

Abstract
We report the case of a 59-year-old female who developed facial edema together with hypoproteinemia. On the basis of 99mTc-human serum albumin scintigraphy and a1-antitrypsin clearance, she was diagnosed with protein-losing gastroenteropathy. Furthermore, she was diagnosed with Sjögren syndrome on the basis of eye and oral dryness, positive result with anti-SSA antibody, and salivary gland biopsy. Her symptoms improved with the use of immunosuppressive agents following steroid pulse therapy. Therefore, steroid pulse therapy and immunosuppressive agents should be considered as possible effective treatment strategies for protein-losing gastroenteropathy associated with autoimmune diseases.
Key Words: Protein-losing gastroenteropathy, Sjögren syndrome, Steroid, Immunosuppressive agents, Cyclophosphamide, Mizoribine
Introduction
Protein-losing gastroenteropathy (PLGE) is a disease that induces excessive plasma protein loss from digestive organs into the gastrointestinal (GI) tract. Causes of PLGE include primary diseases, such as Ménétrier disease or intestinal lymphangiectasia and secondary diseases that develop along with other diseases [1]. Although several reports have been published recently on PLGE associated with collagen diseases [2, 3, 4, 5], it is relatively rare for PLGE to develop together with Sjögren syndrome (SS). Few patients with SS require steroid pulse or immunosuppressive therapy because of resistance to the moderate or high amounts of steroid, as in this case. Here we report a patient who achieved remission with steroid pulse and immunosuppressive therapies.
Case Report
The patient was a 59-year-old female with a medical history of rheumatoid arthritis developed at the age of 40 and Hashimoto thyroiditis developed at the age of 51. Since a few years, she has been free from rheumatoid arthritis symptoms such as morning stiffness, fatigue, and joint pain, and on examination active arthritis was absent. She noticed facial edema around March 2010 and was referred to our hospital in April 2010 for further examination and treatment.
At admission her height was 153.2 cm, weight was 74.4 kg, body temperature was 35.8°C, blood pressure was 121/90 mm Hg, and she had a regular pulse rate of 72/min. Furthermore, no cardiac murmurs or pulmonary rales were heard, and edema on the upper eyelids and lower legs with pitting was observed. Laboratory findings at admission including levels of total protein and albumin were 5.9 g/dl and 2.8 g/dl, respectively, suggesting hypoproteinemia. Urinalysis showed a positive result (+1) for urinary protein. However, the quantitative urinary protein was 34.2 mg/dl, and no abnormal protein leakage into the urine was observed. Hepatic and renal functions were normal. Thyroid hormones were controlled within the normal range (TSH 2.812 μIU/ml, FT3 1.71 pg/ml, and FT4 1.23 ng/dl) with 100 μg/day levothyroxine sodium. The level of the anti-thyroglobulin antibody was 1,630 U/ml and that of the anti-microzome antibody was positive at a dilution of 1:1,280. The levels of complement C3 and C4 were 35.6 mg/dl (normal 75-130 mg/dl) and 20 mg/dl (normal 16-31 mg/dl), respectively. Serum IgG level was 658 mg/dl (normal 932-1,976 mg/dl), and stool fat was detected by Sudan III staining. Chest X-ray on admission revealed a right-sided pleural effusion, and an echocardiogram revealed a pericardial effusion, although no abnormality was found in cardiac function.
After admission, the patient was concomitantly treated with intravenous albumin infusion and diuretic injection (fig. 1), and further examinations were performed to detect the cause of her symptoms. Upper and lower GI endoscopic examinations were performed to find the cause of protein leakage from the digestive organs. Upper GI endoscopy revealed mild swelling of the entire gastric mucosa without any tumor or erosive ulcerative lesions or giant folds such as those manifested in Ménétrier disease and other conditions. Although the mucosal surface appeared normal, lower GI endoscopy revealed mild edema and congestion of the entire colon; however, no tumor formation or erosive ulcerative lesion was observed. 99mTc-human serum albumin (99mTc-HSA) scintigraphy revealed early radioisotope accumulation in the stomach (fig. 2) and a1-antitrypsin (a1-AT) clearance was high at 205 ml/day (normal <13 ml/day), and thus a diagnosis of PLGE was made.
Fig. 1.

Clinical course during hospitalization. PSL = Prednisolone; M-PSL = methylprednisolone; CPA = cyclophosphamide; MZR = mizoribine. Parameters indicate total protein (•) and albumin (▪). PSL (50 mg/day) was administered after the diagnosis of PLGE. However, hypoproteinemia was aggravated with total serum protein at 2.8 g/dl and albumin at 1.0 g/dl (point A). Although steroid pulse therapy was partially effective, it did not adequately improve symptoms (point B). The patient's symptoms improved with the use of CPA pulse therapy, and consequently, oral administration of MZR followed the steroid pulse therapy.
