

Abstract
A robust, reproducible, and high throughput method was developed for the relative quantitative analysis of glycoprotein abundances in human serum. Instead of quantifying glycoproteins by glycopeptides in conventional quantitative glycoproteomics, glycoproteins were quantified by nonglycosylated peptides derived from the glycoprotein digest, which consists of the capture of glycoproteins in serum samples and the release of nonglycopeptides by trypsin digestion of captured glycoproteins followed by two-dimensional liquid chromatography-tandem MS analysis of released peptides. Protein quantification was achieved by comparing the spectrum counts of identified nonglycosylated peptides of glycoproteins between different samples. This method was demonstrated to have almost the same specificity and sensitivity in glycoproteins quantification as capture at glycopeptides level. The differential abundance of proteins present at as low as nanogram per milliliter levels was quantified with high confidence. The established method was applied to the analysis of human serum samples from healthy people and patients with hepatocellular carcinoma (HCC) to screen differential glycoproteins in HCC. Thirty eight glycoproteins were found with substantial concentration changes between normal and HCC serum samples, including α-fetoprotein, the only clinically used marker for HCC diagnosis. The abundance changes of three glycoproteins, i.e. galectin-3 binding protein, insulin-like growth factor binding protein 3, and thrombospondin 1, which were associated with the development of HCC, were further confirmed by enzyme-linked immunosorbent assay. In conclusion, the developed method was an effective approach to quantitatively analyze glycoproteins in human serum and could be further applied in the biomarker discovery for HCC and other cancers.
Mass spectrometry (MS) based quantitative proteomics has become the most commonly used approach for studying expression changes of proteins in large-scale studies (1). A variety of mass spectrometry-driven protein quantification methods have been proposed involving stable isotope labeling of proteins or peptides coupled with tandem mass spectrometry (MS/MS)1 sequencing, e.g. isotope-coded affinity tags (2), stable isotope labeling by amino acids in cell culture (3), and multiplexed quantification using isobaric tagging reagents (4). On the contrary, label-free methods have received increasing attention as promising alternatives that automatically waive some of the disadvantages of using stable isotope labeling methods. Various label-free quantification strategies have also been developed in recent years which were based on comparing the direct mass spectrometric signal intensity for a given peptide (5), or the number of acquired spectra to a protein (6), which is referred as the spectral counting method. With the advances of these quantification methods, the mass spectrometry-based quantitative proteomics is becoming an emerging technology in biomarker discovery of human diseases (7).
The human serum is generally considered as the primary clinical specimen in disease diagnosis and therapeutic monitoring (8). It represents the largest and deepest version of human proteome present in any samples. Beside the classical serum proteins, it contains various tissue proteins (as leakage markers) plus numerous distinct immunoglobulin sequences. However, it is challenging to perform the comprehensive serum proteome analysis because of the extraordinary dynamic range and complexity. Several separation methods have been developed to reduce the complexity of the serum sample. Because most of the disease-related proteins, always secreted or shed from cell surfaces or released from tissue, are glycosylated (9), the enrichment of glycoproteins has become one of the key issues in biomarker discovery and various discovery methods have been developed, such as the capture of glycoproteins with lectins (10). Glycoprotein capture with either single lectin or a combination of lectins with affinity to different types of glycan linkages, coupled with mass spectrometry based quantitative proteomics methods, have been widely applied. However, the major drawback of the lectin capture method is the relatively weak binding of lectins to oligosaccharides, which may introduce large amounts of nonspecific adsorption of abundant proteins such as human serum albumin, which accounts for as much as 55% of total serum protein. For example, it was reported that only 34 of the 152 proteins (22.3%) bound to a concanavalin A (Con A) column in a membrane protein study of breast tumor cell were found to be glycoproteins (11). This nonspecific adsorption would affect the accuracy and reproducibility of glycoprotein quantification. In addition, glycoproteins of low abundance are not likely to be quantified with lectin capture methods because the isolation ability of lectins is limited. Although a depletion of some abundant proteins before lectin capture may reduce nonspecific adsorption and enhance analytical performance (12), this procedure may also deplete some proteins of low abundance, which are possible potential biomarkers because it has been found that immunosorbent columns and antibodies would bind to more than 100 proteins nonspecifically, some of which are glycosylated (13).
Another popular method for the isolation, identification, and quantification of glycoprotein is the capture of oxidized glycopeptides by hydrazide chemistry (14). This method shows high specificity for the solid phase extraction of N-linked glycopeptides. The captured glycopeptides can be released by PNGase F and labeled at N-terminal sides, which makes quantification of N-glycosites feasible. However, isotopic labeling increases sample complexity because of the differential labeling and combining of the samples (15). For glycoprotein with only one glycosite, the quantification accuracy may be affected by the labeling efficiency of different samples because there is only one peptide to be quantified. Moreover, because there exits great heterogeneity in the occupancy of different N-glycosites in glycoproteins with multiple glycosylated sites, quantification of glycoprotein by glycopeptides would lead to ambiguous results.
Compared with isotope labeling, label-free methods have received increasing attention as promising alternatives. Glycoproteins can be quantified by comparing liquid chromatography (LC)-MS maps of deglycosylated peptides that are generated by high resolution linear trap quadrupole-Fourier transform mass spectrometry using bioinformatics tools (16). Although promising reproducibility and sensitivity could be achieved, the variability of signal intensities among replicates increases with decreasing signal intensity, which would introduce ambiguity in the quantification of glycoproteins of low abundance or with few N-glycosylation sites. Instead of quantification by the peak intensity of MS1 in a high accuracy MS instrument, another viable label-free quantitative strategy is spectral counting, where the number of spectra matched to peptides from a protein is used as a surrogate measure of protein abundance (6). Recent studies have demonstrated that spectral counting can be as sensitive as ion peak intensities in terms of detection range, while retaining linearity and is highly reproducible (17). However, there are a few studies in which spectral counting was applied to quantitatively analyze glycoproteins in a high throughput manner (11) and only one study combined spectral counting with the hydrazide chemistry approach, which used the spectrum number of glycopeptides as the index of glycosylation level (18).
