

Abstract
Alzheimer's disease is rapidly growing worldwide and yet there is no cure for it. Currently available drugs only provide symptomatic relief and do not intervene in disease process sufficiently enough to prevent or cure it. Characteristic features of this disease are decline in neuronal mass and cognitive functions. The most dominant hypothesis proposed for pathogenesis of this disease is called “amyloid hypothesis". It states that excessive production of amyloid peptides called abeta peptides (Aβ) is the underlying cause of neuronal death and dysfunction. However, recent drugs designed based on amyloid hypothesis have failed in clinical trails, demanding fresh assessment. Early and persistent molecular events in this disease progression are energy deficiency and high oxidative stress in the neurons. Our review will put together a disease model based on known human and animal data with regards to breakdown in neuronal energy generation. The model will integrate energy deficits as the cause of neuronal dysfunction and abeta peptide production culminating in catastrophic loss of cognitive functions. Finally, based on this model, we will also suggest enzyme targets in neuronal bioenergetics pathway for design and development of new disease modifying therapies.
Keywords: Alzheimer's disease, amyloid hypothesis, neuronal energy, new therapeutics
Introduction
Alzheimer's disease is a degenerative central nervous system disorder associated with extensive loss of specific neuronal cells, and characterized clinically by progressive loss of memory, cognition, reasoning, judgment and emotional stability that leads to profound mental deterioration. The disease currently affects as many as four million individuals in the United States alone. To date, there is no treatment that stops or reverses the disease and it presently causes up to 100,000 deaths annually.
There are two forms of the disease, a genetics based early onset familial Alzheimer's disease and a more prevalent age-dependent form called sporadic Alzheimer's' disease. Brains of individuals with Alzheimer's exhibit neuronal degeneration and characteristic lesions referred to as amyloid plaques and neurofibrillary tangles [1,2]. Currently, the only definitive diagnosis of Alzheimer's is presence of these plaques in post-mortem brains. According to the dominant scientific hypothesis for Alzheimer's, called amyloid hypothesis, progressive cerebral deposition of amyloidogenic peptides Aβ peptides, plays a detrimental role in the pathogenesis of Alzheimer's and can cause cognitive symptoms and onset of dementia [3]. The hypothesis comprises of the amyloid cascade of events: increased production of Aβ peptides, their aggregation leading to plaque and neurofibrillary tangle formation, tau phosphorylation, and finally cognitive decline (Figure 1).
Figure 1.

Amyloid hypothesis of Alzheimer's Disease. The classic amyloid hypothesis cascade of events is shown here. The main features are overproduction of Aβ peptides by secretase cleavage, peptide aggregation followed by neurotoxicity, tau phosphorylation, plaque and neurofibrillary tangle (NFT) formation with end result of cognitive loss and Alzheimer's disease.
The Aβ peptides are produced as a result of excessive processing of the amyloid precursor protein (APP), the parent trans-membrane protein found in neurons and other cells [4]. Amyloid plaques are composed primarily of 40 and 42 amino acid peptides (called Aβ40 and Aβ42, respectively) derived from amyloid precursor protein (APP) by sequential proteolysis catalyzed by the aspartyl protease, β-secretase for beta-site APP cleaving enzyme-1 (BACE-1), followed by presenilin-dependent gamma-secretase cleavage [4] (Figure 1). Aβ42 is more hydropho-bic and less soluble than Aβ40 and is the predominant species in amyloid plaques. Aβ42 is more prone to aggregation and deposition and therefore the cause of neurotoxicity as well as synaptic loss (1, Ref).
Established risk factors for Alzheimer's may trigger neuronal dysfunction [3]. In particular ageing, head injury/trauma/stroke, genetics such as Apo E4/4 allele carriers, cardiovascular disease and type 2 diabetes all predispose an individual towards onset of Alzheimer's disease [5]. Two molecular events, which are closely tied to onset, progression and pathogenesis of the disease, are high oxidative stress and reduced energy production by neurons [6,7]. As major focus of this review we will cite existing data and develop a new energy failure based paradigm for genesis of Alzheimer's disease, followed by nomination of targets for new therapies.
