

Abstract
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant syndrome caused by mutations in the MEN1 tumor suppressor gene. Loss of the functional second copy of the MEN1 gene causes individuals to develop multiple endocrine tumors, primarily affecting the parathyroid, pituitary, and pancreas. While it is clear that the protein encoded by MEN1, menin, suppresses endocrine tumors, its biochemical functions and direct downstream targets remain unclear. Recent studies have suggested that menin may act as a scaffold protein to coordinate gene transcription, and that menin is an oncogenic cofactor for homeobox (HOX) gene expression in hematopoietic cancer. The role of HOX genes in adult cell differentiation is still obscure, but growing evidence suggests that they may play important roles in the development of cancer. Therefore, we hypothesized that specific HOX genes were regulated by menin in parathyroid tumor development. Utilizing quantitative TaqMan RT-PCR, we compared expression profiles of the 39 HOX genes in human familial MEN1 (fMEN1) parathyroid tumors and sporadic parathyroid adenomas with normal samples. We identified a large set of 23 HOX genes whose deregulation is specific for fMEN1 parathyroid tumors, and only 5 HOX genes whose misexpression are specific for sporadic parathyroid tumor development. These findings provide the first evidence that loss of the MEN1 tumor suppressor gene is associated with deregulation of specific HOX gene expression in the development of familial human parathyroid tumors. Our results strongly reinforce the idea that abnormal expression of developmental HOX genes can be critical in human cancer progression.
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant syndrome caused by mutations in the MEN1 tumor suppressor gene (Chandrasekharappa et al. 1997, Guru et al. 1998). MEN1 patients are predisposed to develop multiple endocrine tumors, primarily affecting the parathyroid, pituitary, and pancreas. Tumorigenesis is initiated when loss of heterozygosity (LOH) occurs in patients with inherited missense, insertion, or truncation mutations in the MEN1 gene. Somatic inactivation and LOH of the MEN1 alleles have also been found in a variety of sporadic endocrine tumors (Debelenko et al. 1997, Heppner et al. 1997, Wang et al. 1998). Several independent mouse genetic experiments provide further support for MEN1 as a tumor suppressor gene (Crabtree et al. 2001, Bertolino et al. 2003a,b, Libutti et al. 2003, Loffler et al. 2007). Though mice deficient in both Men1 alleles are embryonic lethal, mice heterozygous for Men1 mutation exhibit tumor development in a spectrum of endocrine tissues, similar to those found in human MEN1 patients (Crabtree et al. 2001, Bertolino et al. 2003a,b).
Menin, the protein product of MEN1, has been shown to function as a transcription regulator by interacting with several nuclear proteins, such as JunD (Agarwal et al. 1999), NF-κB (Heppner et al. 2001), and Smad3 (Kaji et al. 2001). Recent biochemical studies implicate menin in regulating chromatin modifications by interacting with a yeast SET-like complex containing mixed-lineage leukemia (MLL) protein (Hughes et al. 2004, Yokoyama et al. 2004). MLL is a proto-oncogene encoding a large protein (431 kDa) that is often fused with other proteins as a result of chromosomal translocations. These MLL chimeric fusion proteins and the wild-type MLL protein both regulate transcription of homeobox (HOX) genes, which are the master regulators of differentiation, proliferation, and morphogenesis (Krumlauf 1994). Specifically, it has been demonstrated that menin is an oncogenic cofactor required for maintenance of Hox gene expression by the MLL fusion proteins in hematopoietic cancer (Yokoyama et al. 2005). Menin has also been shown to directly regulate Hoxa9 expression during hematopoiesis and myeloid transformation (Chen et al. 2006).
