

Abstract
Background/Aim:
Polymorphisms in the promoter of microsomal triglyceride transfer protein (MTP) lead to decreased MTP transcription, less export of triglyceride from hepatocytes, and greater intracellular triglyceride accumulation. Therefore, functional polymorphisms in MTP may be involved in determining susceptibility to nonalcoholic steatohepatitis (NASH). The aim of this study is to examine the effect of some genetic influences among a group of obese Egyptian children.
Patients and Methods:
A cross-sectional study was conducted on 76 overweight and obese children presenting to the Pediatric Endocrinology Unit, Cairo University Children's Hospital, Egypt, as well as on 20 healthy controls. Anthropometric measurements were taken for all the patients and they underwent clinical examination, ultrasonographic examination of the liver, and liver biopsy when appropriate. Liver functions, blood glucose, serum insulin, C-peptide, and lipid profile were assessed and HOMA-IR calculated. Blood samples from biopsy-proven NASH patients and controls were analyzed by polymerase chain reaction (PCR) and restriction fragment length polymorphism for the –493 G/T polymorphism in the promoter of MTP and the 1183 T/C polymorphism in the mitochondrial targeting sequence of manganese superoxide dismutase (MnSOD).
Results:
Eight had biopsy-proven simple steatosis and 7 had NASH. NASH patients had a much higher incidence of the MTP G/G genotype (P = 0.002, CI: 2.9–392) compared with the controls. NASH patients also had a 100% prevalence of the MnSOD T/T genotype.
Conclusion:
Certain genotypes in MTP and MnSOD are significantly more prevalent among obese children with NASH and may be responsible for such a phenotype.
Keywords: Egyptian children, microsomal triglyceride transfer protein, manganese superoxide dismutase, nonalcoholic steatohepatitis, obesity
The “two-hit” hypothesis has been elaborated to explain the pathogenesis of nonalcoholic fatty liver disease (NAFLD).[1] Genetic variation in lipid metabolism is the first hit. One enzyme is the microsomal triglyceride transfer protein (MTP). It regulates synthesis, storage, and export of hepatic triglyceride content. Low levels of MTP result in failure to excrete triacylglycerol from the liver and hepatic steatosis. A genetic variation of the MTP gene affects the susceptibility for the development of NAFLD.[2] A common genetic variation of the MTP gene is –493 G/T polymorphism.[3]
An excessive free fatty acid oxidation leads to oxidative stress causing hepatocyte apoptosis and liver injury is the second hit. Manganese superoxide dismutase (MnSOD) is the main reactive oxygen species (ROS) scavenger in mitochondria. A 1183 T/C polymorphism in the mitochondrial targeting sequence of MnSOD gene leads to a less efficient transport of MnSOD to the mitochondria and increases the susceptibility for the development of NAFLD.[3,4]
The aim of the current study was to examine the frequency of the MTP gene –493 G/T polymorphism and the 1183 T/C polymorphism in the mitochondrial targeting sequence of MnSOD among biopsy-proven NASH patients.
PATIENTS AND METHODS
This cross-sectional study included 76 children that were referred to the Pediatric Endocrinology Unit at Cairo University Children's Hospital for medical assessment of obesity. The study was approved by the university ethical committee and the patients were enrolled after obtaining an informed consent from their parents.
Patients
Inclusion criteria
Simple obesity, both sexes, age 2–15 years
Exclusion criteria
Patients with known disorders to cause fatty liver (eg, HCV, diabetes, glycogen storage disease, and Wilson's disease), long-term use of drugs known to cause steatosis (eg, glucocorticoids, aspirin) and any case with syndromatic obesity
Controls: Twenty healthy age- and sex-matched control subjects were included for insulin and C-peptide estimation as well as molecular studies.
Methods
All patients were subjected to the following
Full medical history with special emphasis on nutritional history, drug intake, symptoms of liver disease, diabetes, and family history of liver disease.