In this study, we propose an approach that combines the specificity of hydrazide chemistry and the convenience of the spectral counting method to quantify the abundance changes of glycoprotein in human serum sample. Unlike the existing glycopeptides-based methods, only nonglycosylated peptides were released by trypsin digestion after glycoprotein capture and the spectral counts of the identified nonglycopeptides were used to determine the abundance of the glycoproteins. We demonstrate that this approach is accurate, sensitive, and highly reproducible in a wide dynamic range of protein concentrations. Most importantly, unlike the measurement of the glycosylation level of N-glycosites in the conventional approach using glycopeptides, the application of this approach for the analysis of pooled human serum of both healthy people and patients with HCC indicated that this approach quantifies glycoprotein expression. A total number of 38 glycoproteins were found to have differences in concentration, including α-fetoprotein (AFP), which is the only current clinical biomarker for HCC and results of most identified differential glycorpoteins were in accordance with previously published studies. Moreover, three selected glycoproteins were validated by ELISA with another HCC cohort, which proved that this method is an effective approach for high-throughput differential glycoproteins screening.
EXPERIMENTAL PROCEDURES
Sample Collection
The study was approved by the Institutional Review Board at the Second Military Medical University. Informed consent was obtained from every patient enrolled in this study. Sera samples were collected from April to December, 2008, in the Eastern Hepatobiliary Surgery Hospital, Second Military Medical University (Shanghai, China). Serum samples were processed from each individual using a 12G BD Vacutainer safety-Lok™ blood collection system. Blood samples were collected in “gold top” tubes containing gel separator and clot activator. After collection they were immediately placed on ice and allowed to stand for 30 min. After this the samples were centrifuged at 3000 rpm for 15 min. Serum samples were stored at −80 °C until analysis. One sera cohort that composed of 27 HCC and 20 cases of an age and gender-matched normal control cohort, respectively, were pooled for quantitative glycoproteomic analysis. Another sera cohort, including 58 HCC and 30 normal controls were collected. HCC diagnoses were confirmed by histopathlogic study. Normal controls were selected from those without history of liver disease, with normal liver biochemical function and without hepatitis B or hepatitis C infection. The clinical information of HCC samples and normal controls for proteomics analysis and ELISA validation are listed in Table I.
Table I. Clinical information of the HCC samples and normal controls.
| HCC samples for proteomics analysis | Normal control samples for proteomics analysis | HCC samples for ELISA analysis | Normal control samples for ELISA analysis | |
|---|---|---|---|---|
| Sample No. | 27 | 20 | 58 | 30 |
| Age (yr) | 49.9 ± 10.3 | 47.8 ± 9.9 | 51.5 ± 11.7 | 48.7 ± 11.5 |
| Sex (F/M) | 4/23 | 4/16 | 10/48 | 5/25 |
| ALT (U/L) | 38.7 ± 20.4 | - | 40 ± 26.2 | - |
| AST (U/L) | 42.4 ± 28.4 | - | 44.2 ± 41 | - |
| HBsAg (yes/no) | 24/3 | 0/20 | 50/8 | 0/30 |
| HCV (yes/no) | 0/27 | 0/20 | 1/57 | 0/30 |
| AFP (μg/L) | ||||
| <20 | 6 | 22 | ||
| 20∼400 | 13 | 18 | ||
| >400 | 8 | 18 | ||
| Edmondson stage | ||||
| I, II | 6 | 17 | ||
| III, IV | 21 | 41 | ||
| Tumor diameter (cm) | ||||
| <2 | 2 | 3 | ||
| 2∼5 | 13 | 26 | ||
| >5 | 12 | 29 |
Materials and Chemicals
All the water used in this experiment was prepared using a Milli-Q system (Millipore, Bedford, MA); iodoacetamide was purchased from AMERESCO (Solon, OH); sodium periodate, dithiothreitol, ammonium bicarbonate, urea, trifluoroacetic acid (TFA), formic acid, and acetonitrile were all obtained from Aldrich (Milwaukee, WI). All the chemicals are of analytical grade except acetonitrile, which is of high performance liquid chromatography grade. Affi-Gel® Hz Hydrazide Gel was purchased from Bio-Rad (Hercules, CA).
Capture of Glycoprotein by Hydrazide Resin
The procedure for the capture of glycoproteins from pooled serum samples was based on the method reported previously with minor modification (19). Briefly, 10 μl of human serum was first diluted with 30 μl oxidation buffer (100 mm NH4Ac, 150 mm NaCl, pH = 5) and exchanged to the oxidation buffer using a Spin P-6 (Bio-Rad) protein desalting column, then diluted to 180 μl with the same buffer and 20 μl 100 mm sodium periodate was added. The reaction was kept in the dark for one h at room temperature. The oxidant was removed with YM-3 ultrafiltration kit (Millipore) with a molecular weight cut off of 3000 Da. The oxidized glycosylated proteins were coupled to 50 μl hydrazide resin (bead volume) overnight in coupling buffer at room temperature. After supernatants were removed, proteins were denatured by incubation with 20 mm dithiotreitol and 8 m urea in 100 mm ammonium bicarbonate at 37 °C for 2 h. After that, the resin was washed with 100 mm ammonium bicarbonate and 20 mm iodoacetamide was added. The alkylation was carried out at 25 °C in dark for 30 min. The resin was washed with 0.5 ml 100 mm ammonium bicarbonate containing 8 m urea, 1.5 m NaCl, and 100 mm ammonium bicarbonate three times to remove unbound nonglycosylated proteins. The nonglycopeptides of coupled glycoproteins were released by incubation with 5 μg proteomics grade trypsin (Sigma-Aldrich, Milwaukee, WI) in 100 μl of 50 mm NH4HCO3 overnight at 37 °C. Released nonglycopeptides were collected, lyophilized with SpeedVac, and reconstituted in 100 μl 0.1% formic acid solution for further analysis. Glycopeptides were released by further incubating the beads with 2 μl PNGase F in 100 μl of 50 mm NH4HCO3 overnight at 37 °C. Collected glycopeptides were also lyophilized with SpeedVac and reconstituted in 10 μl 0.1% formic acid solution for further analysis.
