPV peptide vaccines. Vaccination against E6 and E7 allows cytotoxic T-cell recognition of infected cells.
mTOR: Mammalian target of rapamycin; STAT3: Signal transducer and activator of transcription-3.
The five drugs, out of the myriad of potential EGFR targeted therapies (Table 2), which have reached Phase III evaluation in the US for HNSCC are the mAbs cetuximab and panitumab and the TKIs erlotinib, gefitinib and lapatinib. Additionally, the mAbs zalutumumab and nimotuzumab are currently in Phase III trials outside the US. Despite existing regulatory approval, currently active studies including several international Phase III trials in new patient populations are continuing to broaden and diversify the base of evidence supporting the use of cetuximab. In contrast, panitumumab, which is also an EGFR targeted mAb, has just recently entered Phase III evaluation in HNSCC. Unlike cetuximab, which is a human–murine chimera IgG1 antibody, panitumumab is a fully humanized IgG2 antibody. As a result, panitumumab may not elicit ADCC as strongly as cetuximab but may reduce the production of neutralizing antibodies against the drug as well as reducing the incidence of life-threatening hypersensitivity reactions [90]. Currently, two Phase III trials of panitumumab are underway. The first is a study evaluating the addition of panitumumab to cisplatin and 5-FU in metastatic and/or recurrent HNSCC (SPECTRUM, NCT00460265) and is expected to complete primary analysis by August of 2010. Positive results from this trial would pave the way for the approval of a second EGFR targeted therapy for recurrent or metastatic HNSCC. Furthermore, SPECTRUM may reveal differences in efficacy and tolerability between panitumumab and cetuximab that have already been observed in colorectal cancer for which both mAbs are currently approved [91]. The second active Phase III study of panitumumab is a comparison of its use with concurrent radiotherapy versus cisplatin and concurrent radiotherapy in locally advanced HNSCC (NCT00820248). Several large Phase II studies of panitumumab in first- and second-line settings are also underway. Zalutumumab is a fully human IgG1 mAb currently being studied in large trials at sites in Denmark (NCT00496652) and Belgium (NCT00382031) and has been reported to be especially effective at inducing ADCC [92]. Nimotuzumab, like cetuximab, is a humanized murine IgG1 mAb that is noted for mild or absent skin toxicity [92]. It is currently being studied in Phase III trials at locations in Singapore (NCT00957086) and China (NCT01074021). Two other anti-EGFR mAbs in development for other malignancies, necitumumab (IMC-11F8) and mAb 806, may also be promising agents in treating HNSCC. Necitumumab is a fully humanized IgG1 mAb which has been evaluated in a variety of solid tumors in a recently published Phase I study [93] and is actively in Phase III investigation in squamous NSCLC (NCT00981058). mAb 806 (ch806) is a chimeric human–mouse antibody targeting an epitope of EGFR accessible only when EGFR is activated [94]. Moreover, ch806 is capable of binding to both wild-type and mutated (EGFRvIII) EGFR and its specificity for activated EGFR allows ch806 to be relatively specific for tumors overexpressing EGFR [95].
Table 2.
Selected EGFR targeted therapies.
| Compound | Mechanism of EGFR blockade | Trade name | Manufacturer | Developmental stage for HNSCC |
|---|---|---|---|---|
| Cetuximab | mAb | Erbitux | Eli Lilly (ImClone), Bristol-Myers Squibb, Merck KgaA | Marketed |
| Panitumumab | mAb | Vectibix | Amgen | Phase III |
| Zalutumumab | mAb | HuMax-EGFr | Genmab | Phase III |
| Nimotuzumab | mAb | None | YM Biosciences | Phase III |
| Necitumumab | mAb | None | Eli Lilly (ImClone) | Phase I* |
| ch806 | mAb | None | Ludwig Institute | Phase I* |
| Erltoinib | TKI | Tarceva | Genentech, OSI Pharmaceuticals, Roche | Phase III |
| Gefitinib | TKI | Iressa | AstraZeneca, Teva | Phase III |
| Lapatinib | TKI | Tykerb/Tyverb | GlaxoSmithKline | Phase III |
Phase I evaluation in HNSCC as part of a study of multiple solid tumor types.
HNSCC: Head and neck squamous cell carcinoma; TKI: Tyrosine kinase inhibitor.
Unlike the mAbs, the TKIs utilize an entirely different mechanism of action to block EGFRs and have not progressed as well in terms of clinical development. Gefitinib was the first TKI to reach a Phase III investigation in HNSCC, but recent study failures in several SCCs have led to its withdrawal from new drug consideration in the US. Early Phase II trials in metastatic or recurrent HNSCC patients demonstrated that gefitinib produced results similar to those of early cetuximab trials [96,97]. However, subsequent Phase II studies have not demonstrated a clinical benefit associated with the addition of gefitinib to standard therapies [98-100]. Furthermore, a large, multi-center Phase III study comparing 250 mg gefitinib, 500 mg gefitinib and methotrexate in treatment refractory HNSCC found that the addition of gefitinib did not improve overall survival (5.6 vs 6 vs 6.7 months respectively) [70]. Another large, multi-center Phase III study examining the addition of gefitinib to docetaxel was terminated early in 2008 due to unlikelihood of meeting its primary end point, increased overall survival. Interim results of this study did not yield a significant overall survival benefit; however, gefitinib was found to favorably increase time to progression [101]. Thus, clinical development of gefitinib for HNSCC is unlikely to proceed further.
Erlotinib, however, remains in active development for HNSCC because it has demonstrated encouraging results in several studies. In a large, multi-center Phase II study of erlotinib as a single agent in 115 patients with refractory HNSCC, median overall survival (6 months) was found to be comparable with that of palliative chemotherapy [102]. Likewise, a Phase I/II study of the addition of erlotinib to cisplatin in metastatic or recurrent HNSCC patients found results (overall response rate (ORR) 21%, progression-free survival 3.3 months, overall survival 7.9 months) that were comparable to those of cetuximab in the same population [103]. Additionally, another Phase II study examining the addition of erlotinib to cisplatin and docataxel provided further encouraging results [104]. Despite ample evidence to justify further study, a Phase III trial of erlotinib in addition to first-line therapy with cisplatin was recently terminated (NCT00448240). Moreover, a trial examining daily administration of erlotinib as maintenance therapy to prevent recurrence in fully resected HNSCC was also terminated (NCT00412217). The last remaining Phase III study of erlotinib in HNSCC is an ongoing examination of the use of erlotinib as a chemopreventative agent in high risk patients with oral intraepithelial neoplasia (IEN) that has not yet progressed to cancer or patients with previously treated IEN (NCT00402779).
Unlike erlotinib and gefitinib, which are EGFR specific TKIs, lapatinib has dual specificity for EGFR and HER-2. In HNSCC, lapatinib is currently being evaluated as an adjuvant to postoperative chemoradiation in a Phase III trial (NCT00424255). This large, multi-center study of ~ 600 patients is currently still open to enrollment with the estimated completion being 2012. Several Phase II trials evaluating the use of lapatinib, including a double-blind, placebo-controlled study of ~ 100 patients examining the addition of lapatinib to platinum based chemoradiotherapy (NCT00387127), are also underway. Importantly, a recent placebo-controlled Phase II study of 88 patients demonstrated that lapatinib improved overall response to subsequent chemoradiotherapy (ORR 86% for lapatinib vs 63% for placebo) [105]. While this study, did not achieve its primary end point of demonstrating increased apoptosis in lapatinib treated tumor tissue, the authors reported decreased proliferation in lapatinib treated tumors and this study justifies continued investigation of lapatinib in HNSCC.
6.2 Angiogenesis
While EGFR is the only proven biological target in HNSCC, agents targeting several other pathways are currently in clinical development (Table 3 and Figure 1). As a general therapeutic approach, the hypothesis that an adequate vascular supply is required for tumor progression has long been recognized [106,107]. While > 3 decades of research have yielded some success, the development of antiangiogenic therapies has been limited by only partial understanding of the complex mechanisms involved in developing tumor vasculature [108]. At present, it is understood that hypoxia in the tumor microenvironment is able to trigger an intricate and well-regulated series of cytokine mediated signaling events between tumor cells and endothelial cells [109,110]. Chief among these cytokines are members of the VEGF family [111]. VEGF-A (VEGF) is a proangiogenic factor secreted by tumor cells that mediates angiogenesis by binding to receptors expressed on endothelial cells [107]. Disruption of VEGF signaling has been shown to limit tumor growth and metastatic potential in an array of different cancers and preclinical models [107]. In HNSCC, VEGF and VEGF receptor expression has been demonstrated in tumor tissue and associated with a worse prognosis [112-115]. Moreover, in animal and in vitro models, VEGF signaling has been shown to mediate tumor growth, vasculature, invasiveness and radioresistance [116-120]. In other aerodigestive tract squamous cell carcinomas, VEGF and its receptors have also been demonstrated to be both highly expressed and having prognostic value [121,122]. Finally, VEGF autocrine and paracrine signaling has been demonstrated to play a role in a variety of cancer processes [123].
Table 3.