The HOX genes are a family of transcription factors that contain the highly conserved DNA-binding HOX domain. The four clusters of HOX genes, HOXA, HOXB, HOXC, and HOXD, are located on different chromosomes at 7p15, 17p21, 12q13, and 2q31, respectively. Based on HOX sequence similarity and chromosome structural organization, 9–11 paralogous HOX genes are assigned in each cluster, giving a total of 39 genes (Krumlauf 1994, Kondo & Duboule 1999, Brend et al. 2003, Spitz et al. 2003). The HOX gene network is involved in body axis patterning, stem cell renewal, cell fate, and cell differentiation in many tissue types including the parathyroid and thyroid glands (Manley & Capecchi 1998). In addition, accumulating evidence suggests that abnormal HOX gene expression plays a central role in oncogenesis (Chen & Sukumar 2003, Grier et al. 2005). Misexpressions of HOX genes have been associated with breast (Raman et al. 2000), cervical (Shim et al. 1998), prostate (Waltregny et al. 2002, Miller et al. 2003), and thyroid cancers (Takahashi et al. 2004). Although HOX genes have well-established roles during embryogenesis, little is known about their upstream regulators and downstream target genes in tumor development.
The menin-dependent mechanisms for HOX gene expression in leukemia and the growing evidence of differential HOX gene expression in cancers have led us to hypothesize that the loss of menin might lead to deregulation of specific HOX gene expression in the endocrine neoplasia observed in MEN1 patients. As an initial step to better understand HOX genes in parathyroid tumor development, we compared the HOX gene expression patterns between familial MEN1 (fMEN1) parathyroid tumors and sporadic parathyroid tumors. Using real-time quantitative RT-PCR, we demonstrated that parathyroid tumor development is associated with deregulation of developmental HOX genes. We identified a large set of HOX genes whose misexpressions are associated with familial parathyroid tumor development. In addition, we revealed a set of common HOX genes that are involved in parathyroid tumor development and five HOX genes whose deregulation is specific for sporadic parathyroid adenoma. These results suggest that there exists a menin-dependent and a menin-independent regulation of HOX genes, which are critical for human parathyroid tumor development.
Materials and methods
Tissue samples
A total of five normal human parathyroid tissues, ten sporadic parathyroid adenomas, and ten fMEN1 parathyroid tumors were obtained from patients under National Institutes of Health Institutional Review Board (NIH IRB) approved protocols. None of the patient was related to each other. The samples were flash-frozen in liquid nitrogen after surgical removal and stored at −80 °C. Frozen parathyroid tissues were embedded in optimal cryo temperature compound (Tissue Tek II, Miles, Elkhart, ID, USA) on dry ice. Tissues were sectioned to 8–10 μm thickness using a cryostat, and scraped off from slides for RNA processing.
Sequence analysis of MEN1
Sequence analysis of the MEN1 gene was performed in our laboratory or Macrogen Corp. (Rockville, MD, USA). Briefly, patient's frozen sectioned parathyroid tissues were processed for genomic DNA isolation. All coding exons of MEN1 were sequenced using the primers listed in Supplementary Table 1, which can be viewed online at http://erc.endocrinology-journals.org/supplemental/, with an annealing temperature of 60 °C. PCR primers designed with Primer Express (Applied Biosystems, Foster City, CA, USA) were appended with M13 forward or reverse tags. Genomic DNA was amplified by PCR using the following conditions: 10 ng genomic DNA, 0.2 mM each primer, 200 mM each dNTP, 2 mM MgCl2, 0.5 units AmpliTaq Gold DNA polymerase (Applied Biosystems), and the manufacturer's buffer. Bidirectional sequencing of amplified DNA using the Big-Dye Terminator method (Applied Biosystems) and M13 forward and reverse primers was analyzed on ABI-Perkin–Elmer platforms (models 3100 and/or 3700). Sequencing results were analyzed by H-C J S and L M Y using Sequencher 4.0.5 software (Gene Codes Corporation, Ann Arbor, MI, USA). MEN1 mutation status and clinical pathological findings of the patient specimens are shown in Table 1. Germ line status of the MEN1 gene was obtained from a database maintained by SJ Marx's laboratory at National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) under NIH IRB approved protocols.