Fig. 2.
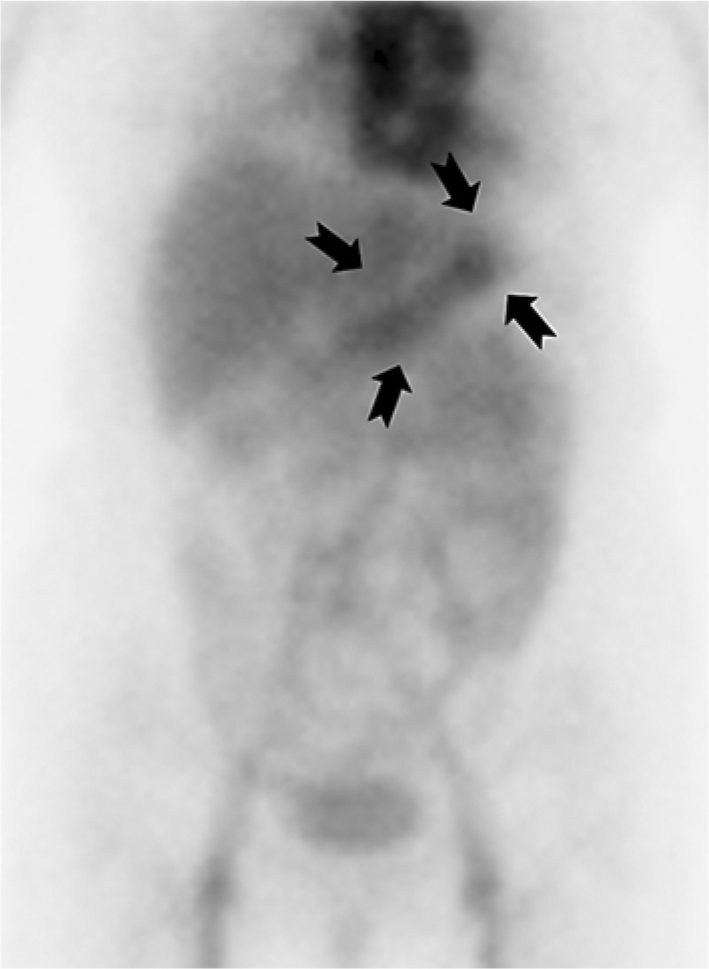
99mTc-HSA scintigraphy. Accumulated radioactivity was detected in areas of the stomach (arrows) 60 min after the intravenous injection of 99mTc-HSA.
Furthermore, complication of her condition by SS was suspected because she had eye and oral dryness for more than a month before admission. Using the gum test, 23 ml of saliva was collected and Schirmer's test results were 6 and 8 mm for the right and left eyes, respectively. According to the diagnostic criteria for SS in Japan, diagnosis of SS was confirmed by a positive result for anti-Ro/SSA antibodies (anti-Ro/SSA antibody level was 355 U/ml and anti-La/SSB antibody level was <7 U/ml) and salivary gland biopsy.
The diagnosis of PLGE complicated by SS was made as described above and 50 mg/day prednisolone (PSL) was administered (fig. 1). However, treatment was not successful. Hypoproteinemia was aggravated with total serum protein at 2.8 g/dl and albumin at 1.0 g/dl, and worsening of edema and development of diarrhea were observed. Therefore, steroid pulse therapy (1.0 g/day methylprednisolone for 3 days) was initiated as an alternative treatment. The levels of total serum protein and albumin increased and facial edema and digestive tract symptoms disappeared. The dose of PSL was tapered off after reducing it to 60 mg/day. However, a flare-up was observed. We obtained informed consent from the patient and performed steroid pulse therapy for the second time together with cyclophosphamide (CPA) pulse therapy and oral administration of 150 mg/day mizoribine (MZR). No relapse occurred, and she was discharged when her total serum protein and albumin had been maintained at nearly normal levels. After treatment, radioisotope accumulation previously observed in 99mTc-HSA scintigraphy disappeared and a1-AT clearance improved to 17.3 ml/day with reduced protein loss from the digestive organs into the GI tract. In addition, upper and lower GI endoscopies revealed disappearance of intestinal edema.