-
Thorough clinical examination including
- Abdominal examination with focus on liver examination
- Anthropometric assessment including the following: Height (Ht), weight (Wt), body mass index (BMI) (Wt in kg and Ht in m2) were plotted on Standard Egyptian growth curves.[5] In children and adolescents, BMI > 85th percentile is defined as overweight and BMI > 95th percentile is defined as obesity.[6] Subscapular skin fold thickness was measured by skin fold caliper (Holtain LTD, UK) and was plotted on Egyptian growth curves. Waist circumference (WC) and hip circumference (HC) were measured and waist/hip (W/H) ratio was calculated.[7] The W/H ratio was considered abnormal if >0.86.[8]
Ultrasonographic abdominal examination was performed for all the enrolled patients by a single sonographer following not less than 8 h fasting using FFsonic (UF-4100, Fukuda Denshi Co, Tokyo, Japan) UF-4100. Liver echopattern was graded according to Mottin et al.[9]
Percutaneous liver biopsy
Forty-one patients were indicated for liver biopsy: clinical hepatomegaly and/or elevated liver enzymes and/or increased liver echogenicity by ultrasonography. However, it was obtained in 33 patients only due to parental refusal. All patients were examined by a single pathologist.
The main histologic features commonly described in NALFD/NASH, including steatosis, inflammation (portal and lobular), hepatocyte ballooning, and fibrosis, were scored according to the scoring system for NAFLD.[10]
Laboratory investigations
Sampling
Laboratory investigations were performed in the morning, following a not less than 12-h fasting period. Blood samples were collected as follows: fasting clotted samples for lipid profile and liver functions and EDTA blood for DNA analysis.
Routine laboratory investigations
Serum triglycerides, total cholesterol (total-C), and high-density lipoprotein cholesterol, fasting blood sugar, total bilirubin, direct bilirubin, alanine transaminase, aspartate aminotransferase, alkaline phosphatase (AP), and gamma-glutamyltransferase, were estimated using commercial kits on the synchron CX5 analyzer from Beckman Instruments Inc., California, USA. Low-density cholesterol was calculated using Friedewald's formula.[11]
Special laboratory investigations
Fasting serum insulin and C-peptide measurement using a commercial solid phase chemiluminescent enzyme immunometric assay (IMMULITE 2000, Diagnostic Products Corp. IL, USA).[12]
Insulin resistance was calculated for both patients and controls using the following equation:
The homeostasis model assessment method (HOMA-IR) = [fasting insulin (µU/mL) × fasting glucose (mg/dL)/405].[13]
PCR-restriction fragment length polymorphism
Determination of MTP and MnSOD genotypes was performed for 8 and 7 patients with histologically proven steatosis and NASH, respectively, as well as for the controls.
Genomic DNA was isolated using the standard salting out technique.[14] The following primers were used, 5’-GGATTTAAATTTAAACTGTTAATTCATATCAC (forward)5’-AGTTTCACACATCAAGGACAATCATCTA (reverse) for the MTP polymorphism and 5’-ACCAGCAGGCAGCTGGCGCCGG-3’ (forward) and 5’ GCGTTGATGTGAGGTTCCAG-3’ (reverse) for the MnSOD polymorphism.This was followed by HphI and NgoMIV restriction enzymes digestion for MTP and MnSOD polymorphisms, respectively. Restriction fragmentswere then separated on 3% agarose gel and sized by comparisonto a DNA step ladder Puc 18 (Promega. WI, USA). This restriction digestion, in case of –493 G/T MTP polymorphismgives rise to one full-length fragment of 109 base pairs (bp) for homozygotesfor the T variant, two fragments of 89 and 20 bp for homozygotesfor the G variant, and three fragments of 109, 89, and 20 bpfor G/T heterozygotes. As for the 1183 T/C MnSOD polymorphism 107 bp with C allele (Ala) was digested into two fragments (89 and 18 bp), whereas the PCR product with T allele (Val) cannot be cut by NgoMIV.[3,15]
Statistical methods
The statistical package for social sciences (SPSS, version 10.0; Chicago, IL, USA) was used for data management and analysis and Microsoft power point for charts. Parametric quantitative data were presented as mean ± SD and compared by Student's t test. Nonparametric quantitative data were expressed as median (range) and compared by Mann–Whitney U test. Qualitative data were expressed as frequency and percentage. Risk estimate was done by odds ratio. P value was considered significant at 0.05.