Energy defects as driver of Alzheimer's initiation and progression
Brain's energy needs are enormous as indicated by the fact that the brain weighs only 2% of total body weight but yet consumes 20% of total energy produced [8]. The high-energy needs of neurons are required for neurotransmisson, synthesis of neuro-transmitters, myelination and other housekeeping functions. It is mainly dependent on glucose utilization for energy generation and is able to use very little of other sources such as fatty acids etc [9]. Ketone bodies can also be used but less efficiently than glucose. The high energy needs can produce a conundrum for the brain especially in Alzheimer's disease. Indeed, defects in energy production by neurons are observed very early in this disease process [10]. Energy deficits in neuron are defined as reduction in production of ATP, the major form of energy in cells. Below we will review functional and molecular data from human and animal studies that support failing energy production as a plausible cause of disease initiation and progression.
Functional data showing reduced glucose utilization by the brain, also known as hypo-metabolism, has been demonstrated in humans using positron emission tomography (PET), an imaging technique using 2-[18F] fluoro-2-deoxy-D-glucose (FDG-PET) [11,12]. FDG, is a non-degradable analog of glucose and suitable as tracer to study energy metabolism. The concentrations of FDG indicate tissue metabolic activity, in terms of regional glucose uptake. Such studies have demonstrated consistent, early and progressive reductions in glucose utilization in Alzheimer's patients [13]. Furthermore, the extent and regional location of hypo-metabolism in the brain correlate with severity of symptoms. Increasing evidence suggests that reductions in brain glucose utilization occur at the pre-symptomatic stages of the disease and observed before the onset of disease [12].
What is the underlying cause of these energy deficits during disease genesis? Interestingly, both human and animal data show that Alzheimer's pathology is preceded and accompanied by significant events as follows that can account for energy deficiency:
Depletion of nicotinamide adenine dinucleotide (NAD+),a coenzyme involved in ATP generating redox reactions. Depletion in levels of this key coenzyme may occur due to high oxidative stress blocking its regeneration as well as consumption by enzymes such as PARP which use NAD as a substrate [14,15].
Decrease in activities of important enzymes involved in glycolysis namely hexokinase and phosphofructokinase and consequent reduction in energy production [16,17].
Decrease in activity of enzymes shown below involved in mitochondrial tricarboxylic acid cycle (TCA cycle) and oxidative phosphorylation processes, which generate most of cellular ATP.
Decrease in activity of alpha-ketoglutarate dehydrogenase complex, a rate-limiting enzyme in TCA cycle [18].
Decreased expression and activity of cytochrome c oxidase, an important component of oxidative phosphorylation via electron transport chain [19].
Decrease in activity of pyruvate dehydrogenase (PDH), a key rate-limiting enzyme in mitochondria to convert pyruvate, the end product of glycolysis, into acetyl-CoA, to initiate the TCA cycle. PDH is the first enzyme component of pyruvate dehydrogenase complex [19].
The putative mechanism of decrease in PDH activity during Alzheimer's may be its phosphorylation by glycogen synthase kinase 3b (GSK-3b) [20,21]. GSK-3b is a serine/threonine protein kinase that mediates phosphorylation of serine and threonine amino acids in enzymes or regulatory proteins. Phosphorylation by GSK-3b usually inhibits the activity of target protein, as in the case of glycogen synthase, the enzyme that controls conversion of glucose to glycogen. The brain has very little stores of glycogen, making it unlikely that GSK-3b plays that role in that tissue. Rather it is now believed that this enzyme may play a regulatory role in Alzheimer's via at least two activities: a) its ability to phos-phorylate microtubule-associated protein tau. Hyperphosphorylation of tau disrupts its interaction with tubulin within microtubules and leads to accumulation of neurofibrillary tangles and b) Phosphorylate PDH and inactivate it [22].
Another enzyme whose expression is modulated directly by neuronal energy deficit is BACE-1. Studies have demonstrated that energy deprivation can cause posttranscriptional induction of BACE-1 [23,24]. This is an important finding because it connects neuronal energy crisis directly to amyloid hypothesis, i.e. increase in BACE-1 activity would lead to increased Aβ42 and initiate the amyloid cascade.