Selected targeted therapies in clinical development for HNSCC.
| Compound | Targeted pathway | Mechanism of action | Manufacturer | Developmental stage for HNSCC |
|---|---|---|---|---|
| Bevacizumab | Angiogenesis, VEGF | mAb against VEGF ligand | Genentech, Roche | Phase III |
| Vandetanib | Angiogenesis, VEGFR, EGFR | TKI specific for VEGFR and EGFR | AstraZeneca | Phase II |
| Sorafenib | Angiogenesis, kinases | Multi-TKI | Bayer, Onyx | Phase II |
| Sunitinib | Angiogenesis, kinases | Multi-TKI | Pfizer | Phase II |
| STAT3 Decoy | Intracellular signaling | STAT3 transcription factor decoy | University of Pittsburgh | Phase 0 |
| Dasatinib | Intracellular signaling | TKI specific for Src family kinases | Bristol-Myers Squibb | Phase II |
| Saracatinib | Intracellular signaling | TKI specific for Src family kinases | AstraZeneca | Phase II |
| Everolimus | Intracellular signaling | mTor kinase inhibitor | Novartis | Phase II |
| Temsirolimus | Intracellular signaling | mTor kinase inhibitor | Wyeth | Phase II |
| HPV-16 vaccine | HPV-16 | Peptide epitope vaccine | University of Maryland | Phase I |
| MAGE-A3 HPV-16 vaccine | HPV-16 | Peptide epitope vaccine | University of Maryland | Phase I |
HNSCC: Head and neck squamous cell carcinoma; mTOR: Mammalian target of rapamycin; STAT3: Signal transducer and activator of transcription-3; TKI: Tyrosine kinase inhibitor.
Currently, the two dominant approaches to targeting angiogenesis are inhibition of VEGF ligand itself and small molecule inhibition of VEGF receptor tyrosine kinases. Bevacizumab is a humanized mAb that binds and sequesters all five isoforms of VEGF [124], reducing the total amount of circulating VEGF. The effect of bevacizumab on autocrine and paracrine signaling, however, remains unclear. Bevacizumab is approved, in combination with other therapies, as a first- or second-line treatment in several solid tumors including NSCLC. In HNSCC, a Phase III clinical trial studying the addition of bevacizumab to chemotherapy for recurrent or metastatic disease is currently underway (NCT00588770). Additionally, several Phase II studies of bevacizumab in combination with other therapies have reported encouraging results. Generally, these studies and studies in other solid tumors have reported that bevacizumab is efficacious but comes with a serious risk of bleeding [125,126]. Another potential approach to targeting VEGF is through the use of soluble VEGF receptors which, in theory, may provide the same type of sequestration effect [127].
Small molecule inhibition of VEGFR is complicated by the fact that the agents closest to commercialization for HNSCC are generally thought to be less specific multikinase inhibitors. At this time, the three most promising VEGFR targeted TKIs are vandetanib, sunitinib and sorafenib. Vandetanib is a dual EGFR and VEGFR inhibitor that is awaiting marketing approval for NSCLC. Phase II studies in HNSCC are underway evaluating the combination of vandetanib with docetaxel (NCT00459043) and cisplatin (NCT00720083). Sorafenib and sunitinib are multikinase inhibitors with specificity for a broad array of tyrosine kinases including VEGFR [128]. Both agents are currently approved to treat other solid tumors and are currently under investigation for use in HNSCC. As a single agent for recurrent or metastatic HNSCC, sorafenib has been reported to be well tolerated but lacking significant tumor response [129]. Despite a low objective response rate, median overall survival of 8 months in this relatively small trial (34 patients evaluated for response) [129] was comparable to other single agent trials and sorafenib continues to be evaluated in combination with carboplatin and paclitaxel (NCT00494182) or with cetuximab (NCT00939627, NCT00815295). Likewise, sunitinib has also been reported to be well tolerated but lacking in single agent efficacy [130]. Importantly, another recent Phase II trial has reported that sunitinib demonstrated moderate efficacy (using stable disease as a metric), but with a high rate of complications including significant grade 3 – 5 bleeding (16%) and grade 3 – 4 fatigue (32%) [131]. These discordant results suggest that single agent sorafenib or sunitinib may not be an effective treatment for HNSCC and that patient selection criteria will be critical for limiting toxicity in future studies.
6.3 Downstream signaling networks: STAT3, SFKs and Akt
Another group of promising targeted therapies in late stage clinical development are agents that target molecules involved in intracellular signaling networks downstream of EGFR, VEGFR and other receptors. The STAT3 is a member of a family of transcription factors that regulates the expression of many critical genes in tumor growth and survival [132]. It has been demonstrated to be a bona fide oncogene [133] that acts as a downstream mediator between EGFR and target gene expression in HNSCC [134,135]. In HNSCC models, activated STAT3 has also been shown to play a role in cell growth [136,137], migration [138] and inhibiting apoptosis [138,139] which may allow STAT3 to contribute to both tumorigenesis and treatment resistance [140-142]. Specifically targeting STAT3 has proven to be relatively difficult using traditional small molecules and clinical development has progressed the furthest with a soluble nucleic acid decoy for the STAT3 promoter. Currently, a Phase 0 proof of concept trial (NCT00696176) is underway to evaluate the effect of intratumorally injected STAT3 decoy on target gene expression in HNSCC.
The nine known Src family kinases (SFKs) are intracellular tyrosine kinases that can mediate and enhance the downstream signaling of receptor tyrosine kinases including EGFR. Signaling via SFKs can activate the MAPK and Akt pathways leading to increased cell proliferation and survival. Additionally, SFKs can activate a variety of transcription factors, including STAT3, that play key roles in angiogenesis, migration, invasion and a host of other cancer associated processes [140]. In HNSCC preclinical models, increased c-Src has been shown to increase invasiveness and proliferation of cancer cells [141] while inhibition of c-Src has been shown to block invasion [141,142] and induce apoptosis [143]. It was also shown that inhibition of both EGFR and c-Src produced a synergistic effect [141]. Accordingly, studies of cetuximab resistance in NSCLC and colorectal cancer models have revealed that cetuximab resistance can be mediated by constitutively active SFKs [144-146]. Currently, two small molecule inhibitors of SFK tyrosine kinase function, dasatinib and saracatinib (AZD0530), are in clinical evaluation for safety and efficacy in HNSCC. An interim analysis of one Phase II dasatinib study [147] has revealed a similar overall toxicity profile to that of other dasatinib patient populations including a dose-limiting risk of pleural effusion [148]. Studies of both dasatinib and saracatinib are ongoing and will soon yield data not only on progression-free survival but a variety of biomarkers that may be useful in designing future trials.
Akt, which is also known as protein kinase B, is a serine–threonine kinase that is downstream of EGFR and other receptor tyrosine kinases. Akt is activated by PI3Ks which are themselves activated by EGFR activation and dimerization with HER3 [58]. PI3K-Akt signaling is antagonized by the phosphatase activity of phosphatase and tensin homolog (PTEN). Together, PI3K and PTEN balance the activity of Akt which, in turn, mediates signals to a variety of pathways involved in cell survival, growth and metabolism [149]. Alterations of the PI3K-PTEN-Akt pathway are incredibly common in many solid tumors as well as HNSCC and lead to increased Akt signaling [150-152]. Moreover, resistance to drugs targeting EGFR has been found to be mediated by a compensatory increase in Akt signaling such as via a decreased expression of PTEN [153,154]. One of the downstream effectors of Akt is the mammalian target of rapamycin (mTOR), a kinase that is involved in regulating protein translation [149]. The development of anticancer agents targeting the Akt pathway has primarily focused on the inhibition of mTOR. In preclinical models of HNSCC, rapamycin, a potent inhibitor of mTOR, was found to have synergistic effects with carboplatin and paclitaxel [155], inhibit tumor growth, limit angiogenesis and induce apoptosis [152]. Currently, the rapamycin analogs everolimus and temsirolimus are being evaluated in combination with other therapies in several Phase II studies to treat HNSCC. Temsirolimus is FDA approved as a first-line and everolimus is approved as a second-line treatment for renal cell carcinoma.
6.4 HPV
HPVs are a family of > 200 non-enveloped double-stranded DNA viruses with tropism for the squamous epithelium [156]. While the majority of HPVs have no known symptoms in humans, some low risk types, particularly HPV-6 and -11, have been shown to cause various forms of warts [156]. High risk HPV types, such as HPV-16 and -18, have been linked to carcinogenesis in the cervix, vagina, anal canal and epithelium of the penis [156]. Several case-controls studies have demonstrated significantly increased risk of developing HNSCC, particularly of the orpharynx, in HPV positive patients [157-162]. Moreover, while overall HNSCC incidence has declined slightly over the last 40 years, subsite analysis has recently revealed a gradual increase in incidence for sites commonly associated with HPV infection in adults younger than 45 [6]. Accordingly, a decade by decade analysis of archived tonsillar cancer tissue revealed an increase in HPV positive cases from 23.3 to 68% between the 1970s and the 2000s [163].
While the molecular pathogenesis of HPV infection has not yet been fully elucidated in HNSCC, using cervical cancer as a model, the framework for understanding the transformation of normal squamous epithelium to carcinoma has been set. Briefly, HPV infects the basal cells via micro-abrasions in the superficial epithelium and in regions where anatomical features, such as invaginations and thinner squamous epithelium of the oropharynx, allow easier access to basal cells. Within the basal cells, HPV undergoes low level episomal replication prior to integration into the host genome during the normal progression of basal cells to terminally differentiated squamous epithelium. On integration, expression of HPV oncoproteins E6 and E7 are upregulated and virion assembly and release occurs from the terminally differentiated layer of squamous epithelium [164]. HPV E6 and E7 proteins, which have increased activity in high risk HPV types [156], interfere with the tumor suppressor proteins p53 and Rb, respectively. HPV E6 causes the ubiquitination and subsequent degradation of p53 which leads to dysregulation of the cell cycle in response to genomic damage and, thus, facilitates genomic instability [165-167]. HPV E7 similarly induces the ubiquitination and degradation of Rb leading to uncontrolled cellular proliferation via aberrant expression of S-phase genes [168,169]. In HNSCC, studies in in vitro models have demonstrated that shRNA mediated silencing of E6 and E7 in HPV-16 positive HNSCC cell lines can restore p53 and Rb activity as well as induce apoptosis [170] and that HPV-16 infection can cause transformation of normal oral keratinocytes. Importantly, in a dual E6 and E7 transgenic mouse model, it was shown that E6 and particularly E7 play roles in carcinogenesis of HNSCC [171]. More specifically, mice singly expressing either E6 or E7 developed chemically induced tumors at a higher rate than control mice but E7 expressing mice developed tumors at a much higher rate. Interestingly, mice simultaneously expressing both E6 and E7 developed more aggressive tumors suggesting that E6 may play a supportive role in HNSCC carcinogenesis.