RESULTS
Our study included 76 patients: 37 were overweight (17 males and 20 females) and 39 were obese (21 males and 18 females). Their age ranged between 2 and 15 years with a mean of 7.7 ± 3.5 years.
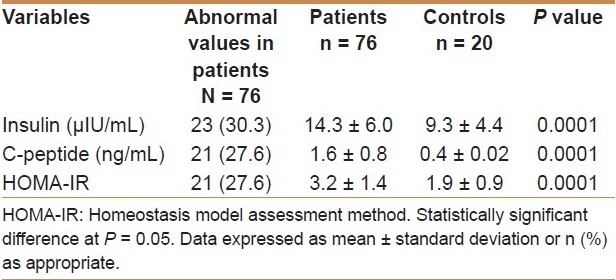
Table 1 shows the comparison of insulin, C-peptide, and HOMA-IR between patients and controls. The patients had higher values of fasting serum insulin, insulin C peptide, and insulin resistance using HOMA ratio.
Table 1.
Comparison between patients and controls with regard to insulin, C-peptide, and HOMA-IR
Five patients (6.5%) had clinical hepatomegaly and 22 patients (29%) had elevated aminotransferases levels. Otherwise, gamma glutamyltranspeptidase (GGT), alkaline phosphatase, serum total protein, albumin, and total bilirubin levels of all the patients were within normal limits. Forty-one patients (54%) had echogenic liver parenchyma by ultrasound.
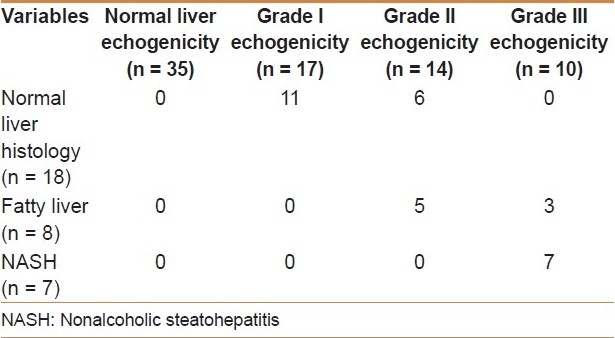
A percutaneous liver biopsy was obtained in 33 patients: 18 cases (54.6%) had normal liver histology, 8 cases (24.2%) were diagnosed as fatty liver (simple steatosis), and 7 cases (21.2%) as NASH. A significant association between the grade of hepatic echogenicity by ultrasonography and the degree of hepatic steatosis as determined by biopsy (P <0.0001) was found [Table 2]. Similarly, a significant association between insulin resistance and the degree of hepatic steatosis was found (P < 0.0001). Six out of 7 children (86%) with NASH had insulin resistance.
Table 2.
Relation between grade of liver echogenicity and result of liver biopsy
The frequency of the polymorphism resulting in a G/T substitution in the sequence of MTP as well as that resulting in a T/C substitution in the MnSOD mitochondrial targeting sequence was studied among the NAFLD patients as well as in the healthy controls [Tables 3 and 4]. As regards MTP (–493 G/T), no T/T genotype was detected in either the NASH or the simple steatosis groups. Combining the MTP G/G and the MnSOD T/T genotypes, a significant risk for developing NASH was obtained when compared with the control group (odds ratio [OR]: 54; 95% confidence interval [CI]: 4.1–707; P = 0.001).
Table 3.
Odds ratio of MTP and MnSOD genotype in simple steatosis and healthy controls
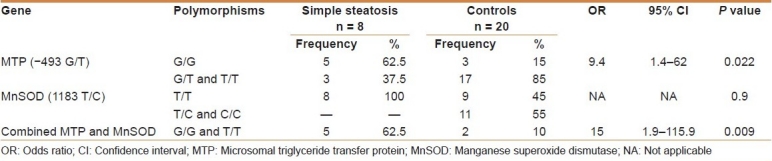
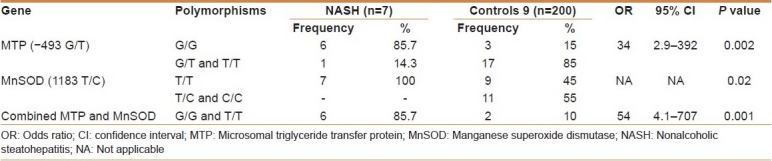
Table 4.