Analysis of Isolated Peptides
The analysis of the released nonglycopeptide samples was carried out by two-dimensional nano-LC-MS/MS according to our previous method (20). The 25 μl of reconstituted peptide sample was loaded onto a phosphate strong cation exchange monolithic column (150 μm i.d. × 7 cm) by air pressure. After that, the SCX column with loaded peptides was manually connected to a C18 column (75 μm i.d. ×10 cm) in tandem by a union. The peptides were eluted onto the analytical reversed phase column using stepwise salt solutions generated with 1000 mm ammonium acetate (pH = 2.7) and 0.1% formic acid. The salt concentrations of the buffer used for ten stepwise elutions were 50, 100, 150, 200, 250, 300, 350, 400, 500, and 1000 mm, respectively. Each salt step lasts 10 min followed by equilibrium with 0.1% formic acid for additional 10 min. After each elution step, a subsequent reverse phase (RP)LC-MS/MS was executed with a 120 min gradient from 5% to 35% acetonitrile. The RPLC-MS/MS was performed on a nano-RPLC-MS/MS system. A Finnigan surveyor MS pump (Thermo Finnigan, San Jose, CA) was used to deliver mobile phase. For the C18 capillary separation column, one end of the fused-silica capillary was manually pulled to a fine point of ∼5 μm with a flame torch. The columns were in-house packed with C18 AQ beads (5 μm, 120 Å) from Michrom BioResources (Auburn, CA) using a pneumatic pump. The nano-RPLC column was directly coupled to a linear trap quadrupole linear ion trap mass spectrometer from Thermo Finnigan (San Jose, CA) with a nanospray electrospray ionization source. The mobile phase consisted of mobile phase A containing 0.1% formic acid (v/v) in water, and mobile phase B containing 0.1% (v/v) formic acid in acetonitrile. The ESI voltage was 1.8 kV and the linear trap quadrupole instrument was operated at positive ion mode. Normalized collision energy was 35.0%. One microscan was set for each MS and MS/MS scan. All MS and MS/MS spectra were acquired in the data dependent mode. The mass spectrometer was set that one full MS scan was followed by six MS/MS scans on the six most intense ions. The dynamic exclusion function was set as follows: repeat count 2, repeat duration 30 s, and exclusion duration 90 s. System control and data collection were done by Xcalibur software version 1.4 (Thermo). The scan range was set from m/z 400 to m/z 2000.
Database Searching and Data Processing
The MS/MS spectra were generated by Extract_msn in Bioworks 3.2 (Thermo) and searched with SEQUEST (version 2.7) against a composite database including both original human protein database of International Protein Index (ipi.human.3.17.fasta, including 60234 entries, http://www.ebi.ac.uk/IPI/IPIhuman.html) and the reversed version of the forward one. For the database search of spiked samples, the sequences of bovine ribonuclease B and alpha-2-HS glycoprotein were added. The enzyme cleavage site was set as trypsin, KR/P. Enzyme limits was set fully enzymatic, cleaving at both end. Maximum of two missed cleavage was allowed. For nonglycopeptides search, carboxyamidomethylation (+57) was set as static modification and dynamic modification was used: oxidation of methionine (+16). For glycopeptides search, dynamic modification of PNGase F catalyzed deamination of asparagine (+1 Da) was added. MS tolerance for precursor ion and fragment ion was 2 Da and 1 Da, respectively. The database search results were filtered with criteria Xcorr≥1.9, 2.2 and 3.75 for +1, +2, and +3 charge states respectively, DeltaCn was set ≥0.1 for the three charge states. Validated peptides sequences were assembled into protein identifications with the nomenclature that provided a minimal list of proteins sufficient to explain all observed peptides, accounting for groups of isoforms and splice variants that share common peptides (21). Glycoproteins identified by nonglycopeptides were considered positive if at least two unique peptides matched or a total spectral count of more four spectra was evident (17). The false detection rate of peptide identification was determined by doubling the number of peptides from the reversed database and dividing by the total number of peptides. The false discovery rate of nonglycopeptides for glycoprotein quantification was below 1% with the filtering criteria described above. Glycopeptides were identified with N*XS/T motif where X could be any amino acid except proline as previously described (22). For the analysis of clinical samples, both the isolated peptides from healthy and cancerous serum were run two replicates in two-dimensional LC-MS/MS and the spectral counts were averaged. A spectral count fold-change of more than two was considered differently expressed between normal and cancer samples.
ELISA
Serological measurement of galectin-3 binding protein (Mac-2 BP or S90K), insulin-like growth factor binding protein 3 (IGFBP-3), and thrombospondin 1 (TSP-1) was performed using an enzyme linked immunosorbance assay (ELISA), from R & D Systems (Minneapolis, MN) according to the suggested protocols with another cohorts consisting of 58 HCC and 30 normal controls. The differences between HCC and control groups were evaluated by unpaired t test with Welch's correction using GraphPad Prism version 4 for Windows (GraphPad software, San Diego, CA).