The emergence of a distinct risk factor profile and significantly better outcomes [172-177] for HPV positive HNSCC suggest that a clinical approach tailored to this growing subset of HNSCC patients may be prudent. More specifically, this difference in clinical outcomes may be due to distinct molecular aberrations in HPV positive compared to HPV negative tumors. For example, while p53 is generally inactivated by HPV E6 protein in HPV positive tumors, p53 is not mutated as is commonly the case in HPV negative tumors [178]. Accordingly, this difference in p53 mutational status may be the cause of the differential responses to treatment seen between HPV positive and HPV negative patients. This observation not only suggests that HPV status may alter conventional treatment selection, but also that HPV positive tumors may possess additional therapeutic molecular targets such as the E6 and E7 proteins.
Currently, two preventative HPV vaccines, Gardasil and Cervarix, have been approved by the FDA for use in preventing cervical cancer. Gardasil, a tetravalent vaccine effective against HPV types 6, 11, 16 and 18, was approved in 2006 for the prevention of cervical cancer in women and was recently approved for prevention of genital warts in both men and women [179]. Cervarix is a bivalent vaccine against HPV-16 and -18 and is approved for the prevention of cervical cancer. The role of these two vaccines, in both men and women, has not yet been studied in HNSCC. However, as the use of these vaccines increases, epidemiological data including HNSCC risk will become available. In addition to prevention, vaccines targeted against HPV may find a therapeutic role in HNSCC. At present, there are a multitude of different approaches for inducing cell mediated immunity against HPV positive, E6 and E7 expressing tumor cells. These include live viral or bacterial, peptide and nucleic acid vaccines. While the majority of these vaccines are being studied in cervical cancer or other solid tumors, several are currently in clinical testing for HNSCC. Two Phase I studies of ~ 140 total patients are examining the use of HPV-16 peptide epitopes in recurrent HPV-16 positive HNSCC (NCT00257738, NCT00704041). It may also be possible to disrupt the action of either HPV E6 or E7 with a small molecular inhibitor. Such an inhibitor could potentially sensitize HPV positive tumor cells to other therapies and/or be used to treat premalignant lesions. Several studies have identified screening techniques to search for compounds that can inhibit the protein–protein interactions of E6 and E7 [180,181]. Moreover, blocking peptides specific for E6 [182,183] and organic disulfide compounds that disrupt the zinc binding domains of E6 have been identified as potential lead compounds [184,185]. Importantly, it was found that binding of peptide aptamers to HPV E6 protein induced apoptosis in HPV positive tumor cells [182].
7. Competitive environment
Cetuximab is currently the only molecular targeted HNSCC therapy approved by regulatory agencies. However, with a plethora of potential new drugs in development, the evolution of the market may soon force competition between new drugs and combinations of drugs based not only on efficacy but tolerability as well. For example, while both cetuximab and cisplatin provide approximately the same 5-year overall survival benefit (9.2 vs 8%, respectively) when combined with radiotherapy, cetuximab generally has less severe acute and chronic toxicities [33,186,187]. Moreover, cetuximab continues to be beneficial to patients that are unable to tolerate or complete a regimen of platinum based chemotherapy. Thus, emerging drugs that target EGFR and other pathways may not necessarily need to be more efficacious.
As a fully humanized IgG2 mAb, panitumumab may differ from cetuximab in both efficacy and tolerability which may allow it to coexist with cetuximab in different HNSCC patient subpopulations. A large Phase III trial directly comparing cetuximab and panitumumab in > 1000 colorectal cancer patients (ASPECCT, NCT01001377) is currently underway. Although this study is not in HNSCC, it may provide some insights regarding the difference in side effects between the two drugs. Moreover, the two current Phase III trials of panitumumab in HNSCC, on which potential FDA approval is likely to be based, use cisplatin in their control arms, which may provide justification for wider use of panitumumab. The absence of potentially severe allergic reactions may establish a niche market for panitumumab. Many EGFR targeted therapies are also being evaluated in chemopreventative and maintenance settings and TKIs may be particularly suited to this use because of their more convenient oral administration.
8. Potential development issues
The primary problem in the development of both EGFR targeted and other molecular targeted HNSCC therapies is the lack of proven biomarkers for predicting clinical outcomes and response to treatment. Head and neck cancers are generally accessible for biopsy and, thus, have the potential for rapid analysis by histology and molecular diagnostics. Despite the availability of material, the development of prognostic tests has not progressed in conjunction with new therapies. During early development of EGFR targeted therapies, it was thought that higher EGFR expression, whether through gene amplification or increased transcriptional activity, would be a predictor of better response to treatment. However, while EGFR overexpression has been linked to better response to conventional treatment [188,189], a relationship between increased EGFR in tumor cells and outcomes from treatment with molecular targeted therapies has not yet been established in HNSCC. Conversely, in NSCLC, the presence of activating EGFR mutations has been demonstrated to predict response to EGFR targeted TKIs [190-192]. Likewise, the presence of K-ras mutations which are frequent in colorectal cancer but rare in HNSCC [193,194] have been shown to predict response to cetuximab [195]. A recent retrospective analysis of cetuximab clinical trial data has suggested that the development of an acute acneiform rash in HNSCC patients treated with cetuximab may be a predictor of response. Specifically, patients who developed a moderate or severe rash had an increased survival benefit (68.8 months median overall survival) when compared to those who had a mild or no rash (25.6 months) [33]. While the exact mechanism of this reaction remains unresolved, this correlation with better outcomes provides a promising lead from which to develop a diagnostic test and an early indicator in successful treatment.
In conjunction with discovering new biomarkers, the elucidation of mechanisms of resistance to targeted therapies may further improve outcomes by identifying new potential drug and diagnostic targets as well as providing evidence to support the use of combinations of molecular targeted therapies. Resistance to EGFR targeted therapies is complex and probably multifaceted. However, several important factors have been identified. The ligand independent, constitutively active type III variant of EGFR (EGFRvIII) has been reported to be present in 42% of HNSCC patients [196] as well as in other malignancies [197]. In HNSCC models, EGFRvIII has been shown to mediate decreased response to cetuximab as well as cisplatin [196]. Additionally, a recent study in an in vitro HNSCC model of cetuximab resistance demonstrated that cetuximab resistance was mediated by defective endocytosis and trafficking of EGFR from the plasma membrane [198]. In colorectal cancer, sensitivity or resistance to cetuximab has been found to be mediated by K-Ras mutations, decreased PTEN activity leading to a hyperactive PI3K-Akt pathway, high amphiregulin and epiregulin expression, and presence or absence of the R521K polymorphism in EGFR [199]. These factors are currently being investigated in HNSCC and their utility as diagnostic markers, as well as EGFRvIII and EGFR trafficking defects, may soon be elucidated.
Finally, the emergence of HPV infection as an etiological agent and risk factor for HNSCC has introduced a slew of new questions in the development of emerging targeted therapies. First, given the drastic differences in response rate and survival between HPV positive and HPV negative patients using conventional therapies, stratifying these two groups of patients seems warranted. Moreover, if tumor material is available, data from previous studies underlying the use of cetuximab and other biological agents should continue to be retrospectively analyzed with respect to HPV status. Analysis of patient response to radiotherapy may be particularly important due to the fact that functionally intact p53 often mediates radiation induced cell death and p53 is less commonly mutated in HPV positive tumors [200]. Specifically, the developmental focus on drug candidates that act as radiosensitizers may be prudent if HPV positive tumor cells are shown to be especially sensitive to radiotherapy. Indeed, the pivotal Phase III trial [32] underlying the approval of cetuximab as an adjunct to radiotherapy was conducted in a cohort where > 50% of the primary tumors were located in the oropharynx. Given the increased HPV positivity in tumors of the orpharynx and the evidence that EGFR targeted therapies may act as radiosensitizers, this distribution of tumor sites suggests that HPV status may underlie recent cetuximab results. Thus, it is critically important to account for HPV status in all active and planned clinical trials.
Overall, HPV status may become a facet, albeit an important one, of a larger individual molecular profile of every HNSCC patient and tumor. Technological advances may soon permit rapid and widespread profiling of genetic and epigenetic inherited factors, gene expression and a variety of other measurable biomarkers that will result in an exponential growth of correlative data. The addition of HNSCC to the Cancer Genome Atlas (TCGA) in September 2009 is an important step in building a map of the genomic changes important in HNSCC as well as correlating these changes to clinical variables. For example, results from a pilot TCGA study in glioblastoma multiforme (GBM) have not only confirmed major subgroups of GBM based on genomic changes, but have also identified methylation and mutational changes of the gene MGMT that will have immediate clinical implications [201]. This groundswell of information may cause further stratification of the HNSCC patient population according to predicted response to biological agents, resulting in increasingly individually tailored treatment regimens.