Odds ratio of MTP and MnSOD genotype in NASH and healthy controls
Sixty percent of NAFLD patients having a G/G genotype for MTP (–493 G/T) had a high triglyceride level (>150 mg/dL). A significant association between the G/G genotype and high triglycerides level was found (P = 0.01).
DISCUSSION
Childhood obesity is the new pandemic of the new millennium, with significant adverse effects on childhood health. The prevalence of NAFLD is increasing in parallel with the growing proportions of childhood obesity. The prevalence of NAFLD among children is unknown, but some data indicate that 2.6%–9.6% of children have NAFLD, increasing up to 38%–53% among obese children.[16] NAFLD prevalence in this study was 20% (10.5% simple steatosis and 9.2% NASH). This prevalence could have increased if biopsy was performed in all biopsy-indicated patients. Similarly, de Silva et al.[17] reported NASH prevalence of 18% among obese patients. Higher frequencies were reported by other authors.[18–20]
Insulin resistance was assessed by measuring fasting serum insulin level, glucose, and calculating HOMA-IR. Eighty-six percent of our NASH patients were found to have insulin resistance, which plays a key role in the development of NASH.[21,22]
The pathogenesis and progress of NASH remain unclear and the most advocated theory is the “two-hit hypothesis.”[1] Briefly, the first hit is the deposition of fatty acid in hepatocytes triggered by different factors, whereas the second hit is the concomitant liver damage induced by oxidative stress and lipid peroxidation. The fact that NASH is observed only in a fraction of patients with NAFLD suggests genetic predisposition to this disease. Remarkably, the NASH patients included in this study had both an increased frequency of the G allele (92.8%) and of the G/G genotype (85.7%) of MTP, indicating a genetic background favorable for the development of steatosis. This is in accordance with the study by Namikawa et al.[4] who showed an increased frequency of the G allele (91%) and of the G/G genotype (83%). No T/T genotype was detected among our NASH patients. This was also observed in the work of Namikawa et al.[4] who did not find any T/T genotypes among their NASH patients, but in partial agreement to the work of Gambino et al.[23] who found a 66% G/G frequency among NASH patients and a T/T genotype of 7%.
The frequency of the G/G genotype among the simple steatosis group was significantly higher than that in healthy controls, which makes them more prone to develop NAFLD (OR: 9.4; 95% CI: 1.4–62; P = 0.022). The G/G genotype in the MTP promoter would render a patient more susceptible to steatosis. Similar results have been described by Bernard et al.[2] who demonstrated that the MTP G allele confers genetic susceptibility to liver steatosis in patients with type 2 diabetes.
The impact of the G/G genotype as a susceptibility factor to steatosis is proved by the fact that 100% of NASH patients carrying the G/G genotype had elevated triglycerides levels (>150 mg/dL) (P = 0.01). These findings are consistent with the current understanding of NASH pathogenesis. The G allele has been previously shown to produce less MTP gene transcription than the T allele.[2,4] Less MTP activity, in turn, would lead to less triglyceride excretion, and greater accumulation of lipid inside the hepatocytes.
Having dealt with the first hit, analyzing another gene that could possibly confer predisposition to develop a second insult, such as oxidative stress, has been performed. One enzyme that is important in detoxifying mitochondrial ROS is MnSOD. A limited number of polymorphisms have been described for MnSOD, including a T/C polymorphism in the mitochondrial targeting sequence leading to a valine-to-alanine amino acid change. In turn, this amino acid substitution may alter the helical structure of the mitochondrial targeting sequence, enhancing transport and increased localization of MnSOD into the mitochondrial matrix.[4,24] This localization pattern may increase the ability of MnSOD to process superoxide anion produced in the mitochondria, and on the contrary, the presence of valine would be associated with increased generation of ROS. In agreement with this hypothesis, a higher frequency of the T/T genotype was observed in our NASH patients in comparison to the control group (100% vs 45%). A higher frequency of the T/T genotype among NASH patients was also observed by Namikawa et al.[4] However, this high frequency observed in our study was not statistically significant. Thus, this group of patients has an increased frequency of genotypes predisposing not only to fatty liver, but also to oxidative stress, one of the factors believed to be involved in severe injury and the development of NASH.