RESULTS
Glycoprotein Identification with Nonglycopeptides
The purpose of this research is to establish a method for the high throughput monitoring of differential abundance of glycoproteins in clinical serum samples. Glycoprotein identification with glycopeptide capture method has been a popular protocol for several years (23). The specificity of glycopeptide isolation with hydrazide chemistry has been well studied in a previous report (19). It has advantages of high specificity and high efficiency of extracting N-glycopeptides from complex samples like human liver tissue (21). In this method, glycoproteins are digested at first and only the glycosylated peptides would be captured and analyzed whereas the nonglycopeptides, which take the most part of a glycoprotein, are discarded and neglected. Recent research showed that the nonglycopeptides can also be used to identify glycoproteins and a good overlap of identifying results exists between that by glycopeptides and by nonglycopeptides (24). This finding prompts us to quantify serum glycoproteins by nonglycopeptides. In a typical procedure of our approach, glycoproteins in serum samples were first oxidized with sodium periodate and then coupled to solid beads with hydrazide chemistry. After thoroughly washing, the nonglycopeptides of captured glycoproteins were released with trypsin digestion and the digest was analyzed with two-dimensional LC-MS/MS. Glycoproteins were identified with matched nonglycopeptides and could be finally quantified with spectra counting approach as shown in Fig. 1. Compared with the method of glycopeptide capture, identification with nonglycopeptides has its own advantages. First, glycoproteins are captured by the same hydrazide chemistry, which can guarantee the specificity and efficiency. We optimized the glycoprotein capture and washing procedure to reduce the nonspecific adsorption of albumin and related proteins that may bind to albumin. After the captured proteins being denatured, the hydrazide beads were washed with 8 m urea in 100 mm NH4HCO3 for another three times. This washing step could wash off albumin and the nonglycoproteins that adhesive to albumin and captured glycoproteins. Serum sample was processed with this optimized procedure. In a randomly chosen two-dimensional LC-MS/MS run, there were more than 1000 proteins identified. Within these proteins, 222 proteins were identified with at least two unique matched peptides or 4 MS/MS spectra, which is highly confident. For comparison, the glycopeptides on the beads were released by PNGase F, after the release of nonglycopeptides, and analyzed by LC-MS/MS. Fig. 2 shows the overlap of glycoprotein identifications between the glycopeptide method and nonglycopeptide method, which demonstrated that serum glycoproteins can also be identified by nonglycopeptides. It was observed that some glycoproteins identified by glycopeptides have not been identified by nonglycopeptides. This is because in glycoprotein identification with nonglycopeptides, much stricter criteria was applied, which needs two unique peptideidentifications. Actually, the coverage of serum glycoprotein identification by nonglycopeptides was better than that with glycopeptides as more glycoproteins were identified than that with glycopeptides. We investigated the 222 proteins identified by nonglycopeptides in the Swiss-prot database. A total number of 174 proteins were annotated and 120 (67.0%) of them were found to be glycoproteins. Although the percentage of glycoproteins was still lower than that in glycopeptides identification, this specificity is much better than that in lectin chromatography method, which is only 22.3% as reported by Wang et al. (11). The percentage of glycoproteins was also higher than the previous work using nonglycopeptides for glycoprotein identification, in which 191 of 589 proteins (32.4%) identified in Hela cell are glycosylated (24). Low specificity would reduce the accuracy and sensitivity in identification and quantification of low abundance glycoprotein in serum biomarker discovery because the most abundant serum albumin is nonglycosylated and some proteins would bind to albumin to form protein complexes. However, nonspecific adsorptions such as this could be removed with additional washing steps after protein being denatured. Moreover, the spectrum number of glycoproteins took more than 95% in the total spectrum number, which demonstrated that the nonspecific proteins had little affects on glycoprotein identification and quantification. Second, there are much more nonglycopeptides than glycopeptides in a glycoprotein, thus the increased number of peptides may increase the sensitivity and confidence in identifying low abundance glycoproteins, especially for the proteins with only one glycosylated site, e.g. α-fetoprotein. Third, in a glycoproteomics study of human liver tissue, it has been found that some glycoproteins can not be identified by glycopeptide capture with only trypsin digestion because the tryptic glycopeptides are beyond the detection range of mass spectrometry (21). However, it would not be a problem by using nonglycopeptides to identify glycoproteins. Forth and importantly, identifying with nonglycopeptides facilitates the quantification of glycoproteins by spectral counting method, which is a more sensitive method for detecting protein abundance changes (17).
Fig. 1.

Scheme for the relative quantification of glycoproteins in human serum by a glycoprotein capture based label-free approach.
Fig. 2.

Overlap of the identified glycoproteins in human serum by nonglycopeptides and by N-glycopeptides.
Relative Quantification of Glycoproteins with Nonglycopeptides
As a method developed to find potential biomarkers, it must be capable of accessing the relative abundance of proteins from various concentration levels in reproducible and high throughput way, from abundant glycoproteins like antitrypsin (mg/ml) to typical biomarker like α-fetoprotein (ng/ml). To investigate the capability of our method, the linearity and sensitivity have been evaluated with processing of commercially available human serum spiked with different amount of standard glycoproteins, i.e. rebonuclease B and α-2-HS glycoprotein from bovine. The spiked concentrations of glycoprotein standard ranged over two orders of magnitude. The linearity of the spectral counts and the amount of spiked standard glycoproteins was given in Fig. 3. It could be seen that the spectral counts correlated well with the amount of spiked standard glycoproteins with a linear range of at least 102 orders of degree, which demonstrated that the method could quantify the difference of glycoprotein abundance sensitively in a wide concentration range. Similar linearity and sensitivity with ordinary spectral counting approach have also been reported (6). It should be noted that when calculating the spectral counts, the glycosylated peptides that released by PNGase F were not taken into account because compared with the number of nonglycopeptides, the spectral counts of glycopeptides could be neglected and the ratio of spectral counts would not be influenced. Quantification by nonglycopeptides should be more sensitive than by glycopeptides theoretically because there will be much more nonglycosylated peptides than glycosylated peptides generated from a glycoprotein. However, increased number of peptides may also complicate the sample loaded on to MS instrument. To overcome the limitation of peak capacity of 1D nano-LC column, a two-dimensional LC-MS/MS system was used to analyze the complex tryptic peptides. The increased number of peptides facilitated the quantification by spectral counting method, which is an advance over quantification by MS peak intensity.