Discussion
PLGE is a disease that induces excessive loss of plasma proteins into the GI tract. The causes of PLGE include various diseases such as digestive tract diseases, cardiac diseases, or collagen diseases. For autoimmune PLGE, the number of reports regarding PLGE cases associated with collagen disease has been increasing in Japan [3, 4, 5], especially after the study of Pachas et al. in 1971 of systemic lupus erythematosus (SLE) complicated by PLGE [2]. Primary collagen diseases include SLE – which is observed most commonly -, mixed connective tissue diseases, SS and scleroderma [3].
To the best of our knowledge, from 1986 until 2010, only 22 cases of PLGE associated with SS were reported in Japan (including minutes in a conference and our case) [3, 5]. Among these, 86.4% of patients, including our case, were females with a mean age of 45.3 years. In most cases (90.9%), edema was the main symptom and digestive tract symptoms, such as diarrhea, were rare (22.7%). PLGE has recently been diagnosed using 99mTc-HSA scintigraphy. The most common site of protein loss, if detectable, is the small intestine. Protein loss from the stomach, as presented in this case, is rare; only 4 cases have been reported until date. The mean a1-AT clearance was 219.1 ml/day; the level was likely to be higher than the 100 ml/day in PLGE cases that developed repeatedly along with inflammatory bowel disease with mucosal disorders [6].
Upper GI endoscopic examination performed before treatment revealed neither giant folds at the greater curvature of the body of the stomach nor viscous gastric fluid secretion as that manifested in Ménétrier disease, indicating that abnormality in the mucosal surface was absent. Furthermore, lymphangiectasia was not detected in biopsies of the stomach and duodenum, from where protein loss was suspected.
The mechanisms of protein loss from the GI wall involve abnormality of the intestinal lymphatic system [7], increased papillary permeability [8], or abnormality of gastric mucosal epithelium [9]. However, the exact mechanism responsible for PLGE in association with collagen disease remains unclear. A majority of the GI endoscopic findings in previously reported cases were edematous changes of lesions with a very mild redness, and the biopsy findings were often mild inflammatory cell infiltration and mild lymphangiectasia.
With regard to pathological observations, deposition of immunoglobulins and complement components, such as C3, were observed with immunostaining on the capillary walls in the lamina propria. It has been reported that complement activation may induce increased capillary permeability [3, 4, 10] resulting in protein loss. In this case, deposition of IgG, C3, C1q, and fibrinogen was detected on submucosal capillaries during immunostaining of biopsy samples having no lymphangiectasia, suggesting that the condition was induced by increased capillary permeability.
With regard to treatment methods, all 22 patients with PLGE associated with SS have been reported to be treated with steroid therapy [3, 5]. Among the available data, in 13 cases, the patients were treated with moderate to high levels of steroid and the treatment was effective in 10 patients (76.9%). Thus, the outcome was relatively satisfactory. However, in the present case, the patient was resistant to high levels of steroid (50 mg/day PSL) and required steroid pulse therapy (fig. 1, point A). Although steroid pulse therapy was partially effective, it did not adequately improve symptoms in our patient (fig. 1, point B). Finally, her symptoms improved with the use of immunosuppressive agents (CPA pulse therapy and oral administration of MZR) that followed the two-fold steroid pulse therapy; the steroid dose was eventually reduced. Therefore, more aggressive immunosuppression may increase the overall response of patients with PLGE associated with collagen disease resistant to a moderate to high dose of PSL. In some cases, it has been reported that CPA pulse therapy or administration of oral azathioprine (AZA) was effective in autoimmune PLGE cases associated with mixed connective tissue disease or SLE [11, 12].
CPA is an alkylating agent and the most widely used agent in this class. Although it has been used in various non-neoplastic autoimmune diseases, some reports have shown an association of immunosuppressive drugs with an increased risk of hematological and lymphoreticular malignancies [13]. Thus, patients must be informed of these potential side effects. We employed intravenous CPA pulse therapy to treat patients because some reports have suggested the efficacy of this therapy for PLGE associated with SLE [14]. In addition, other reports suggest that treatment of PLGE with a combination of PSL and AZA is effective and well tolerated by the patients [12].
We used MZR (an imidazole nucleoside), which has frequently been used instead of AZA as a component of immunosuppressive drug regimens (renal transplantation patients, severe lupus nephritis, steroid-resistant nephritic syndrome, and rheumatoid arthritis patients). Furthermore, the efficacy of MZR for ameliorating glandular symptoms has been reported through improvements in immune abnormalities in patients with SS [15].
We present the first report of a patient with PLGE associated with SS that shows a favorable therapeutic effect of MZR. This patient showed improvement with the use of CPA pulse therapy, and consequently, MZR followed the steroid pulse therapy. Therefore, the dose of the steroid administered could be reduced. She has been free from facial edema as well as hypoproteinemia and has maintained complete remission after the combination therapy of low-dose PSL and MZR.