Table 1. Clinical specimens.
| Patient ID | Tissue type | Age at surgery | Gender | Diagnosis | MEN1 mutation in parathyroid | Germ line mutations of MEN1 |
|---|---|---|---|---|---|---|
| 1 | LU parathyroid | 56 | F | Adenoma | None detected | Not done |
| 2 | LL parathyroid | 55 | M | Adenoma | None detected | None detected |
| 3 | RU parathyroid | 61 | M | Adenoma | None detected | Not done |
| 4 | L carotid sheath | 74 | M | Adenoma | None detected | Not done |
| 5 | RU parathyroid | 49 | F | Adenoma | None detected | Not done |
| 6 | Parathyroid | 14 | M | Adenoma | None detected | None detected |
| 7 | RL parathyroid | 57 | F | Adenoma | None detected | Not done |
| 8 | LU parathyroid | 56 | F | Adenoma | None detected | Not done |
| 9 | LU parathyroid | 66 | F | Adenoma | None detected | Not done |
| 10 | Thymic parathyroid | 59 | F | Adenoma | None detected | Not done |
| 11 | Parathyroid | 43 | F | Familial MEN1 | 416delC | 416delC |
| 12 | Thymic parathyroid | 45 | M | Familial MEN1 | None detected | None detected |
| 13 | RL parathyroid | 50 | F | Familial MEN1 | Y327X (TAC>TAG) | Y327X (TAC>TAG) |
| 14 | Parathyroid | 39 | F | Familial MEN1 | 357del4 | 357del4 |
| 15 | RL parathyroid | 44 | F | Familial MEN1 | 357del4 | 357del4 |
| 16 | Parathyroid | 30 | F | Familial MEN1 | None detected | None detected |
| 17 | Parathyroid | 25 | F | Familial MEN1 | None detected | Not done |
| 18 | L carotid sheath | 40 | M | Familial MEN1 | 512delC | 512delC |
| 19 | Parathyroid | 21 | F | Familial MEN1 | 357del4 | 357del4 |
| 20 | LU parathyroid | 22 | M | Familial MEN1 | 512delC | 512delC |
LU, left upper; RU, right upper; LL, left lower; RL, right lower.
Reverse transcription and quantitative real-time PCR
Total RNA samples were purified using the PicoPure system (Arcturus, Mountain View, CA, USA) in a final volume of 11 μl RNase-free water. RNA quality was tested by spectrophotometry and by evaluation on BioAnalyzer minichips (Agilent Technologies, Palo Alto, CA, USA). Total RNA was used as a template to generate single-stranded cDNA with random hexamer primers. The final volume of the reaction was brought to 500–750 μl using RNase-free water and stored at −20 °C until use.
For the quantitative analysis of mRNA expression, ABI Prism 7500 Sequence Detection System and Applied Biosystems 7500 System SDS Software (Applied Biosystems) were utilized. All primers and probes were designed against GenBank-published sequences in association with Primer Express (Applied Biosystems). The majority of HOX primers were synthesized as previously described (Takahashi et al. 2004) and listed in Supplementary Table 2, which can be viewed online at http://erc.endocrinology-journals.org/supplemental/. Standards were generated using gel-extracted PCR products with QIAquick Gel Extraction Kit (Qiagen), except for the glyceraldehyde 3-phosphate dehydrogenase (G3PDH) standard, which is plasmid DNA (a kind gift from Dr Tetsuya Moriuchi, HokkaidoUniversity, Japan). For the PCR, 12.5 μl TaqMan Universal PCR Master Mix (Applied Biosystems), 0.5 μl of 80 μM for each of the forward and reverse primers, 0.5 μl of 10 μM dual labeled fluorescent probe (FAM reporter dye and TAMARA quencher dye), and 6 μl RNase-free water were combined. To each well, 5 μl cDNA were added resulting in a final reaction volume of 25 μl. The PCR was performed for 40 cycles with a two-step program: denaturation at 95 °C for 15 s, and annealing and extension at 60 °C for 1 min. The fluorescence signal was read at the completion of the 60 °C step. All PCRs were run on 2% agarose gel to verify the size of PCR products. For each experiment, non-template reactions were included as a negative control. Standard curve absolute quantification was used to quantify copy number for each gene of interest. Briefly, for each gene of interest, a standard curve was generated using a tenfold serial dilution of four different known concentrations of standards. The number of PCR cycles required for threshold detection of the fluorescence signal (cycle threshold or Ct) was determined for each sample and standard. The Ct values for all unknown samples were compared with the standard curve to determine the exact copy numbers in unknown samples. For each 96-well plate, duplicates of standards for G3PDH and gene of interest, and triplicates of cDNA for each patient sample were amplified for G3PDH and the gene of interest. Standard curves from each experiment were compared to ensure accurate, precise, and reproducible results.