Fig. 3.

Spectral counts of glycoproteins versus its spiked concentration, A, α-2-HS glycoprotein, from 5 μg/ml to 500 μg/ml, B, Rebonuclease B, from 5 μg/ml to 100 μg/ml. Different amounts of glycoprotein standards were spiked into 10 μl human serum of healthy donors and the spiked samples were processed with the procedure described above.
In the quantification of cell surface proteome changes by analysis of LC-MS maps of isolated N-glycopeptides (16), high Squared-Pearson-Correlation R2 (0.986–0.989) of technical replicates and R2 = (0.925–962) for biological replicates has been found. However, the variability of signal intensities among replicates increases with decreasing signal intensity. High variability for peptide quantification of low-intensity peptides was likely due to the low signal-to-noise ratio as well as the interference of higher abundance peptides (25), which is more prominent in serum sample analysis. Although the reproducibility can be improved by using more than one peptide for quantification, it is not practical for quantification by glycopeptides because it has been found that average of less than two glycopeptides were identified for one glycoprotein in large-scale glycoproteomics study (19, 21).To assess the reproducibility of quantifying glycoproteins with glycoprotein capture and spectral counting, the spectral counts of identified glycoproteins in two technical replicates and two sample replicates were plotted with each other. For technical replicates, the same nonglycopeptides mixtures were analyzed in replicates with the online two-dimensional LC-MS/MS system. For sample replicates, two serum samples of the same amount were processed in parallel with the glycoprotein capture and nonglycopeptides releasing. Released nonglycopeptides were analyzed with two-dimensional LC-MS/MS respectively. The scatter plots and R-squared values for the two-dimensional LC-MS/MS run of the technical runs and two sample replicates were given in Fig. 4. The spectral count had a high reproducibility (R2 = 0.9942, 0.9843, respectively). This could be attributed to the high efficiency and specificity of simplified glycoprotein capture procedure, which need no immunodepletion. Beside, as there are many more nonglycopeptides identified compared with glycopeptides in one glycoprotein, the influence of the variances in instrument condition, glycoprotein capture procedure, and the other experimental processes can be offset, which will certainly compromise the reproducibility and accuracy in quantification with peak intensity of glycopeptides. The spectrum counting approach was proved to be more reproducible than that of MS1 ion chromatogram in the analyzes of S. cerevisiae crude membrane fractions (26). It was suggested that quantification with the peak area of a peptides in MS1 is a peptide or locally based quantitative approach whereas spectrum counting is a protein or global based approach. Large standard deviations would occur by using ion chromatograms when post-translational modification exists. Besides, spectrum counting has a wider dynamic range over ion chromatogram ratios.
Fig. 4.

Scatter plots and R-squared values for the two-dimensional LC-MS/MS run of (A) two technical runs and (B) two sample replicates, the linearity of the plots demonstrates the reproducibility of the developed approach in both mass spectrometry identification and sample process.
Briefly, the combination of the selective capture of glycoprotein in human serum and label-free quantification of the nonglycopeptides is quite a feasible way to quantify concentration changes of glycoprotein in human serum samples. This approach demonstrated high selectivity to enrich the glycoproteins in human serum without the need of immunodepletion step. The relative quantification of glycoprotein by comparing the spectral counts of released nonglycopeptides is an accurate, sensitive and reproducible approach to detect the differences in concentration of glycoproteins over a linear range of more than two orders of magnitude.
Application in the Differential Glycoprotein Screening in Hepatocellualr Carcinoma
As this accurate, reproducible and high throughput approach to quantitatively analyze glycoproteins in human serum was developed, it was applied to find diffrentially expressed serum glycoprotein in HCC. The pooled serum samples of 20 healthy people and 27 patients with HCC were analyzed with the established workflow. Each sample digest was analyzed by two-dimensional LC-MS/MS in two replicates. The spectral counts for a protein in each replicate run were averaged. The average of the spectral counts in healthy and HCC sample were compared. Totally 148 glycoproteins were quantified with high confidence when the strict criteria were applied to filter the dataset as described in experimental section. All information about the spectral counts and identified peptides of these proteins is given in supporting materials. Fig. 5 illustrates the changes of spectral counting of these proteins. The majority of identified glycoproteins except some high abundant glycoproteins like antitrypsin did not show substantial concentration changes with log2 spectrum count ratio within the range of –1 to +1, which suggested there was no analytical bias toward different samples. However, there are 38 glycoproteins found with significant differences in concentration in two sets of samples which were listed in Table II. The good accordance of the results with previous publications demonstrated the reliability of our approach in differential glycoprotein identification. AFP, the only clinically used marker for HCC diagnosis, was only detected in HCC serum. Cholinesterase, formed in liver, is widely measured as a liver function test and the activity was reported to decrease in liver cirrhosis or hepatocellular carcinoma (27)'which is in accordance with our results. Vitronectin, which is a multifunctional plasma glycoprotein that participates in the regulation of coagulation, fibrinolysis, and the complement cascade (28), was also found in decreased concentration in serum of HCC patients. The plasma vitronectin level was low in all liver disease groups as compared with the healthy controls. The difference from the controls was significant in patients with hepatocellular carcinoma and decompensated cirrhosis. Moreover, the plasma vitronectin level was positively correlated with the levels of serum cholinesterase (29). The function and interaction of this protein has attracted extensive attentions. It has been demonstrated that increased expression of vitronectin associated with areas of lymphocyte infiltration in chronically inflamed liver and in primary and metastatic tumors, supporting the adhesion and migration of tumor-infiltrating lymphocytes (TIL), which suggested that vitronectin play an important role in recruiting and positioning lymphocytes with inflamed and malignant liver tissue (30). The 8900 Da C-terminal part of the V10 fragment of vitronectin was found with increased concentration in sera samples of HCC patients as compared with in the sera of patients with cirrhosis (31). All these results demonstrate that vitronectin need to be further studied to find its role and function in HCC. Some other glycoproteins that interact with vitronectin were also found with different concentration between healthy and cancerous samples. It has been found that vitronectin associates with the γ-chain of fibrinogen during coagulation, and may thereby modulate hemostasis and inflammation (32). In our results the significant up-regulation of the γ-B chain of fibrinogen was found, which was in accordance with the results reported by Comunale et al. (33). The increased level of complement factor B was also in consistent with their results.