Energy Targets and approaches to new Therapies
It's clear that several players may contribute to neuronal energy deficiency observed in Alzheimer's. We have integrated above data on energy deficiency into a hypothesis around which drugs targets can be identified and pursued for discovery of new therapies. We propose (Figure 2) that risk factors associated with Alzheimer's disease including ageing, traumatic brain injury/stroke, genetics and metabolic diseases (e.g. type 2 diabetes) can combine to trigger detrimental molecular pathways in neurons. Specifically brain hypoxia and inflammatory cytokine production which occur in stroke or traumatic brain injury, insulin resistance and dysregulation of insulin signaling in diabetes and genetics in patients with apoE4 alleles act as switches to bring about gene expression or enzyme activity changes. The resulting chronic hypo-metabolic state by itself can trigger neuronal dysfunction and cognitive decline. This coupled with induced up-regulation of BACE-1 can further lead to Aβ42 accumulation and fatal entry into amyloid cascade culminating in hallmark features of Alzheimer's disease. Since the core cause of neuronal dysfunction in our model is energy driven we have termed it as bioenergetics of neurodegeneration model (BEND model) (Figure 2).
Figure 2.

Bioenergetics of neurodegeneration model (BEND model). An “energy deprivation” based model of Alzheimer's is proposed. The model states that known disease risk factors trigger molecular pathways, which leads to decrease in enzyme activities important in cellular energy generation. Energy defects can result in neuronal dysfunction/loss, BACE-1 induction and cognitive decline.
Based on above model we selected enzyme targets in our bioenergetics breakdown paradigm. The choice of targets was based on druggability factors, i.e. low possibility of toxicity when target is modulated, likelihood of efficacy when target is regulated, information available on crystal structure and availability of existing chemistry scaffolds to design compounds. The selected targets are discussed below and listed in Table 1:
Table 1.
Proposed energy sensitive targets for new therapeutics
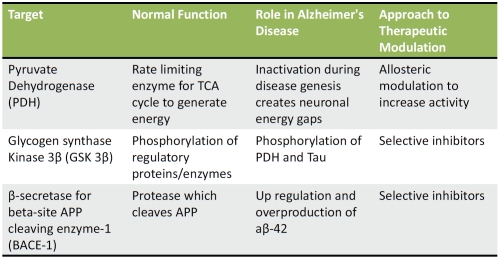 |
PDH: As the enzyme, which is conduit between glycolysis and TCA cycle, PDH is pivotal in energy generation. There has been interest in this target from oncology perspective [25]. Because of work already done in oncology, there is a lot of information available about regulating this target. In case of Alzheimer's, the objective would be to increase PDH's activity and enhance energy production. Pharmacologically it's more difficult to activate an enzyme than to inhibit is activity. But in case of PDH there are avenues available to activate it through allosteric approaches. An example of this is that the enzyme uses alpha lipoic acid as a cofactor and its mimetics can be designed to boost activity [26,27]. Secondly, PDH activity can be modulated by either pyruvate dehydrogenase kinase [28] or GSK-3b. It would be efficient to design drugs around these kinases and GSK-3b inhibition is especially attractive and will be discussed below.
-
GSK-3b: This enzyme is an especially attractive target for several reasons. Its involvement in Alzheimer's is at least at two separate levels-one which fits well with our energy paradigm-blocking GSK-3b activity should result in continued activation of PDH and second its role in phosphorylation of tau protein. In addition, inhibition of GSK-3b has also been shown to decrease Aβ42 production [29,30]. Thus inhibition of this target could improve energy production as well as tie into the amyloid plaque formation and tau phosphorylation.
GSK-3b has been studied extensively as a target for diabetes because of its role in insulin signaling related pathway [31,32]. There is vast published literature on highly potent inhibitors for this enzyme and some of those chemical structures can be starting points for a brain penetrable inhibitor for Alzheimer's [33,34].
BACE-1: This enzyme is already being pursued vigorously for design of new therapeutics for Alzheimer's as a way to decrease Aβ42 [35]. There are preclinical and clinical data available for some early stage drug candidates as well [36,4]. The reason for including this target in our list of “energy sensitive” targets is because of its regulation by cellular energy deprivation. While direct inhibition continues to be a favored approach for this target, because of this enzyme's involvement with other substrates, selectivity and toxicity will always be a burden. In contrast, if one could modulate cellular energy and indirectly lower BACE-1 activity, it could accomplish dual objectives of safety and efficacy.