Statistical analysis
Quantitative mRNA expression data were analyzed with a Mann–Whitney non-parametric test using InStats (GraphPad Software, San Diego, CA, USA). A P value of <0.05 was considered statistically significant.
Results
MEN1 mutation status in familial and sporadic parathyroid tumors
To confirm the MEN1 alleles' status from patient samples used for HOX expression profiles, sequencing analyses were performed on DNA isolated from parathyroid tumors. As shown in Table 1, seven of the fMEN1 patients have mutations in the MEN1 coding exon in their parathyroid tumors, validating the heterozygous MEN1 mutation status with DNA isolated from peripheral blood leukocytes. LOH was confirmed at the site of their exonic mutations, which was demonstrated by the presence of only the mutated MEN1 allele in their parathyroid tumors. In patient numbers 13, 15, and 18, LOH was further verified utilizing the D418D polymorphism marker for fMEN1. The three fMEN1 patients (numbers 12, 16, and 17) without detectable mutations of the MEN1 alleles all have a family history of fMEN1 syndrome, where mutation might lie within regulatory regions of the MEN1 gene. All patients with the sporadic parathyroid tumors and patients providing normal parathyroid tissue do not have mutations in the MEN1 coding exons, nor do they have a family history of fMEN1 syndrome.
Expression profiles of HOX genes in fMEN1 and sporadic parathyroid tumors
Using qRT-PCR analysis, expression patterns of the 39 HOX genes in normal parathyroid, fMEN1 and sporadic parathyroid tumors are shown in Fig. 1. When compared with normal parathyroid, expression of HOX genes was largely upregulated in both fMEN1 and sporadic parathyroid tumors. Only two HOX gene expressions, HOXA4 and HOXB9, were downregulated in both fMEN1 and sporadic samples. We were unable to group the 39 HOX gene expression patterns by diagnosis using unsupervised hierarchical clustering (data not shown).
Figure 1.

Quantitative real-time PCR analysis of (A) HOXA cluster, (B) HOXB cluster, (C) HOXC cluster, and (D) HOXD cluster. Relative mRNA expression of 39 HOX genes in 10 familial MEN1 parathyroid tumors, 10 sporadic parathyroid adenomas, and 5 normal parathyroids. Each circle indicates an individual parathyroid sample, and horizontal bars represent the gene expression average within each diagnosis group (x-axis). A P value of < 0.05 was considered statistically significant, and indicated by the asterisk. The y-axis represents relative expression ratio of target HOX gene to internal reference gene (G3PDH).
Using normal samples as the reference, we performed statistical analysis to determine whether HOX gene expression is significantly different for fMEN1 and sporadic parathyroid tumor samples. We revealed that 28 HOX genes in fMEN1 samples were significantly different from normal samples, whereas 10 HOX genes in sporadic samples were significantly different from normal samples (Fig. 2). Using a Venn diagram, deregulation of five HOX genes (A4, A5, B2, B9, and C4) was found to be common for both fMEN1 and sporadic parathyroid tumor development (Fig. 2). Interestingly, expression of the 23 HOX genes that are specific for fMEN1 samples were all upregulated in fMEN1 samples, with the exception of HOXC12. In contrast, expressions of the five HOX genes (A1, D3, D4, D8, and D13) specific for sporadic parathyroid tumors were all downregulated. Among the 23 HOX genes that are overexpressed in fMEN1 parathyroid tumors, we did not observe a significant functional link utilizing Ingenuity Pathway Analysis (data not shown). We further noted that HOXA3 and HOXB3 genes critical for parathyroid development during embryogenesis (Manley & Capecchi 1998) were highly expressed in fMEN1 tumors.
Figure 2.