9. Conclusion
The continuing success of cetuximab has validated the molecular targeted approach to HNSCC therapy. While, the overall market for HNSCC therapies remains small relative to other malignancies such as colorectal, lung and breast cancer, this may be due in part to the fact that cetuximab was only recently approved and remains the sole targeted therapy available for HNSCC. As new biological agents targeting both EGFR and other pathways gain regulatory approval, the overall market is expected to expand considerably. Within the realm of EGFR targeted therapies, newer mAbs may replace cetuximab within a few years as results from ongoing clinical trials become available. Importantly, results from a nearly completed Phase III study (SPECTRUM) of panitumumab may shed some light on the differences in efficacy, tolerability and potentially even mechanism of action between IgG1 (cetuximab) and IgG2 (pantiumumab) anti-EGFR antibodies. With the exception of lapatinib, the clinical development of TKIs has been less active. Gefitinib is no longer in active development for HNSCC and erlotininb is not currently being studied in Phase III trials. While the targeting of other pathways remains investigational in HNSCC, agents such as bevacizumab are in late stage clinical development and positive Phase III results may soon provide another proven HNSCC therapeutic target. Biological agents including cetuximab have thus far not demonstrated outstanding efficacy as monotherapies. Accordingly, a multitude of currently active clinical trials are investigating combinations of targeted therapies in addition to combinations of targeted therapies with conventional chemoradiotherapy.
10. Expert opinion
The emergence of HPV as a significant etiological and prognostic factor has and will continue to shape the development of therapies for HNSCC. Unlike studies conducted only 5 years ago, current clinical trials now universally collect HPV status data as part of standard protocols and resulting data may establish HPV positive HNSCC as a distinct disease. Already, retrospective analyses of conventional chemoradiotherapy studies have identified HPV positive patients as a subgroup with significantly better treatment outcomes and HPV negative patients as a subgroup with poorer outcomes. This finding may soon necessitate reexamination of treatment plans based on HPV status with the likely outcome being a deintensification of therapy for HPV positive patients and intensification for HPV negative patients. Whether outcomes will remain better with respect to molecular targeted therapies remains unanswered.
Ultimately, the future expansion of molecular targeted therapies may in fact depend on the parallel development of useful biomarkers such as HPV. Drugs in development and targets being considered for clinical development may be found to be particularly effective or ineffective in different subgroups of HNSCC patients. In this increasingly stratified environment, the head and neck cancer market may be able to sustain many more molecular targeted therapies than at present allowing patients to receive the targeted therapy or, more likely, the combination of therapies that best suit their needs.
Footnotes
Declaration of interest The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Bibliography
- 1.Horner M, Ries L, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2006. [31 March 2010];2009 Available from: http://seercancergov/csr/1975_2006/
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24(14):2137–50. doi: 10.1200/JCO.2005.05.2308. [DOI] [PubMed] [Google Scholar]
- 4.Boyle P, Levin B, editors. World Cancer Report 2008. WHO Press; Lyon: 2008. [Google Scholar]
- 5.Curado MP, Hashibe M. Recent changes in the epidemiology of head and neck cancer. Curr Opin Oncol. 2009;21(3):194–200. doi: 10.1097/CCO.0b013e32832a68ca. [DOI] [PubMed] [Google Scholar]
- 6.Shiboski CH, Schmidt BL, Jordan RC. Tongue and tonsil carcinoma: increasing trends in the U.S. population ages 20-44 years. Cancer. 2005;103(9):1843–9. doi: 10.1002/cncr.20998. [DOI] [PubMed] [Google Scholar]
- 7.Hashibe M, Brennan P, Benhamou S, et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007;99(10):777–89. doi: 10.1093/jnci/djk179. [DOI] [PubMed] [Google Scholar]
- 8.Hashibe M, Brennan P, Chuang SC, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. 2009;18(2):541–50. doi: 10.1158/1055-9965.EPI-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lubin JH, Purdue M, Kelsey K, et al. Total exposure and exposure rate effects for alcohol and smoking and risk of head and neck cancer: a pooled analysis of case-control studies. Am J Epidemiol. 2009;170(8):937–47. doi: 10.1093/aje/kwp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purdue MP, Hashibe M, Berthiller J, et al. Type of alcoholic beverage and risk of head and neck cancer – a pooled analysis within the INHANCE Consortium. Am J Epidemiol. 2009;169(2):132–42. doi: 10.1093/aje/kwn306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Negri E, Boffetta P, Berthiller J, et al. Family history of cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Int J Cancer. 2009;124(2):394–401. doi: 10.1002/ijc.23848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marron M, Boffetta P, Zhang ZF, et al. Cessation of alcohol drinking, tobacco smoking and the reversal of head and neck cancer risk. Int J Epidemiol. 2010;39(1):182–96. doi: 10.1093/ije/dyp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev. 2005;14(2):467–75. doi: 10.1158/1055-9965.EPI-04-0551. [DOI] [PubMed] [Google Scholar]
- 14.Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12–9. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 15.Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer. Cancer. 2004;101(2):270–80. doi: 10.1002/cncr.20365. [DOI] [PubMed] [Google Scholar]
- 16.Heck JE, Berthiller J, Vaccarella S, et al. Sexual behaviours and the risk of head and neck cancers: a pooled analysis in the International Head and Neck Cancer Epidemiology (INHANCE) consortium. Int J Epidemiol. 2009;39(1):166–81. doi: 10.1093/ije/dyp350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 18.Carvalho AL, Nishimoto IN, Califano JA, Kowalski LP. Trends in incidence and prognosis for head and neck cancer in the United States: a site-specific analysis of the SEER database. Int J Cancer. 2005;114(5):806–16. doi: 10.1002/ijc.20740. [DOI] [PubMed] [Google Scholar]
- 19.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371(9625):1695–709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong RJ, Shah JP. The role of the head and neck surgeon in contemporary multidisciplinary treatment programs for advanced head and neck cancer. Curr Opin Otolaryngol Head Neck Surg. 2010;18(2):79–82. doi: 10.1097/MOO.0b013e32833782f0. [DOI] [PubMed] [Google Scholar]
- 21.Haigentz M, Jr, Silver CE, Corry J, et al. Current trends in initial management of oropharyngeal cancer: the declining use of open surgery. Eur Arch Otorhinolaryngol. 2009;266(12):1845–55. doi: 10.1007/s00405-009-1109-2. [DOI] [PubMed] [Google Scholar]
- 22.Pignon JP, le Maitre A, Maillard E, Bourhis J. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): an update on 93 randomised trials and 17,346 patients. Radiother Oncol. 2009;92(1):4–14. doi: 10.1016/j.radonc.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Dirix P, Nuyts S. Evidence-based organ-sparing radiotherapy in head and neck cancer. Lancet Oncol. 2010;11(1):85–91. doi: 10.1016/S1470-2045(09)70231-1. [DOI] [PubMed] [Google Scholar]
- 24.Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008;359(11):1143–54. doi: 10.1056/NEJMra0707975. [DOI] [PubMed] [Google Scholar]
- 25.Jones AS, Fish B, Fenton JE, Husband DJ. The treatment of early laryngeal cancers (T1-T2 N0): surgery or irradiation? Head Neck. 2004;26(2):127–35. doi: 10.1002/hed.10361. [DOI] [PubMed] [Google Scholar]
- 26.NCCN Clinical Practice Guidelines in Oncology – Head and Neck Cancers V.1.2010. 2010 [Google Scholar]
- 27.Adelstein DJ, Li Y, Adams GL, et al. An intergroup phase III comparison of standard radiation therapy and two schedules of concurrent chemoradiotherapy in patients with unresectable squamous cell head and neck cancer. J Clin Oncol. 2003;21(1):92–8. doi: 10.1200/JCO.2003.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Brizel DM, Albers ME, Fisher SR, et al. Hyperfractionated irradiation with or without concurrent chemotherapy for locally advanced head and neck cancer. N Engl J Med. 1998;338(25):1798–804. doi: 10.1056/NEJM199806183382503. [DOI] [PubMed] [Google Scholar]
- 29.Bensadoun RJ, Benezery K, Dassonville O, et al. French multicenter phase III randomized study testing concurrent twice-a-day radiotherapy and cisplatin/5-fluorouracil chemotherapy (BiRCF) in unresectable pharyngeal carcinoma: results at 2 years (FNCLCC-GORTEC) Int J Radiat Oncol Biol Phys. 2006;64(4):983–94. doi: 10.1016/j.ijrobp.2005.09.041. [DOI] [PubMed] [Google Scholar]