Conclusions
Because of urgent need for new safe and efficacious therapies for Alzheimer's, large amounts of investments are being made in identifying new approaches and drugs. Targeting amyloid hypothesis continues to be the favored approach, but other approaches must be considered since amyloid based drug candidates to date have failed in the clinic [37,38,39,40]. Based on existing literature and evolving ideas [5,41] we have reiterated energy crisis as the central theme and as basis of novel drug discovery approaches. We propose three targets: a) PDH, which is upstream of amyloid cascade; b) GSK-3b, a bridge between energy crisis and amyloid cascade; and c) BACE-1, an integral part of amyloid cascade but may be modulated by energy improvements. Evolution of new ideas and targets will enhance the likelihood of generating successful drugs in the near future.
Acknowledgments
The author is a compensated consultant to Kareus therapeutics, SA. The author would like to thank Drs. Patrick Doyle and Dr.Venkateswarlu for their reading of the manuscript and comments.
References
- 1.Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 3.Saxena U. Alzheimer's disease amyloid hypothesis at crossroads: where do we go from here? Expert Opinion Therapeutic Targets. 2010;14(12):1273–1277. doi: 10.1517/14728222.2010.528285. [DOI] [PubMed] [Google Scholar]
- 4.De Strooper B, Vassar R, Golde T. The secreta-ses: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6(2):99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Struble RG, Ala T, Patrylo PR, Brewer GJ, Yan XX. Is brain amyloid production a cause or a result of dementia of the Alzheimer's type? J Alzheimers Dis. 2010;22(2):393–399. doi: 10.3233/JAD-2010-100846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferreira IL, Resende R, Ferreiro E, Rego AC, Pereira CF. Multiple defects in energy metabolism in Alzheimer's disease. Curr Drug Targets. 2010;11(10):1193–1206. doi: 10.2174/1389450111007011193. [DOI] [PubMed] [Google Scholar]
- 7.Reddy VP, Zhu X, Perry G, Smith MA. Oxidative stress in diabetes and Alzheimer's disease. J Alzheimers Dis. 2009;16(4):763–774. doi: 10.3233/JAD-2009-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saxena U. Nature India. Is Alzheimer's A Lifestyle Disease? doi: 10.1038/nindia.2011.10; Published Online 31 January 2011. [Google Scholar]
- 9.Costantini LC, Barr LJ, Vogel JL, Henderson ST. Hypometabolism as a therapeutic target in Alzheimer's disease. BMC Neurosci. 2008;9(Suppl 2):S16. doi: 10.1186/1471-2202-9-S2-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetics deficit precedes alzheimer's pathology in female mouse model of alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106(34):14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in pre-clinical Alzheimer's disease. Ann NY Acad Sci. 2008;1147:180–195. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Pirraglia E, De Santi S, Reisberg B, Wisniewski T, de Leon MJ. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2009;36(5):811–822. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer's disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging. 2005;32(4):486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- 14.Braidy N, Guillemin G, Grant R. promotion of cellular NAD(+) anabolism: therapeutics potential for oxidative stress in ageing and Alzheimer's disease. Neurotox Res. 2008;13(3-4):173–184. doi: 10.1007/BF03033501. [DOI] [PubMed] [Google Scholar]
- 15.Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly (ADP-ribose) poly-merase-1-mediated neuronal death. J Neurosci. 2010;30(8):2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liguri G, Taddei N, Nassi P, Latorraca S, Nediani C, Sorbi S. Changes in Na□,K□-ATPase, Ca2□-ATPase and some soluble enzymes related to energy metabolism in brains of patients with Alzheimer's disease. NeurosciLett. 1990;112:338–342. doi: 10.1016/0304-3940(90)90227-z. [DOI] [PubMed] [Google Scholar]
- 17.Sims NR, Blass JP, Murphy C, Bowen DM, Neary D. Phospho- fructokinase activity in the brain in Alzheimer's disease. Ann Neurol. 1987;21:509–510. doi: 10.1002/ana.410210517. [DOI] [PubMed] [Google Scholar]
- 18.Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm. 1998;105:855–870. doi: 10.1007/s007020050099. [DOI] [PubMed] [Google Scholar]