Fig. 5.

Log2 ratio of spectra counts (HCC vs. normal) for each glycoprotein versus its average spectra counts, which demonstrated the distribution of identified glycoproteins.
Table II. List of differently expressed glycoproteinsa found with quantitative proteomics method in human serum of patients with hepatocellular carcinoma.
| IPI Number | Swiss-prot | Protein Name | SpCb_Cancer | SpC_Normal |
|---|---|---|---|---|
| IPI00004573 | P01833 | Polymeric-immunoglobulin receptor | 6.5 | NDc |
| IPI00007240 | P05160 | Coagulation factor XIII B chain | 8 | 16.5 |
| IPI00008556 | P03951 | Splice Isoform 1 of Coagulation factor XI | 1 | 5.5 |
| IPI00009793 | Complement component 1, r subcomponent-like variant (Fragment) | 2.5 | 7 | |
| IPI00010295 | P15169 | Carboxypeptidase N catalytic chain | 17.5 | 7 |
| IPI00018305 | P17936 | Insulin-like growth factor-binding protein 3 | 3 | 7 |
| IPI00021857 | P02656 | Apolipoprotein C-III | 16 | 81.5 |
| IPI00021891 | P02679 | Splice Isoform Gamma-B of Fibrinogen gamma chain | 20 | ND |
| IPI00022295 | P10720 | Platelet factor 4 variant | ND | 4 |
| IPI00022394 | P02747 | Complement C1q subcomponent subunit C | 21.5 | 12.5 |
| IPI00022443 | P02771 | Alpha-fetoprotein | 76.5 | 0.5 |
| IPI00022731 | P55056 | Apolipoprotein C-IV | 1 | 4.5 |
| IPI00023014 | P04275 | Von Willebrand factor | 33 | 9.5 |
| IPI00023673 | Q08380 | Galectin-3-binding protein | 21.5 | 6.5 |
| IPI00025864 | P06276 | Cholinesterase | 1.5 | 6.5 |
| IPI00027410 | P40197 | Platelet glycoprotein V | ND | 4.5 |
| IPI00027507 | Q02985 | Complement factor H-related protein 3 | 9 | ND |
| IPI00029193 | Q04756 | Hepatocyte growth factor activator | 1 | 7.5 |
| IPI00029260 | P08571 | Monocyte differentiation antigen CD14 | 16 | 7 |
| IPI00029717 | P02671 | Splice Isoform 2 of Fibrinogen alpha chain | 19.5 | 4 |
| IPI00030739 | O95445 | Apolipoprotein M | 5 | 11.5 |
| IPI00031411 | Q14517 | Cadherin-related tumor suppressor homolog | ND | 4.5 |
| IPI00064667 | Q96KN2 | Glutamate carboxypeptidase-like protein 2 | 3 | 11 |
| IPI00186903 | O14791 | Splice Isoform 2 of Apolipoprotein-L1 | 6 | 15.5 |
| IPI00215894 | P01042 | Splice Isoform LMW of Kininogen-1 | 47 | 112 |
| IPI00218413 | P43251 | biotinidase | 5 | 11.5 |
| IPI00291262 | P10909 | Clusterin | 38 | 82 |
| IPI00296099 | P07996 | Thrombospondin-1 | 7.5 | 23.5 |
| IPI00296176 | P00740 | Coagulation factor IX | 4.5 | 12 |
| IPI00298497 | P02675 | Fibrinogen beta chain | 10.5 | ND |
| IPI00298860 | P02788 | Growth-inhibiting protein 12 | 2 | 10.5 |
| IPI00298971 | P04004 | Vitronectin | 56.5 | 115 |
| IPI00299307 | P48740 | Complement-activating component of Ra-reactive factor | 2.5 | 5.5 |
| IPI00299503 | P80108 | Phosphatidylinositol-glycan-specific phospholipase D 1 | 6.5 | 14 |
| IPI00385264 | P04220 | Ig mu heavy chain disease protein | 194.5 | 94.5 |
| IPI00385509 | Q15127 | Phosphatidylinositol-glycan-specific phospholipase D 2 | ND | 9.5 |
| IPI00478809 | P12259 | Coagulation factor V | 2.5 | 8 |
| IPI00639937 | P00751 | Complement factor B | 137.5 | 68 |
a Proteins were confirmed as glycoproteins by identification of N-glycopeptides in this study and previous large-scale studies of human liver N-glycoproteome (21).
b The spectral counting was the average of spectral counts of three replicated LC-MS/MS runs.
c ND denotes no mass spectra matched to this protein in this sample set.