- 30.Budach V, Stuschke M, Budach W, et al. Hyperfractionated accelerated chemoradiation with concurrent fluorouracil-mitomycin is more effective than dose-escalated hyperfractionated accelerated radiation therapy alone in locally advanced head and neck cancer: final results of the radiotherapy cooperative clinical trials group of the German Cancer Society 95-06 Prospective Randomized Trial. J Clin Oncol. 2005;23(6):1125–35. doi: 10.1200/JCO.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Huguenin P, Beer KT, Allal A, et al. Concomitant cisplatin significantly improves locoregional control in advanced head and neck cancers treated with hyperfractionated radiotherapy. J Clin Oncol. 2004;22(23):4665–73. doi: 10.1200/JCO.2004.12.193. [DOI] [PubMed] [Google Scholar]
- 32.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 33.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21–8. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 34.Clark J, Li W, Smith G, et al. Outcome of treatment for advanced cervical metastatic squamous cell carcinoma. Head Neck. 2005;27(2):87–94. doi: 10.1002/hed.20129. [DOI] [PubMed] [Google Scholar]
- 35.Ferrari D, Codeca C, Fiore J, et al. A review on the treatment of relapsed/metastatic head and neck cancer. Expert Opin Pharmacother. 2009;10(16):2625–32. doi: 10.1517/14656560903232645. [DOI] [PubMed] [Google Scholar]
- 36.Jacobs C, Lyman G, Velez-Garcia E, et al. A phase III randomized study comparing cisplatin and fluorouracil as single agents and in combination for advanced squamous cell carcinoma of the head and neck. J Clin Oncol. 1992;10(2):257–63. doi: 10.1200/JCO.1992.10.2.257. [DOI] [PubMed] [Google Scholar]
- 37.Forastiere AA, Metch B, Schuller DE, et al. Randomized comparison of cisplatin plus fluorouracil and carboplatin plus fluorouracil versus methotrexate in advanced squamous-cell carcinoma of the head and neck: a Southwest Oncology Group study. J Clin Oncol. 1992;10(8):1245–51. doi: 10.1200/JCO.1992.10.8.1245. [DOI] [PubMed] [Google Scholar]
- 38.Adamo V, Ferraro G, Pergolizzi S, et al. Paclitaxel and cisplatin in patients with recurrent and metastatic head and neck squamous cell carcinoma. Oral Oncol. 2004;40(5):525–31. doi: 10.1016/j.oraloncology.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 39.Basaran M, Bavbek SE, Gullu I, et al. A phase II study of paclitaxel and cisplatin combination chemotherapy in recurrent or metastatic head and neck cancer. J Chemother. 2002;14(2):207–13. doi: 10.1179/joc.2002.14.2.207. [DOI] [PubMed] [Google Scholar]
- 40.Forastiere AA, Leong T, Rowinsky E, et al. Phase III comparison of high-dose paclitaxel + cisplatin + granulocyte colony-stimulating factor versus low-dose paclitaxel + cisplatin in advanced head and neck cancer: Eastern Cooperative Oncology Group Study E1393. J Clin Oncol. 2001;19(4):1088–95. doi: 10.1200/JCO.2001.19.4.1088. [DOI] [PubMed] [Google Scholar]
- 41.Gibson MK, Li Y, Murphy B, et al. Randomized phase III evaluation of cisplatin plus fluorouracil versus cisplatin plus paclitaxel in advanced head and neck cancer (E1395): an intergroup trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2005;23(15):3562–7. doi: 10.1200/JCO.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 42.Janinis J, Papadakou M, Xidakis E, et al. Combination chemotherapy with docetaxel, cisplatin, and 5-fluorouracil in previously treated patients with advanced/recurrent head and neck cancer: a phase II feasibility study. Am J Clin Oncol. 2000;23(2):128–31. doi: 10.1097/00000421-200004000-00005. [DOI] [PubMed] [Google Scholar]
- 43.Shin DM, Glisson BS, Khuri FR, et al. Phase II trial of paclitaxel, ifosfamide, and cisplatin in patients with recurrent head and neck squamous cell carcinoma. J Clin Oncol. 1998;16(4):1325–30. doi: 10.1200/JCO.1998.16.4.1325. [DOI] [PubMed] [Google Scholar]
- 44.Shin DM, Khuri FR, Glisson BS, et al. Phase II study of paclitaxel, ifosfamide, and carboplatin in patients with recurrent or metastatic head and neck squamous cell carcinoma. Cancer. 2001;91(7):1316–23. doi: 10.1002/1097-0142(20010401)91:7<1316::aid-cncr1134>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 45.Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–27. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 46.Burtness B, Goldwasser MA, Flood W, et al. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol. 2005;23(34):8646–54. doi: 10.1200/JCO.2005.02.4646. [DOI] [PubMed] [Google Scholar]
- 47.Herbst RS, Arquette M, Shin DM, et al. Phase II multicenter study of the epidermal growth factor receptor antibody cetuximab and cisplatin for recurrent and refractory squamous cell carcinoma of the head and neck. J Clin Oncol. 2005;23(24):5578–87. doi: 10.1200/JCO.2005.07.120. [DOI] [PubMed] [Google Scholar]
- 48.Baselga J, Trigo JM, Bourhis J, et al. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol. 2005;23(24):5568–77. doi: 10.1200/JCO.2005.07.119. [DOI] [PubMed] [Google Scholar]
- 49.Mesia R, Rivera F, Kawecki A, et al. Quality of life of patients receiving platinum-based chemotherapy plus cetuximab first line for recurrent and/or metastatic squamous cell carcinoma of the head and neck. Ann Oncol. 2010 doi: 10.1093/annonc/mdq077. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eli Lilly and Co. Fourth-Quarter and Full-Year 2009 Results. Indianapolis, Indiana: 2010. Jan 28, [Google Scholar]
- 51.Bristol-Myers Squibb Fourth Quarter Earnings 2009 Results. 2010 January 28 [Google Scholar]
- 52.Merck KgGA Business Results for 4th Quarter/Fiscal Year 2009. 2010 February 23 [Google Scholar]
- 53.Zeng G. LLY: growth challenged over the near-term. Zacks Equity Research; Chicago, IL: 2010. Mar 30, [Google Scholar]
- 54.Rowan K. Should cetuximab replace cisplatin in head and neck cancer? J Natl Cancer Inst. 2010;102(2):74–6. 8. doi: 10.1093/jnci/djp531. [DOI] [PubMed] [Google Scholar]
- 55.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53(15):3579–84. [PubMed] [Google Scholar]
- 56.Bei R, Budillon A, Masuelli L, et al. Frequent overexpression of multiple ErbB receptors by head and neck squamous cell carcinoma contrasts with rare antibody immunity in patients. J Pathol. 2004;204(3):317–25. doi: 10.1002/path.1642. [DOI] [PubMed] [Google Scholar]
- 57.Ongkeko WM, Altuna X, Weisman RA, Wang-Rodriguez J. Expression of protein tyrosine kinases in head and neck squamous cell carcinomas. Am J Clin Pathol. 2005;124(1):71–6. doi: 10.1309/BTLN5WTMJ3PCNRRC. [DOI] [PubMed] [Google Scholar]
- 58.Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77(6):400–10. doi: 10.1159/000279388. [DOI] [PubMed] [Google Scholar]
- 59.Cavalot A, Martone T, Roggero N, et al. Prognostic impact of HER-2/neu expression on squamous head and neck carcinomas. Head Neck. 2007;29(7):655–64. doi: 10.1002/hed.20574. [DOI] [PubMed] [Google Scholar]
- 60.Ekberg T, Nestor M, Engstrom M, et al. Expression of EGFR, HER2, HER3, and HER4 in metastatic squamous cell carcinomas of the oral cavity and base of tongue. Int J Oncol. 2005;26(5):1177–85. doi: 10.3892/ijo.26.5.1177. [DOI] [PubMed] [Google Scholar]
- 61.Xia W, Lau YK, Zhang HZ, et al. Combination of EGFR, HER-2/neu, and HER-3 is a stronger predictor for the outcome of oral squamous cell carcinoma than any individual family members. Clin Cancer Res. 1999;5(12):4164–74. [PubMed] [Google Scholar]
- 62.Xia W, Lau YK, Zhang HZ, et al. Strong correlation between c-erbB-2 overexpression and overall survival of patients with oral squamous cell carcinoma. Clin Cancer Res. 1997;3(1):3–9. [PubMed] [Google Scholar]
- 63.Sheu JJ, Hua CH, Wan L, et al. Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Res. 2009;69(6):2568–76. doi: 10.1158/0008-5472.CAN-08-3199. [DOI] [PubMed] [Google Scholar]
- 64.Braut T, Krstulja M, Kujundzic M, et al. Epidermal growth factor receptor protein expression and gene amplification in normal, hyperplastic, and cancerous glottic tissue: immunohistochemical and fluorescent in situ hybridization study on tissue microarrays. Croat Med J. 2009;50(4):370–9. doi: 10.3325/cmj.2009.50.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim S, Grandis JR, Rinaldo A, et al. Emerging perspectives in epidermal growth factor receptor targeting in head and neck cancer. Head Neck. 2008;30(5):667–74. doi: 10.1002/hed.20859. [DOI] [PubMed] [Google Scholar]
- 66.Grandis JR, Chakraborty A, Zeng Q, et al. Downmodulation of TGF-alpha protein expression with antisense oligonucleotides inhibits proliferation of head and neck squamous carcinoma but not normal mucosal epithelial cells. J Cell Biochem. 1998;69(1):55–62. [PubMed] [Google Scholar]
- 67.Liang K, Ang KK, Milas L, et al. The epidermal growth factor receptor mediates radioresistance. Int J Radiat Oncol Biol Phys. 2003;57(1):246–54. doi: 10.1016/s0360-3016(03)00511-x. [DOI] [PubMed] [Google Scholar]
- 68.Milas L, Fan Z, Andratschke NH, Ang KK. Epidermal growth factor receptor and tumor response to radiation: in vivo preclinical studies. Int J Radiat Oncol Biol Phys. 2004;58(3):966–71. doi: 10.1016/j.ijrobp.2003.08.035. [DOI] [PubMed] [Google Scholar]
- 69.Dai Q, Ling YH, Lia M, et al. Enhanced sensitivity to the HER1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib hydrochloride in chemotherapy-resistant tumor cell lines. Clin Cancer Res. 2005;11(4):1572–8. doi: 10.1158/1078-0432.CCR-04-0993. [DOI] [PubMed] [Google Scholar]