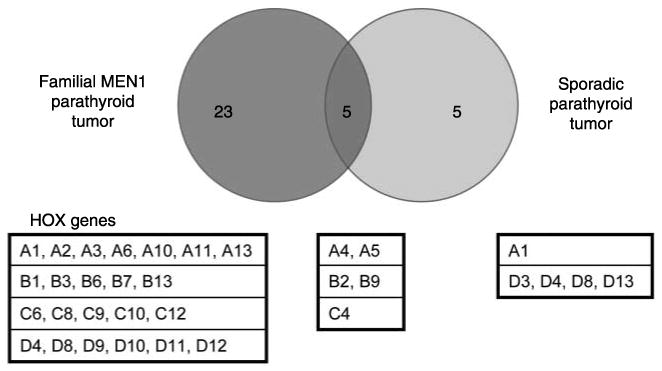
A Venn diagram to illustrate the number and identification of HOX genes whose deregulation are shared between familial and sporadic parathyroid tumors, or the HOX genes whose deregulation are specific for either familial (dark gray circle) or sporadic parathyroid tumors (light gray circle).
Discussion
In the present study, we hypothesized that the loss of menin leads to deregulation of HOX genes in parathyroid tumors from fMEN1 patients. Quantitative RT-PCR analysis of 39 HOX genes was performed in 10 fMEN1 parathyroid tumors, 10 sporadic parathyroid adenomas, and 5 normal parathyroid samples. Although it has been previously demonstrated that familial and sporadic parathyroid tumors do not exhibit an array expression pattern that is unique for each group (Haven et al. 2004), we were able to identify 23 HOX genes whose expressions were significantly deregulated in fMEN1 parathyroid tumors, and only 5 HOX genes whose downregulated expression are specific for sporadic parathyroid tumors. We further identified five HOX genes, the altered gene expressions of which are associated with both familial and sporadic parathyroid tumors. Our findings suggest that there exists a menin-dependent mechanism in regulating HOX gene expression in familial parathyroid tumor, and that a different molecular pathway may be utilized for the development of familial and sporadic parathyroid tumors.
MEN1 and its encoded protein, menin, were originally identified as a tumor suppressor in the human endocrine syndrome of fMEN1 (Chandrasekharappa et al. 1997). Recently, menin has been shown to serve as a stabilizing protein to maintain normal HOX gene expression in non-endocrine contexts. In complex with MLL nuclear proteins, menin positively regulates Hoxc8 in mouse embryonic fibroblast cells (Hughes et al. 2004), and directly activates Hoxa9 in mouse bone marrow cells (Chen et al. 2006). In human HeLa cells or leukemia cells, HOXA7, HOXA9, and HOXA10 are shown to be the direct targets for menin in association with MLL oncoproteins (Yokoyama et al. 2004, 2005). In contrast to published studies, we described here that the loss of menin led to upregulation of HOX gene expression for HOXA9, HOXA10, and HOXC8 in familial parathyroid tumors. Together, these results suggest that menin might be a positive or negative regulator for HOX gene transcription depending on cell type. In addition, a genome-wide analysis of menin binding demonstrates that menin occupies intergenic and intragenic portions of HOX clusters in HeLa cells (Scacheri et al. 2006). Specifically, menin occupancy is reported at HOXA6, A10, A11, A13, B6, B7, C4, C6, C9, C10, and C11, while no significant binding is detected at the HOXD cluster. Consistent with this report, 9 of the above 11 menin-occupied HOX gene clusters were found to be significant for fMEN1 parathyroid tumors in our study, suggesting a direct role for menin in regulating these HOX gene expressions. It remains to be determined whether menin directly regulates all HOX genes identified for fMEN1 parathyroid tumors. Further experiments will also be needed to elucidate if menin modulates HOX gene expression via chromatin methylation and modification in parathyroid tumors, as demonstrated for hematopoietic cancers (Hughes et al. 2004, Yokoyama et al. 2004).
Since somatic inactivation of the MEN1 alleles have been reported in 15–30% of sporadic parathyroid tumors, it has been suggested that molecular pathway utilized in fMEN1 syndrome might be active in the pathogenesis of some sporadic parathyroid tumors (Debelenko et al. 1997, Heppner et al. 1997, Wang et al. 1998). With respect to HOX genes, we did not observe a menin-dependent pathway that is shared between the fMEN1 and sporadic parathyroid tumors. While normal MEN1 gene was not a criterion in selecting sporadic parathyroid adenomas, we noted that none of the sporadic parathyroid tumors analyzed had mutations in the MEN1 gene in this study. Therefore, there remains a possibility that menin-dependent regulation of HOX genes may be important in some sporadic parathyroid tumors containing somatic inactivation of MEN1.