Neither the T/T genotype nor the T alleles of MnSOD (OR: 0.5; 95% CI: 0.3–0.8; P = 0.9 and OR: 0.64; 95% CI: 0.5–0.8; P = 0.2, respectively) were increased among our simple steatosis patients. Although our results were in accordance with the work of Namikawa and coworkers,[4] concerning the T allele, they were contradictory as regards the T/T genotype. The T allele was, therefore, not found to impose an increased risk for NASH in the simple steatosis group in the present study.
The exact significance of the polymorphism in the MnSOD gene is still unclear. It is possible that the presence of more MnSOD in the mitochondrial matrix affords a better protection against pro-oxidant stimuli because of a more efficient conversion of superoxide anion.[3] However, others speculate that under the same conditions, more hydrogen peroxide would be generated, resulting in more severe cell damage. Thus, in many studies investigating genetic polymorphisms it would be important to provide evidence for a genotype–phenotype correlation.[25]
The combined presence of G/G and T/T genotypes of MTP and MnSOD, respectively, in simple steatosis determines their greater susceptibility to developing NAFLD (OR: 15; 95% CI: 1.9–115.9; P = 0.009).
In conclusion, the G/G genotype of MTP may impact NASH by modulating postprandial lipemia and lipoprotein metabolism. Homozygous G/G carriers have a more atherogenic lipid profile than other genotypes. This may mandate establishment of treatment modalities for carriers of this haplotype to prevent progression to NASH.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Day CP, James OF. Steatohepatitis: A tale of two ‘hits’? Gastroenterology. 1998;114:842–4. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 2.Bernard S, Touzet S, Personne I, Lapras V, Bondon PJ, Berthezene F. Association between microsomal triglyceride transfer protein gene polymorphism and the biological features of liver steatosis in patients with type II diabetes. Diabetologia. 2000;43:995–9. doi: 10.1007/s001250051481. [DOI] [PubMed] [Google Scholar]
- 3.Karpe F, Lundahl B, Ehrenborg E, Eriksson P, Hamsten A. A common functional polymorphism in the promoter region of the microsomal triglyceride transfer protein gene influences plasma LDL levels. Atheroscler Thromb Vasc Biol. 1998;18:756–61. doi: 10.1161/01.atv.18.5.756. [DOI] [PubMed] [Google Scholar]
- 4.Namikawa C, Shu-Ping Z, Vyselaar JR, Nozaki Y, Nemoto Y, Ono M, et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J Hepatol. 2004;40:781–6. doi: 10.1016/j.jhep.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 5.Standard Egyptian Growth. Diabetes Endocrine Metabolism Pediatric Unit Cairo University Children's Hospital. [Last accessed on 2009 Aug 13, Last revised 2008 Nov 29]. Available from: http://www.dempuegypt.blogspot.com/.
- 6.Cole TJ, Bellizzi MC, Flegal KM, Dietz WH. Establishing a standard definition for child overweight and obesity worldwide: International survey. BMJ. 2000;320:1240–3. doi: 10.1136/bmj.320.7244.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernández JR, Redden DT, Pietrobelli A, Allison DB. Waist circumference percentiles in nationally representative samples of African-American, European-American, and Mexican-American children and adolescents. J Pediatr. 2004;145:439–44. doi: 10.1016/j.jpeds.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 8.Giugliano R, Melo A. Diagnosis of overweight and obesity in school children: Utilization of BMI international standards. J Pediatr. 2004;80:129–34. [PubMed] [Google Scholar]