- 70.Stewart JS, Cohen EE, Licitra L, et al. Phase III study of gefitinib compared with intravenous methotrexate for recurrent squamous cell carcinoma of the head and neck [corrected] J Clin Oncol. 2009;27(11):1864–71. doi: 10.1200/JCO.2008.17.0530. [DOI] [PubMed] [Google Scholar]
- 71.Rubin Grandis J, Melhem MF, Gooding WE, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90(11):824–32. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 72.Temam S, Kawaguchi H, El-Naggar AK, et al. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol. 2007;25(16):2164–70. doi: 10.1200/JCO.2006.06.6605. [DOI] [PubMed] [Google Scholar]
- 73.Chung CH, Ely K, McGavran L, et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J Clin Oncol. 2006;24(25):4170–6. doi: 10.1200/JCO.2006.07.2587. [DOI] [PubMed] [Google Scholar]
- 74.Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62(24):7350–6. [PubMed] [Google Scholar]
- 75.Maurizi M, Almadori G, Ferrandina G, et al. Prognostic significance of epidermal growth factor receptor in laryngeal squamous cell carcinoma. Br J Cancer. 1996;74(8):1253–7. doi: 10.1038/bjc.1996.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carter CA, Kelly RJ, Giaccone G. Small-molecule inhibitors of the human epidermal receptor family. Expert Opin Investig Drugs. 2009;18(12):1829–42. doi: 10.1517/13543780903373343. [DOI] [PubMed] [Google Scholar]
- 77.Chinnaiyan P, Huang S, Vallabhaneni G, et al. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva) Cancer Res. 2005;65(8):3328–35. doi: 10.1158/0008-5472.CAN-04-3547. [DOI] [PubMed] [Google Scholar]
- 78.Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res. 2000;6(6):2166–74. [PubMed] [Google Scholar]
- 79.Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59(8):1935–40. [PubMed] [Google Scholar]
- 80.Vincenzi B, Zoccoli A, Pantano F, et al. Cetuximab: from bench to bedside. Curr Cancer Drug Targets. 2010;10(1):80–95. doi: 10.2174/156800910790980241. [DOI] [PubMed] [Google Scholar]
- 81.Lopez-Albaitero A, Lee SC, Morgan S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58(11):1853–64. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taylor RJ, Chan SL, Wood A, et al. FcgammaRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. 2009;58(7):997–1006. doi: 10.1007/s00262-008-0613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lopez-Albaitero A, Ferris RL. Immune activation by epidermal growth factor receptor specific monoclonal antibody therapy for head and neck cancer. Arch Otolaryngol Head Neck Surg. 2007;133(12):1277–81. doi: 10.1001/archotol.133.12.1277. [DOI] [PubMed] [Google Scholar]
- 84.Iwase M, Takaoka S, Uchida M, et al. Epidermal growth factor receptor inhibitors enhance susceptibility to Fas-mediated apoptosis in oral squamous cell carcinoma cells. Oral Oncol. 2008;44(4):361–8. doi: 10.1016/j.oraloncology.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 85.Bozec A, Formento P, Ciccolini J, et al. Response of endothelial cells to a dual tyrosine kinase receptor inhibition combined with irradiation. Mol Cancer Ther. 2005;4(12):1962–71. doi: 10.1158/1535-7163.MCT-05-0108. [DOI] [PubMed] [Google Scholar]
- 86.Hirata A, Ogawa S, Kometani T, et al. ZD1839 (Iressa) induces antiangiogenic effects through inhibition of epidermal growth factor receptor tyrosine kinase. Cancer Res. 2002;62(9):2554–60. [PubMed] [Google Scholar]
- 87.Huang SM, Li J, Harari PM. Molecular inhibition of angiogenesis and metastatic potential in human squamous cell carcinomas after epidermal growth factor receptor blockade. Mol Cancer Ther. 2002;1(7):507–14. [PubMed] [Google Scholar]
- 88.Ciardiello F, Caputo R, Troiani T, et al. Antisense oligonucleotides targeting the epidermal growth factor receptor inhibit proliferation, induce apoptosis, and cooperate with cytotoxic drugs in human cancer cell lines. Int J Cancer. 2001;93(2):172–8. doi: 10.1002/ijc.1335. [DOI] [PubMed] [Google Scholar]
- 89.Milas L, Mason K, Hunter N, et al. In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clin Cancer Res. 2000;6(2):701–8. [PubMed] [Google Scholar]
- 90.Saltz L, Easley C, Kirkpatrick P. Panitumumab. Nat Rev Drug Discov. 2006;5(12):987–8. doi: 10.1038/nrd2204. [DOI] [PubMed] [Google Scholar]
- 91.Kim R. Cetuximab and panitumumab: are they interchangeable? Lancet Oncol. 2009;10(12):1140–1. doi: 10.1016/S1470-2045(09)70312-2. [DOI] [PubMed] [Google Scholar]
- 92.Rivera F, Vega-Villegas ME, Lopez-Brea MF, Marquez R. Current situation of Panitumumab, Matuzumab, Nimotuzumab and Zalutumumab. Acta Oncol. 2008;47(1):9–19. doi: 10.1080/02841860701704724. [DOI] [PubMed] [Google Scholar]
- 93.Kuenen B, Witteveen PO, Ruijter R, et al. A phase I pharmacologic study of necitumumab (IMC-11F8), a fully human IgG1 monoclonal antibody directed against EGFR in patients with advanced solid malignancies. Clin Cancer Res. 2010;16(6):1915–23. doi: 10.1158/1078-0432.CCR-09-2425. [DOI] [PubMed] [Google Scholar]
- 94.Jungbluth AA, Stockert E, Huang HJ, et al. A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. Proc Natl Acad Sci USA. 2003;100(2):639–44. doi: 10.1073/pnas.232686499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Scott AM, Lee FT, Tebbutt N, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci USA. 2007;104(10):4071–6. doi: 10.1073/pnas.0611693104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cohen EE, Rosen F, Stadler WM, et al. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003;21(10):1980–7. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 97.Cohen EE, Kane MA, List MA, et al. Phase II trial of gefitinib 250 mg daily in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005;11(23):8418–24. doi: 10.1158/1078-0432.CCR-05-1247. [DOI] [PubMed] [Google Scholar]
- 98.Caponigro F, Romano C, Milano A, et al. A phase I/II trial of gefitinib and radiotherapy in patients with locally advanced inoperable squamous cell carcinoma of the head and neck. Anticancer Drugs. 2008;19(7):739–44. doi: 10.1097/CAD.0b013e32830676a8. [DOI] [PubMed] [Google Scholar]
- 99.Chua DT, Wei WI, Wong MP, et al. Phase II study of gefitinib for the treatment of recurrent and metastatic nasopharyngeal carcinoma. Head Neck. 2008;30(7):863–7. doi: 10.1002/hed.20792. [DOI] [PubMed] [Google Scholar]
- 100.Hainsworth JD, Spigel DR, Burris HA, III, et al. Neoadjuvant chemotherapy/gefitinib followed by concurrent chemotherapy/radiation therapy/gefitinib for patients with locally advanced squamous carcinoma of the head and neck. Cancer. 2009;115(10):2138–46. doi: 10.1002/cncr.24265. [DOI] [PubMed] [Google Scholar]
- 101.Argiris A, Ghebremichael M, Gilbert J, et al. A phase III randomized, placebo-controlled trial of docetaxel (D) with or without gefitinib (G) in recurrent or metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN): a trial of the Eastern Cooperative Oncology Group (ECOG) J Clin Oncol. 2009;27(Suppl):5s. doi: 10.1200/JCO.2012.45.4272. abstract 6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soulieres D, Senzer NN, Vokes EE, et al. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22(1):77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 103.Siu LL, Soulieres D, Chen EX, et al. Phase I/II trial of erlotinib and cisplatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck: a Princess Margaret Hospital phase II consortium and National Cancer Institute of Canada Clinical Trials Group Study. J Clin Oncol. 2007;25(16):2178–83. doi: 10.1200/JCO.2006.07.6547. [DOI] [PubMed] [Google Scholar]
- 104.Kim ES, Kies MS, Glisson BS, et al. Final results of a phase II study of erlotinib, docetaxel and cisplatin in patients with recurrent/metastatic head and neck cancer. 2007 ASCO Annual Meeting Proceedings Part I. J Clin Oncol. 2007;25(18S):6013. June 20 Supplement. [Google Scholar]
- 105.delCampo J, Sebastian P, Hitt R, et al. Results of a phase II randomised study in patients with locally advanced squamous cell carcinoma of the head and neck (SCCHN) 33rd ESMO Congress. 2008 [Google Scholar]
- 106.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 107.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ribatti D. The discovery of antiangiogenic molecules: a historical review. Curr Pharm Des. 2009;15(4):345–52. doi: 10.2174/138161209787315855. [DOI] [PubMed] [Google Scholar]
- 109.Gerber PA, Hippe A, Buhren BA, et al. Chemokines in tumor-associated angiogenesis. Biol Chem. 2009;390(12):1213–23. doi: 10.1515/BC.2009.144. [DOI] [PubMed] [Google Scholar]
- 110.Montag M, Dyckhoff G, Lohr J, et al. Angiogenic growth factors in tissue homogenates of HNSCC: expression pattern, prognostic relevance, and interrelationships. Cancer Sci. 2009;100(7):1210–8. doi: 10.1111/j.1349-7006.2009.01158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cortes-Funes H. The role of antiangiogenesis therapy: bevacizumab and beyond. Clin Transl Oncol. 2009;11(6):349–55. doi: 10.1007/s12094-009-0368-0. [DOI] [PubMed] [Google Scholar]