We suggest that a combination of immunosuppressive agents may be effective in patients with PLGE-associated autoimmune diseases. However, these treatments are still controversial and to support efficacy require more evidence with regards to the types of immunosuppressive agents, dose and duration of the treatment.
Footnotes
This is an Open Access article licensed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs 3.0 License (www.karger.com/OA-license), applicable to the online version of the article only. Distribution for non-commercial purposes only.
References
- 1.Waldmann TA. Protein-losing enteropathy. Gastroenterology. 1966;50:422–443. [PubMed] [Google Scholar]
- 2.Pachas WN, Linscheer WG, Pinals RS. Protein-losing enteropathy in systemic lupus erythematosus. Am J Gastroenterol. 1971;55:162–167. [PubMed] [Google Scholar]
- 3.Tsutsumi A, Sugiyama T, Matsumura R, et al. Protein losing enteropathy associated with collagen disease. Ann Rheum Dis. 1991;50:178–181. doi: 10.1136/ard.50.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakajima A, Ohnishi S, Mimura T, et al. Protein-losing enteropathy with hypocomplementemia and anti-nuclear antibodies. J Gastroenterol. 2000;35:627–630. doi: 10.1007/s005350070063. [DOI] [PubMed] [Google Scholar]
- 5.Nagashima T, Hoshino M, Shimoji S, et al. Protein-losing gastroenteropathy associated with primary Sjögren's syndrome: a characteristic oriental variant. Rheumatol Int. 2009;29:817–820. doi: 10.1007/s00296-008-0794-2. [DOI] [PubMed] [Google Scholar]
- 6.Miura S, Yoshioka M, Tanaka S, et al. Faecal clearance of alpha 1-antitrypsin reflects disease activity and correlates with rapid turnover proteins in chronic inflammatory bowel disease. J Gastroenterol Hepatol. 1991;6:49–52. doi: 10.1111/j.1440-1746.1991.tb01144.x. [DOI] [PubMed] [Google Scholar]
- 7.Tsuchiya M, Miura S. Basic and clinical aspects of intestinal lymphatics. J Gastroenterol Hepatol. 1988;3:261–278. [Google Scholar]
- 8.Stewart RD, Stewart JH. A case of unexplained gastrointestinal protein loss. Gut. 1965;6:146–150. doi: 10.1136/gut.6.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinfeld JL, Davidson JD, Gordon RS, Jr, et al. The mechanism of hypoproteinemia in patients with regional enteritis and ulcerative colitis. Am J Med. 1960;29:405–415. doi: 10.1016/0002-9343(60)90036-x. [DOI] [PubMed] [Google Scholar]
- 10.Itoi K, Sasaki T, Sawai T, et al. Protein-losing enteropathy in association with immune deposits in gastrointestinal mucosal capillaries. Am J Gastroenterol. 1989;84:187–191. [PubMed] [Google Scholar]
- 11.Furuya T, Suzuki T, Onoda N, et al. Mixed connective tissue disease associated with protein losing enteropathy: successful treatment with intravenous cyclophosphamide therapy. Intern Med. 1992;31:1359–1362. doi: 10.2169/internalmedicine.31.1359. [DOI] [PubMed] [Google Scholar]
- 12.Mok CC, Ying KY, Mak A, et al. Outcome of protein-losing gastroenteropathy in systemic lupus erythematosus treated with prednisolone and azathioprine. Rheumatology (Oxford) 2006;45:425–429. doi: 10.1093/rheumatology/kei164. [DOI] [PubMed] [Google Scholar]
- 13.Baltus JA, Boersma JW, Hartman AP, et al. The occurrence of malignancies in patients with rheumatoid arthritis treated with cyclophosphamide: a controlled retrospective follow-up. Ann Rheum Dis. 1983;42:368–373. doi: 10.1136/ard.42.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werner de Castro GR, Appenzeller S, Bértolo MB, et al. Protein-losing enteropathy associated with systemic lupus erythematosus response to cyclophosphamide. Rheumatol Int. 2005;25:135–138. doi: 10.1007/s00296-004-0483-8. [DOI] [PubMed] [Google Scholar]
- 15.Nakayamada S, Fujimoto T, Nonomura A, et al. Usefulness of initial histological features for stratifying Sjogren's syndrome responders to mizoribine therapy. Rheumatology (Oxford) 2009;48:1279–1282. doi: 10.1093/rheumatology/kep228. [DOI] [PubMed] [Google Scholar]