- 9.Mottin CC, Moretto M, Padoin AV. The role of ultrasound in the diagnosis of hepatic steatosis in morbidly obese patients. Obes Surg. 2004;14:635–7. doi: 10.1381/096089204323093408. [DOI] [PubMed] [Google Scholar]
- 10.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 11.Freidewald WT, Levy RJ, Fredrickson DS. Estimation of concentration of LDL-C in plasma without use of the preparative ultracentrifuge. Clin Chem. 1972;8:499. [PubMed] [Google Scholar]
- 12.Wiedmeyer HM, Polonsky KS, Myers GL, Little RR, Greenbaum CJ, Goldstein DE, et al. International Comparison of C-Peptide Measurements. Clin Chem. 2007;53:784–7. doi: 10.1373/clinchem.2006.081570. [DOI] [PubMed] [Google Scholar]
- 13.Yokoyama H, Emoto M, Fujiwara S, Motoyama K, Morioka T, Komatsu M, et al. Quantitative insulin sensitivity check index and the reciprocal index of homeostasis model assessment in normal range weight and moderately obese type 2 diabetic patients. Diabetes Care. 2003;26:2426–32. doi: 10.2337/diacare.26.8.2426. [DOI] [PubMed] [Google Scholar]
- 14.Sambrook J, Fritsch EF, Maniatis T. Cold Spring Harbor. Vol. 1. NY: Cold Spring Harbor Laboratory; 1989. Molecular cloning: A laboratory manual; pp. 1–62. [Google Scholar]
- 15.Cai Q, Shu XO, Wen W, Cheng JR, Dai Q, Gao YT, et al. Genetic polymorphism in the manganese superoxide dismutase gene, antioxidant intake, and breast cancer risk: Results from the Shanghai Breast Cancer Study. Breast Cancer Res. 2004;6:R647–55. doi: 10.1186/bcr929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angulo P. GI Epidemiology: Nonalcoholic fatty liver disease. Aliment Pharmacol Ther. 2007;25:883–9. doi: 10.1111/j.1365-2036.2007.03246.x. [DOI] [PubMed] [Google Scholar]
- 17.De Silva KS, Wickramasinghe VP, Gooneratne IN. Metabolic consequences of childhood obesity--a preliminary report. Ceylon Med J. 2006;51:105–9. doi: 10.4038/cmj.v51i3.1253. [DOI] [PubMed] [Google Scholar]
- 18.Rashid M, Roberts EA. Non alcoholic steatohepatitis in children. J Pediatr Gastro Nutr. 2000;30:48–53. doi: 10.1097/00005176-200001000-00017. [DOI] [PubMed] [Google Scholar]
- 19.Xanthokos S, Miles L, Bucuvalas J, Daniels S, Garcia V, Inge T. Histologic spectrum of nonalcoholic fatty liver disease in morbidly obese adolescents. Clin Gastrol Hepatol. 2006;4:226–32. doi: 10.1016/s1542-3565(05)00978-x. [DOI] [PubMed] [Google Scholar]
- 20.Sagi R, Reif S, Neuman G, Webb M, Phillip M, Shalitin S. Nonalcoholic fatty liver disease in overweight children and adolescents. Acta Paediatr. 2007;96:1209–1. doi: 10.1111/j.1651-2227.2007.00399.x. [DOI] [PubMed] [Google Scholar]
- 21.Dixon JB, Bhathal PS, O’Brien PE. Nonalcoholic fatty liver disease: Predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121:91–100. doi: 10.1053/gast.2001.25540. [DOI] [PubMed] [Google Scholar]
- 22.Finucane FM, Teong L, Pittock S, Fallon M, Hatunic M, Costigan C, et al. Adverse metabolic profiles in a cohort of obese Irish children. Ann Clin Biochem. 2008;45:206–9. doi: 10.1258/acb.2007.007115. [DOI] [PubMed] [Google Scholar]
- 23.Gambino R, Cassader M, Pagano G, Durazzo M, Musso G. Polymorphism in microsomal triglyceride transfer protein: A link between liver disease and atherogenic postprandial lipid profile in NASH? Hepatology. 2007;45:1097–107. doi: 10.1002/hep.21631. [DOI] [PubMed] [Google Scholar]
- 24.Stewart SF, Leathart JB, Chen Y, Daly AK, Rolla R, Vay D. Valine-alanine manganese superoxide dismutase polymorphism is not associated with alcohol-induced oxidative stress or liver fibrosis. Hepatology. 2002;36:1355–60. doi: 10.1053/jhep.2002.36940. [DOI] [PubMed] [Google Scholar]
- 25.Marra F. NASH: Are genes blowing the hits? J Hepatol. 2004;40:853–6. doi: 10.1016/j.jhep.2004.03.005. [DOI] [PubMed] [Google Scholar]