- 112.Kyzas PA, Cunha IW, Ioannidis JP. Prognostic significance of vascular endothelial growth factor immunohistochemical expression in head and neck squamous cell carcinoma: a meta-analysis. Clin Cancer Res. 2005;11(4):1434–40. doi: 10.1158/1078-0432.CCR-04-1870. [DOI] [PubMed] [Google Scholar]
- 113.Kyzas PA, Stefanou D, Batistatou A, Agnantis NJ. Prognostic significance of VEGF immunohistochemical expression and tumor angiogenesis in head and neck squamous cell carcinoma. J Cancer Res Clin Oncol. 2005;131(9):624–30. doi: 10.1007/s00432-005-0003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Neuchrist C, Erovic BM, Handisurya A, et al. Vascular endothelial growth factor C and vascular endothelial growth factor receptor 3 expression in squamous cell carcinomas of the head and neck. Head Neck. 2003;25(6):464–74. doi: 10.1002/hed.10235. [DOI] [PubMed] [Google Scholar]
- 115.Kyzas PA, Geleff S, Batistatou A, et al. Evidence for lymphangiogenesis and its prognostic implications in head and neck squamous cell carcinoma. J Pathol. 2005;206(2):170–7. doi: 10.1002/path.1776. [DOI] [PubMed] [Google Scholar]
- 116.Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362(6423):841–4. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 117.Riedel F, Gotte K, Li M, et al. Abrogation of VEGF expression in human head and neck squamous cell carcinoma decreases angiogenic activity in vitro and in vivo. Int J Oncol. 2003;23(3):577–83. [PubMed] [Google Scholar]
- 118.Bock JM, Sinclair LL, Bedford NS, et al. Modulation of cellular invasion by VEGF-C expression in squamous cell carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg. 2008;134(4):355–62. doi: 10.1001/archotol.134.4.355. [DOI] [PubMed] [Google Scholar]
- 119.Tong M, Lloyd B, Pei P, Mallery SR. Human head and neck squamous cell carcinoma cells are both targets and effectors for the angiogenic cytokine, VEGF. J Cell Biochem. 2008;105(5):1202–10. doi: 10.1002/jcb.21920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Seiwert TY, Cohen EE. Targeting angiogenesis in head and neck cancer. Semin Oncol. 2008;35(3):274–85. doi: 10.1053/j.seminoncol.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 121.Kleespies A, Guba M, Jauch KW, Bruns CJ. Vascular endothelial growth factor in esophageal cancer. J Surg Oncol. 2004;87(2):95–104. doi: 10.1002/jso.20070. [DOI] [PubMed] [Google Scholar]
- 122.Horn L, Sandler AB. Angiogenesis in the treatment of non-small cell lung cancer. Proc Am Thorac Soc. 2009;6(2):206–17. doi: 10.1513/pats.200807-066LC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kyzas PA, Stefanou D, Batistatou A, Agnantis NJ. Potential autocrine function of vascular endothelial growth factor in head and neck cancer via vascular endothelial growth factor receptor-2. Mod Pathol. 2005;18(4):485–94. doi: 10.1038/modpathol.3800295. [DOI] [PubMed] [Google Scholar]
- 124.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 125.Cohen EE, Davis DW, Karrison TG, et al. Erlotinib and bevacizumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck: a phase I/II study. Lancet Oncol. 2009;10(3):247–57. doi: 10.1016/S1470-2045(09)70002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cohen R, Stebbing J, Windsor A. Bevacizumab and surgery: what is the real risk? Future Oncol. 2009;5(7):915–7. doi: 10.2217/fon.09.63. [DOI] [PubMed] [Google Scholar]
- 127.Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99(17):11393–8. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stein MN, Flaherty KT. CCR drug updates: sorafenib and sunitinib in renal cell carcinoma. Clin Cancer Res. 2007;13(13):3765–70. doi: 10.1158/1078-0432.CCR-06-2844. [DOI] [PubMed] [Google Scholar]
- 129.Williamson SK, Moon J, Huang CH, et al. A phase II trial of sorafenib in patients with recurrent and/or metastatic head and neck squamous cell carcinoma (HNSCC): A Southwest Oncology Group (SWOG) trial. 2007 ASCO Annual Meeting Proceedings Part I. J Clin Oncol. 2007;25(18S):6044. June 20 Supplement. [Google Scholar]
- 130.Choong NW, Kozloff M, Taber D, et al. Phase II study of sunitinib malate in head and neck squamous cell carcinoma. Invest New Drugs. 2009 doi: 10.1007/s10637-009-9296-7. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 131.Machiels JP, Henry S, Zanetta S, et al. Phase II study of sunitinib in recurrent or metastatic squamous cell carcinoma of the head and neck: GORTEC 2006-01. J Clin Oncol. 2010;28(1):21–8. doi: 10.1200/JCO.2009.23.8584. [DOI] [PubMed] [Google Scholar]
- 132.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19(21):2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 133.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98(3):295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 134.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003;63(14):3923–30. [PubMed] [Google Scholar]
- 135.Hambek M, Baghi M, Strebhardt K, et al. STAT 3 activation in head and neck squamous cell carcinomas is controlled by the EGFR. Anticancer Res. 2004;24(6):3881–6. [PubMed] [Google Scholar]
- 136.Grandis JR, Drenning SD, Chakraborty A, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J Clin Invest. 1998;102(7):1385–92. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kijima T, Niwa H, Steinman RA, et al. STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ. 2002;13(8):355–62. [PubMed] [Google Scholar]
- 138.Neiva KG, Zhang Z, Miyazawa M, et al. Cross talk initiated by endothelial cells enhances migration and inhibits anoikis of squamous cell carcinoma cells through STAT3/Akt/ERK signaling. Neoplasia. 2009;11(6):583–93. doi: 10.1593/neo.09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grandis JR, Drenning SD, Zeng Q, et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci USA. 2000;97(8):4227–32. doi: 10.1073/pnas.97.8.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6(10):587–95. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 141.Koppikar P, Choi SH, Egloff AM, et al. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin Cancer Res. 2008;14(13):4284–91. doi: 10.1158/1078-0432.CCR-07-5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nozawa H, Howell G, Suzuki S, et al. Combined inhibition of PLC{gamma}-1 and c-Src abrogates epidermal growth factor receptor-mediated head and neck squamous cell carcinoma invasion. Clin Cancer Res. 2008;14(13):4336–44. doi: 10.1158/1078-0432.CCR-07-4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin Cancer Res. 2005;11(19 Pt 1):6924–32. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 144.Wheeler DL, Iida M, Kruser TJ, et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther. 2009;8(8):696–703. doi: 10.4161/cbt.8.8.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li C, Iida M, Dunn EF, et al. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28(43):3801–13. doi: 10.1038/onc.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lu Y, Li X, Liang K, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67(17):8240–7. doi: 10.1158/0008-5472.CAN-07-0589. [DOI] [PubMed] [Google Scholar]
- 147.Brooks HD, Glisson B, Lu C, et al. Phase II study of dasatinib in the treatment of head and neck squamous cell carcinoma (HNSCC) J Clin Oncol. 2009;27(Suppl):15s. abstract 6022. [Google Scholar]
- 148.Johnson FM, Agrawal S, Burris H, et al. Phase 1 pharmacokinetic and drug-interaction study of dasatinib in patients with advanced solid tumors. Cancer. 2010;116(6):1582–91. doi: 10.1002/cncr.24927. [DOI] [PubMed] [Google Scholar]
- 149.Moral M, Paramio JM. Akt pathway as a target for therapeutic intervention in HNSCC. Histol Histopathol. 2008;23(10):1269–78. doi: 10.14670/HH-23.1269. [DOI] [PubMed] [Google Scholar]
- 150.Segrelles C, Moral M, Lara MF, et al. Molecular determinants of Akt-induced keratinocyte transformation. Oncogene. 2006;25(8):1174–85. doi: 10.1038/sj.onc.1209155. [DOI] [PubMed] [Google Scholar]
- 151.Pedrero JM, Carracedo DG, Pinto CM, et al. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005;114(2):242–8. doi: 10.1002/ijc.20711. [DOI] [PubMed] [Google Scholar]
- 152.Amornphimoltham P, Sriuranpong V, Patel V, et al. Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res. 2004;10(12 Pt 1):4029–37. doi: 10.1158/1078-0432.CCR-03-0249. [DOI] [PubMed] [Google Scholar]
- 153.Wu Z, Gioeli D, Conaway M, et al. Restoration of PTEN expression alters the sensitivity of prostate cancer cells to EGFR inhibitors. Prostate. 2008;68(9):935–44. doi: 10.1002/pros.20745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jhawer M, Goel S, Wilson AJ, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68(6):1953–61. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Aissat N, Le Tourneau C, Ghoul A, et al. Antiproliferative effects of rapamycin as a single agent and in combination with carboplatin and paclitaxel in head and neck cancer cell lines. Cancer Chemother Pharmacol. 2008;62(2):305–13. doi: 10.1007/s00280-007-0609-2. [DOI] [PubMed] [Google Scholar]
- 156.McLaughlin-Drubin ME, Munger K. Oncogenic activities of human papillomaviruses. Virus Res. 2009;143(2):195–208. doi: 10.1016/j.virusres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nishioka S, Fukushima K, Nishizaki K, et al. Human papillomavirus as a risk factor for head and neck cancers – a case-control study. Acta Otolaryngol Suppl. 1999;540:77–80. [PubMed] [Google Scholar]