Confirmation of Differential Level of IGFBP-3, S90K and TSP-1 in HCC Serum by ELISA
Apart from the alpha-fetoprotein, which is the only clinically used biomarker for HCC right now, other glycoproteins with substantial expression changes were also found, e.g. insulin-like growth factor binding protein 3 (IGFBP-3), galectin-3 binding protein (S90K,or Mac-2 BP) and thrombospondin 1(TSP-1). To confirm their differential expression of galectin-3 binding protein, IGFBP-3 and TSP-1 other cohorts of serum samples from 58 HCC patients and 30 normal control subjects were analyzed by commercial ELISA kit. Serum levels of galectin-3 binding protein were significantly elevated in HCC patients (median = 4.49 μg/ml), in comparison to healthy individuals (median = 3.43 μg/ml) (p < 0.0001). The IGFBP-3 serum levels in HCC were significantly lower (median = 0.5526 μg/ml) than those in healthy controls (median = 2.143 μg/ml) (p = 0.0085). The TSP-1 serum levels in HCC were also lower (median = 0.7092 μg/ml) than those in healthy controls (median = 1.058 μg/ml) (p < 0.0001)(Fig. 6). All the results of ELISA were in accordance with that achieved with label-free quantitative glycoproteomics approach, which suggested that the developed approach was accurate and reliable in differential glycoprotein identification. In a preliminary assessment of these glycoproteins for the discrimination between HCC patients and healthy people, the diagnostic sensitivity was calculated, which was: galectin-3 binding protein - 72%; IGFBP-3 - 98%; TSP-1 - 78% (Fig. 7).
Fig. 6.
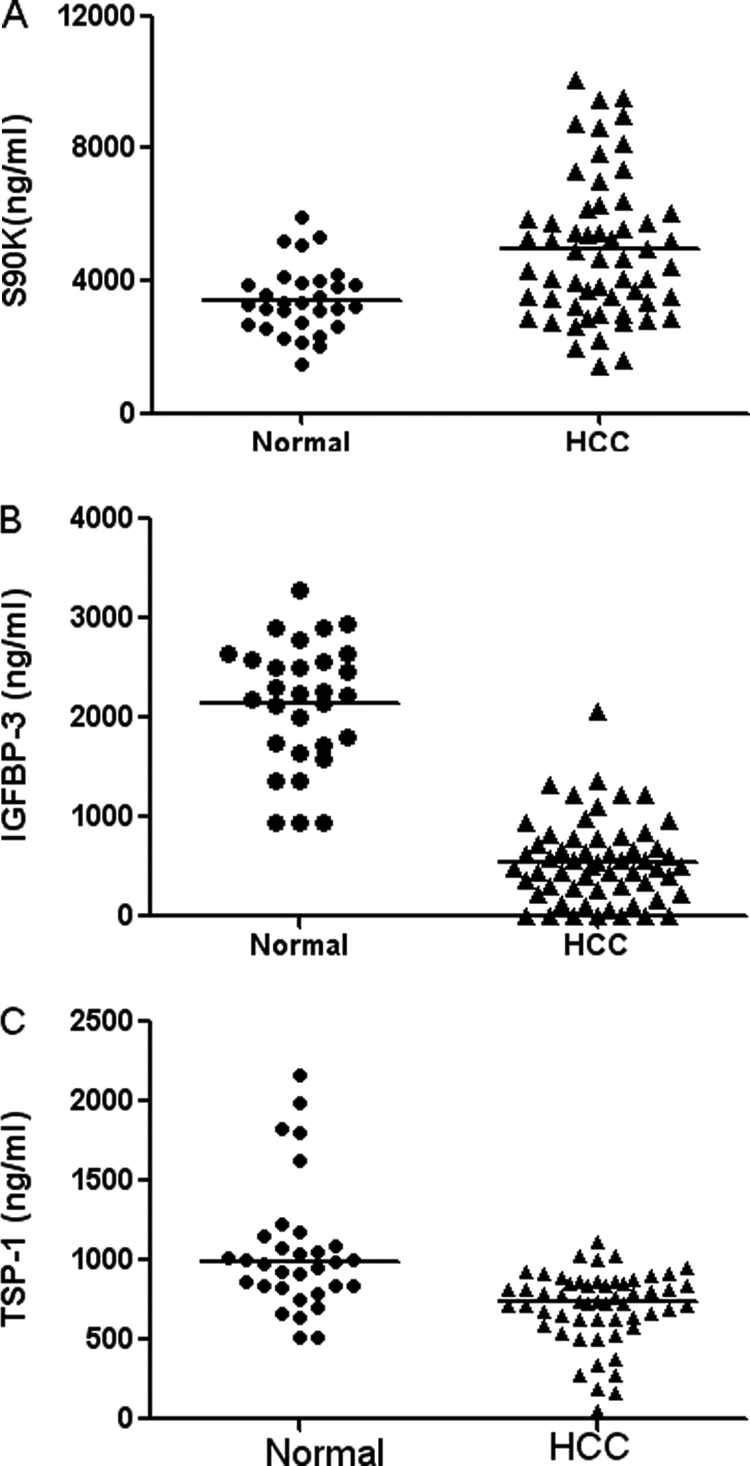
Scatter plots demonstrating the distribution of serum S90K, IGFBP-3, and TSP-1concentrations in 58 serum samples of hepatocellular carcinoma cases and 30 of normal control. Serum levels of S90K (A), IGFBP-3 (B), and TSP-1 (C) were assayed using ELISA tests. Unpaired test with Welch's correction test demonstrates that serum S90K level is significantly higher whereas TSP-1 and IGFBP-3 are significantly lower in HCC patients than normal controls.
Fig. 7.