- 19.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106(34):14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshi M, Takashima A, Noguchi K, Murayama M, Sato M, Kondo S, Saitoh Y, Ishiguro K, Hoshino T, Imahori K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase 3beta in brain. Proc Natl Acad Sci U S A. 1996;93(7):2719–23. doi: 10.1073/pnas.93.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imahori K, Hoshi M, Ishiguro K, Sato K, Takahashi M, Shiurba R, Yamaguchi H, Takashima A, Uchida T. Possible role of tau protein kinases in pathogenesis of Alzheimer's disease. Neurobiol Aging. 1998;19(1 Suppl):S93–98. doi: 10.1016/s0197-4580(98)00025-6. [DOI] [PubMed] [Google Scholar]
- 22.Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15(23):2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velliquette RA, O'Connor T, Vassar R. Energy inhibition elevates ß-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J Neurosci. 2005;25:10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hebert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz L, Abolhassani M, Guais A, Sanders E, Steyaert JM, Campion F, Israël M. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: preliminary results. Oncol Rep. 2010;23(5):1407–1416. doi: 10.3892/or_00000778. [DOI] [PubMed] [Google Scholar]
- 27.Holmquist L, Stuchbury G, Berbaum K, Muscat S, Young S, Hager K, Engel J, Münch G. Lipoic acid as a novel treatment for Alzheimer's disease and related dementias. Pharmacol Ther. 2007;113(1):154–164. doi: 10.1016/j.pharmthera.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31(Pt6):1143–1151. doi: 10.1042/bst0311143. [DOI] [PubMed] [Google Scholar]
- 29.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423(6938):435–439. doi: 10.1038/nature01640. 22. [DOI] [PubMed] [Google Scholar]
- 30.Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, Clarimón J, Lleó A, Gómez-Isla T. A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35(3):359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 31.Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res Clin Pract. 2007;77(Suppl 1):S49–57. doi: 10.1016/j.diabres.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 32.Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol. 2009;156(6):885–898. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khanfar MA, Hill RA, Kaddoumi A, El Sayed KA. Discovery of novel GSK-3β inhibitors with potent in vitro and in vivo activities and excellent brain permeability using combined ligand- and structure-based virtual screening. J Med Chem. 2010;53(24):8534–8545. doi: 10.1021/jm100941j. [DOI] [PubMed] [Google Scholar]
- 34.Wagman AS, Johnson KW, Bussiere DE. Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes. Curr Pharm Des. 2004;10(10):1105–1137. doi: 10.2174/1381612043452668. [DOI] [PubMed] [Google Scholar]
- 35.Evin G, Kenche VB. BACE inhibitors as potential therapeutics for Alzheimer's disease. Recent Pat CNS Drug Discov. 2007;2(3):188–199. doi: 10.2174/157488907782411783. [DOI] [PubMed] [Google Scholar]
- 36.Albert JS. Progress in the development of beta -secretase inhibitors for Alzheimer's disease. Prog Med Chem. 2009;48:133–161. doi: 10.1016/s0079-6468(09)04804-8. [DOI] [PubMed] [Google Scholar]
- 37.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM, AN1792(QS-21)-201 Study Team Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 38.Wilcock GK, Black SE, Hendrix SB, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA, Tarenflurbil Phase II Study investigators Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: a randomised phase II trial. Lancet Neurol. 2008;7:483–493. doi: 10.1016/S1474-4422(08)70090-5. [DOI] [PubMed] [Google Scholar]
- 39.Fleisher AS, Raman R, Siemers ER, Becerra L, Clark CM, Dean RA, Farlow MR, Galvin JE, Peskind ER, Quinn JF, Sherzai A, Sowell BB, Aisen PS, Thai U. Phase 2-safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol. 2008;65(8):1031–1038. doi: 10.1001/archneur.65.8.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lilly Halts Development of Semagacestat for Alzheimer's disease based on preliminary results of Phase III Clinical Trials. Lilly Press release, Indianapolis, Eli Lilly and Company, 17 August 2010. Available from: www.lilly.com (Last accessed 8 October 2010)
- 41.Parihar MS, Brewer GJ. Mitoenergetic failure in Alzheimer disease. Am J Physiol Cell Physiol. 2007;292(1):C8–23. doi: 10.1152/ajpcell.00232.2006. Jan. [DOI] [PubMed] [Google Scholar]