- 158.Strome SE, Savva A, Brissett AE, et al. Squamous cell carcinoma of the tonsils: a molecular analysis of HPV associations. Clin Cancer Res. 2002;8(4):1093–100. [PubMed] [Google Scholar]
- 159.Chen PC, Kuo C, Pan CC, Chou MY. Risk of oral cancer associated with human papillomavirus infection, betel quid chewing, and cigarette smoking in Taiwan – an integrated molecular and epidemiological study of 58 cases. J Oral Pathol Med. 2002;31(6):317–22. doi: 10.1034/j.1600-0714.2002.00129.x. [DOI] [PubMed] [Google Scholar]
- 160.Smith EM, Ritchie JM, Summersgill KF, et al. Human papillomavirus in oral exfoliated cells and risk of head and neck cancer. J Natl Cancer Inst. 2004;96(6):449–55. doi: 10.1093/jnci/djh074. [DOI] [PubMed] [Google Scholar]
- 161.Mork J, Lie AK, Glattre E, et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med. 2001;344(15):1125–31. doi: 10.1056/NEJM200104123441503. [DOI] [PubMed] [Google Scholar]
- 162.Herrero R, Castellsague X, Pawlita M, et al. Human papillomavirus and oral cancer: the International Agency for Research on Cancer multicenter study. J Natl Cancer Inst. 2003;95(23):1772–83. doi: 10.1093/jnci/djg107. [DOI] [PubMed] [Google Scholar]
- 163.Hammarstedt L, Lindquist D, Dahlstrand H, et al. Human papillomavirus as a risk factor for the increase in incidence of tonsillar cancer. Int J Cancer. 2006;119(11):2620–3. doi: 10.1002/ijc.22177. [DOI] [PubMed] [Google Scholar]
- 164.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 165.Scheffner M, Werness BA, Huibregtse JM, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 166.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75(3):495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 167.Kessis TD, Slebos RJ, Nelson WG, et al. Human papillomavirus 16 E6 expression disrupts the p53-mediated cellular response to DNA damage. Proc Natl Acad Sci USA. 1993;90(9):3988–92. doi: 10.1073/pnas.90.9.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 169.Munger K, Werness BA, Dyson N, et al. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8(13):4099–105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rampias T, Sasaki C, Weinberger P, Psyrri A. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J Natl Cancer Inst. 2009;101(6):412–23. doi: 10.1093/jnci/djp017. [DOI] [PubMed] [Google Scholar]
- 171.Strati K, Lambert PF. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res. 2007;67(24):11585–93. doi: 10.1158/0008-5472.CAN-07-3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gillison ML, DSouza G, Westra W, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100(6):407–20. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 173.Kies MS, Holsinger FC, Lee JJ, et al. Induction chemotherapy and cetuximab for locally advanced squamous cell carcinoma of the head and neck: results from a phase II prospective trial. J Clin Oncol. 2010;28(1):8–14. doi: 10.1200/JCO.2009.23.0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fakhry C, Westra WH, Li S, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100(4):261–9. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 175.Nichols AC, Faquin WC, Westra WH, et al. HPV-16 infection predicts treatment outcome in oropharyngeal squamous cell carcinoma. Otolaryngol Head Neck Surg. 2009;140(2):228–34. doi: 10.1016/j.otohns.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 176.Ragin CC, Taioli E. Survival of squamous cell carcinoma of the head and neck in relation to human papillomavirus infection: review and meta-analysis. Int J Cancer. 2007;121(8):1813–20. doi: 10.1002/ijc.22851. [DOI] [PubMed] [Google Scholar]
- 177.Licitra L, Perrone F, Bossi P, et al. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J Clin Oncol. 2006;24(36):5630–6. doi: 10.1200/JCO.2005.04.6136. [DOI] [PubMed] [Google Scholar]
- 178.Westra WH, Taube JM, Poeta ML, et al. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2008;14(2):366–9. doi: 10.1158/1078-0432.CCR-07-1402. [DOI] [PubMed] [Google Scholar]
- 179.Lin K, Doolan K, Hung CF, Wu TC. Perspectives for preventive and therapeutic HPV vaccines. J Formos Med Assoc. 2010;109(1):4–24. doi: 10.1016/s0929-6646(10)60017-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Baleja JD, Cherry JJ, Liu Z, et al. Identification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins. Antiviral Res. 2006;72(1):49–59. doi: 10.1016/j.antiviral.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jung SO, Ro HS, Kho BH, et al. Surface plasmon resonance imaging-based protein arrays for high-throughput screening of protein–protein interaction inhibitors. Proteomics. 2005;5(17):4427–31. doi: 10.1002/pmic.200500001. [DOI] [PubMed] [Google Scholar]
- 182.Butz K, Denk C, Ullmann A, et al. Induction of apoptosis in human papillomaviruspositive cancer cells by peptide aptamers targeting the viral E6 oncoprotein. Proc Natl Acad Sci USA. 2000;97(12):6693–7. doi: 10.1073/pnas.110538897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sterlinko Grm H, Weber M, Elston R, et al. Inhibition of E6-induced degradation of its cellular substrates by novel blocking peptides. J Mol Biol. 2004;335(4):971–85. doi: 10.1016/j.jmb.2003.10.079. [DOI] [PubMed] [Google Scholar]
- 184.Beerheide W, Bernard HU, Tan YJ, et al. Potential drugs against cervical cancer: zinc-ejecting inhibitors of the human papillomavirus type 16 E6 oncoprotein. J Natl Cancer Inst. 1999;91(14):1211–20. doi: 10.1093/jnci/91.14.1211. [DOI] [PubMed] [Google Scholar]
- 185.Beerheide W, Sim MM, Tan YJ, et al. Inactivation of the human papillomavirus-16 E6 oncoprotein by organic disulfides. Bioorg Med Chem. 2000;8(11):2549–60. doi: 10.1016/s0968-0896(00)00193-0. [DOI] [PubMed] [Google Scholar]
- 186.Machtay M, Moughan J, Trotti A, et al. Factors associated with severe late toxicity after concurrent chemoradiation for locally advanced head and neck cancer: an RTOG analysis. J Clin Oncol. 2008;26(21):3582–9. doi: 10.1200/JCO.2007.14.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bentzen SM, Trotti A. Evaluation of early and late toxicities in chemoradiation trials. J Clin Oncol. 2007;25(26):4096–103. doi: 10.1200/JCO.2007.13.3983. [DOI] [PubMed] [Google Scholar]
- 188.Bentzen SM, Atasoy BM, Daley FM, et al. Epidermal growth factor receptor expression in pretreatment biopsies from head and neck squamous cell carcinoma as a predictive factor for a benefit from accelerated radiation therapy in a randomized controlled trial. J Clin Oncol. 2005;23(24):5560–7. doi: 10.1200/JCO.2005.06.411. [DOI] [PubMed] [Google Scholar]
- 189.Eriksen JG, Steiniche T, Askaa J, et al. The prognostic value of epidermal growth factor receptor is related to tumor differentiation and the overall treatment time of radiotherapy in squamous cell carcinomas of the head and neck. Int J Radiat Oncol Biol Phys. 2004;58(2):561–6. doi: 10.1016/j.ijrobp.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 190.Han SW, Kim TY, Hwang PG, et al. Predictive and prognostic impact of epidermal growth factor receptor mutation in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol. 2005;23(11):2493–501. doi: 10.1200/JCO.2005.01.388. [DOI] [PubMed] [Google Scholar]
- 191.Mitsudomi T, Kosaka T, Endoh H, et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J Clin Oncol. 2005;23(11):2513–20. doi: 10.1200/JCO.2005.00.992. [DOI] [PubMed] [Google Scholar]
- 192.Taron M, Ichinose Y, Rosell R, et al. Activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor are associated with improved survival in gefitinib-treated chemorefractory lung adenocarcinomas. Clin Cancer Res. 2005;11(16):5878–85. doi: 10.1158/1078-0432.CCR-04-2618. [DOI] [PubMed] [Google Scholar]
- 193.Ruiz-Godoy RL, Garcia-Cuellar CM, Herrera Gonzalez NE, et al. Mutational analysis of K-ras and Ras protein expression in larynx squamous cell carcinoma. J Exp Clin Cancer Res. 2006;25(1):73–8. [PubMed] [Google Scholar]
- 194.Yarbrough WG, Shores C, Witsell DL, et al. Ras mutations and expression in head and neck squamous cell carcinomas. Laryngoscope. 1994;104(11 Pt 1):1337–47. doi: 10.1288/00005537-199411000-00005. [DOI] [PubMed] [Google Scholar]
- 195.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 196.Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006;12(17):5064–73. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 197.Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995;55(14):3140–8. [PubMed] [Google Scholar]
- 198.Wheeler DL, Huang S, Kruser TJ, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27(28):3944–56. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cooper JB, Cohen EE. Mechanisms of resistance to EGFR inhibitors in head and neck cancer. Head Neck. 2009;31(8):1086–94. doi: 10.1002/hed.21109. [DOI] [PubMed] [Google Scholar]
- 200.Sisk EA, Soltys SG, Zhu S, et al. Human papillomavirus and p53 mutational status as prognostic factors in head and neck carcinoma. Head Neck. 2002;24(9):841–9. doi: 10.1002/hed.10146. [DOI] [PubMed] [Google Scholar]
- 201.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]