An increasing number of published works implicate deregulation of developmental HOX genes in tumorigenesis (Chen & Sukumar 2003, Grier et al. 2005). Consistent with this idea, we showed that expressions of HOX group 3 paralogs, genes required for formation of parathyroid glands during embryogenesis (Manley & Capecchi 1998), were significantly deregulated in both familial and sporadic parathyroid tumors. In particular, elevated expression of HOXA3 and HOXB3 might imply that reactivation of these HOX genes is related to the initiation of familial parathyroid tumor development. In addition, we observed that 12 out of 16 posterior abdominal-B subgroups of HOX genes, which are paralogs 9–11 expressed in the caudal region of the embryo (Krumlauf 1994), were significantly misexpressed in either familial (HOXA10, A11, A13, B13, C9, C10, C12, D9, D10, D11, and D12) or sporadic (HOXD13) parathyroid tumors. Disorganization of the spatiotemporal colinearity order of the HOX gene chromosomal position may also contribute to parathyroid tumorigenesis. Detailed molecular studies will be required to further delineate mechanisms involving deregulation of HOX genes in human parathyroid tumors.
In conclusion, this is the first study employing a comprehensive analysis of the 39 HOX genes in human parathyroid tumors. We identified common HOX genes that are involved in parathyroid tumor development, and HOX genes whose deregulation is specific for either fMEN1 or sporadic parathyroid tumors. These results demonstrate the existence of a menin-dependent and a menin-independent regulation of HOX genes critical for the development of human parathyroid tumors. Further analysis of the expression network among HOX genes and identification of menin-dependent pathways will lead to a better understanding of how HOX genes contribute to parathyroid tumor development.
Supplementary Material
Acknowledgments
We gratefully thank Dominique Lorang for technical assistance and the NIH Fellows Editorial Board for their review of the manuscript. We thank the patients and referring physicians. This research was supported by funds from the Intramural Research Program of the NIH, National Cancer Institute, and National Institute of Diabetes and Digestive and Kidney Diseases. The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
References
- Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Molecular Endocrinology. 2003a;17:1880–1892. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Research. 2003b;63:4836–4841. [PubMed] [Google Scholar]
- Brend T, Gilthorpe J, Summerbell D, Rigby PW. Multiple levels of transcriptional and post-transcriptional regulation are required to define the domain of Hoxb4 expression. Development. 2003;130:2717–2728. doi: 10.1242/dev.00471. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Chen H, Sukumar S. HOX genes: emerging stars in cancer. Cancer Biology and Therapy. 2003;2:524–525. doi: 10.4161/cbt.2.5.525. [DOI] [PubMed] [Google Scholar]
- Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, Brown EJ, Hess JL, Pear WS, Hua X. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. PNAS. 2006;103:1018–1023. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. PNAS. 2001;98:1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelenko LV, Brambilla E, Agarwal SK, Swalwell JI, Kester MB, Lubensky IA, Zhuang Z, Guru SC, Manickam P, Olufemi SE, et al. Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Human Molecular Genetics. 1997;6:2285–2290. doi: 10.1093/hmg/6.13.2285. [DOI] [PubMed] [Google Scholar]
- Grier DG, Thompson A, Kwasniewska A, McGonigle GJ, Halliday HL, Lappin TR. The pathophysiology of HOX genes and their role in cancer. Journal of Pathology. 2005;205:154–171. doi: 10.1002/path.1710. [DOI] [PubMed] [Google Scholar]
- Guru SC, Manickam P, Crabtree JS, Olufemi SE, Agarwal SK, Debelenko LV. Identification and characterization of the multiple endocrine neoplasia type 1 (MEN1) gene. Journal of Internal Medicine. 1998;243:433–439. doi: 10.1046/j.1365-2796.1998.00346.x. [DOI] [PubMed] [Google Scholar]
- Haven CJ, Howell VM, Eilers PH, Dunne R, Takahashi M, van Puijenbroek M, Furge K, Kievit J, Tan MH, Fleuren GJ, et al. Gene expression of parathyroid tumors: molecular subclassification and identification of the potential malignant phenotype. Cancer Research. 2004;64:7405–7411. doi: 10.1158/0008-5472.CAN-04-2063. [DOI] [PubMed] [Google Scholar]
- Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC, Manickam P, Olufemi SE, Skarulis MC, Doppman JL, et al. Somatic mutation of the MEN1 gene in parathyroid tumours. Nature Genetics. 1997;16:375–378. doi: 10.1038/ng0897-375. [DOI] [PubMed] [Google Scholar]
- Heppner C, Bilimoria KY, Agarwal SK, Kester M, Whitty LJ, Guru SC, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, et al. The tumor suppressor protein menin interacts with NF-κB proteins and inhibits NF-κB-mediated transactivation. Oncogene. 2001;20:4917–4925. doi: 10.1038/sj.onc.1204529. [DOI] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Molecular Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. PNAS. 2001;98:3837–3842. doi: 10.1073/pnas.061358098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Duboule D. Breaking colinearity in the mouse HoxD complex. Cell. 1999;97:407–417. doi: 10.1016/s0092-8674(00)80749-7. [DOI] [PubMed] [Google Scholar]
- Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- Libutti SK, Crabtree JS, Lorang D, Burns AL, Mazzanti C, Hewitt SM, O'Connor S, Ward JM, Emmert-Buck MR, Remaley A, et al. Parathyroid gland-specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Research. 2003;63:8022–8028. [PubMed] [Google Scholar]
- Loffler KA, Biondi CA, Gartside M, Waring P, Stark M, Serewko-Auret MM, Muller HK, Hayward NK, Kay GF. Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. International Journal of Cancer. 2007;120:259–267. doi: 10.1002/ijc.22288. [DOI] [PubMed] [Google Scholar]
- Manley NR, Capecchi MR. Hox group 3 paralogs regulate the development and migration of the thymus, thyroid, and parathyroid glands. Developmental Biology. 1998;195:1–15. doi: 10.1006/dbio.1997.8827. [DOI] [PubMed] [Google Scholar]
- Miller GJ, Miller HL, van Bokhoven A, Lambert JR, Werahera PN, Schirripa O, Lucia MS, Nordeen SK. Aberrant HOXC expression accompanies the malignant phenotype in human prostate. Cancer Research. 2003;63:5879–5888. [PubMed] [Google Scholar]
- Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature. 2000;405:974–978. doi: 10.1038/35016125. [DOI] [PubMed] [Google Scholar]
- Scacheri PC, Davis S, Odom DT, Crawford GE, Perkins S, Halawi MJ, Agarwal SK, Marx SJ, Spiegel AM, Meltzer PS, et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genetics. 2006;2:e51. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim C, Zhang W, Rhee CH, Lee JH. Profiling of differentially expressed genes in human primary cervical cancer by complementary DNA expression array. Clinical Cancer Research. 1998;4:3045–3050. [PubMed] [Google Scholar]
- Spitz F, Gonzalez F, Duboule D. A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell. 2003;113:405–417. doi: 10.1016/s0092-8674(03)00310-6. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Hamada J, Murakawa K, Takada M, Tada M, Nogami I, Hayashi N, Nakamori S, Monden M, Miyamoto M, et al. Expression profiles of 39 HOX genes in normal human adult organs and anaplastic thyroid cancer cell lines by quantitative real-time RT-PCR system. Experimental Cell Research. 2004;293:144–153. doi: 10.1016/j.yexcr.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Waltregny D, Alami Y, Clausse N, de Leval J, Castronovo V. Overexpression of the homeobox gene HOXC8 in human prostate cancer correlates with loss of tumor differentiation. Prostate. 2002;50:162–169. doi: 10.1002/pros.10045. [DOI] [PubMed] [Google Scholar]
- Wang EH, Ebrahimi SA, Wu AY, Kashefi C, Passaro E, Jr, Sawicki MP. Mutation of the MENIN gene in sporadic pancreatic endocrine tumors. Cancer Research. 1998;58:4417–4420. [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyl-transferase complex with menin to regulate Hox gene expression. Molecular and Cellular Biology. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.