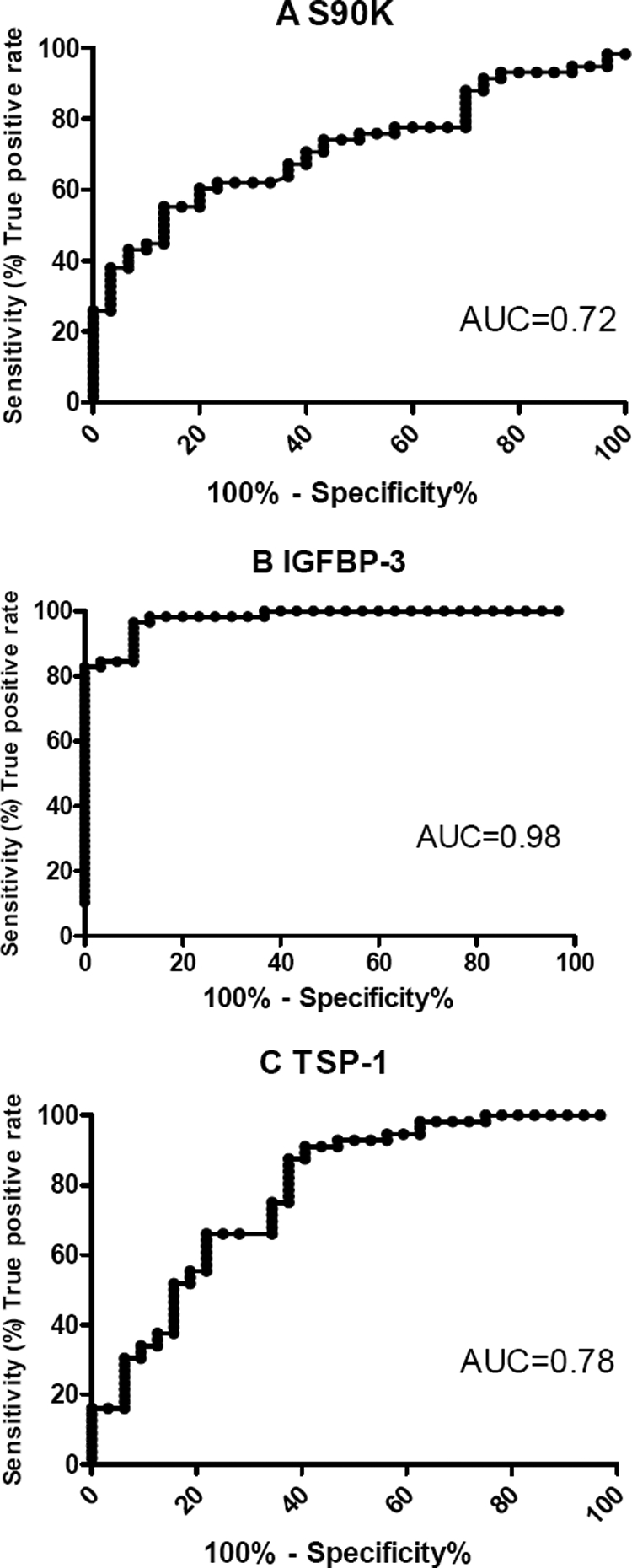
Specificity assessment of serum levels of S90K'IGFBP-3′ and TSP-1 achieved by ELISA for the discrimination of HCC patients from healthy people. ROC curves and the corresponding area under the curve (AUC) are shown for (A) S90K, (B) IGFBP-3, and (C) TSP-1.
DISCUSSION
Although great technical achievements have been made in the past few years to measure the global protein expression with mass spectrometry based quantitative proteomics approach, there are still some unresolved issues for the high-throughput and accurate quantification of proteins with modifications like glycosylation. Glycoprotein quantification with lectin-based approach was very popular in the discovery of serum glycoprotein biomarkers as lectins can recognize certain types of glycan linkages and glycan structure, which may reveal distinct glycosylation pattern changes in human cancers. Unlike lectin-based approach, hydrazide chemistry based approach captures oxidized glycoproteins onto solid beads via covalent linkage. Although the glycan information was lost during periodate oxidation, the efficiency and selectivity for extraction of glycoproteins from complex samples like serum could be increased. In hydrazide chemistry approach, glycoproteins were traditionally identified and quantified with the deglycosylated peptides which were released by PNGase F. It was reported in recent studies that the intensity of the glycopeptides in mass spectrometry were not consistent with the protein abundance, which indicates the quantification of glycopeptides was only a relative reflection of the site specific N-glycosylation occupancy. Generally, most current quantitative glycoproteomics approaches quantify the glycosylation change but not the protein expression. Since the most assay methods in disease diagnosis are the measure of protein expressions, effective methods to measure the actual amount of glycoprotein in clinical samples like serum are needed. We developed this glycoprotein captured based label-free approach which could quantify serum glycoproteins in accurate and high-throughput way. The performance of the developed approach has been demonstrated with control experiments that described in the results section. As most secreted glycoproteins should be glycosylated in the ER and Golgi apparatus and then secreted through the plasma membrane to the blood, it could be assumed that the total amount of the protein in the serum was equal to the glycosylated form of the protein. Because of the high efficiency of the developed approach, which captures both N-linked and O-linked glycoproteins at protein level, this method actually quantifies the protein expression change rather than glycosylation change. In this study, the quantitative results of most identified differential glycoproteins were in accordance with that reported with traditional methods. Moreover, the relative changes of three glycoproteins, IGFBP-3, galectin-3 binding protein and TSP-1, were validated with ELISA assay which were in accordance with that achieved with the quantitative proteomics with our approach. Though these differential glycoproteins were identified, there are still overlaps between HCC and normal samples. As most of the found marker candidates have the problem of overlap, the specificity could be increased by the combination of multiple proteins. Another possible approach to increase the specificity of cancer diagnosis was the combination both the changes of protein expression and glycosylation pattern. For example, the specificity of AFP could be increased using lectins like Lens culinaris agglutinin to detect fucosyaltion change(34). As a result, glycosylation pattern of these identified differential glycoproteins could be analyzed to see if the overlap could be reduced by lectin-based approaches.
Estimation from the year 2000 indicates ∼564,000 new cases of HCC occurs worldwide per year. HCC remains the fifth most common malignancy in men and the eighth in women worldwide (35). Early detection of HCC is of great importance to improve the prognosis as therapeutic options increase. AFP is currently the only clinical marker which has been widely used for serological diagnosis of human HCC (36). However, its specificity is low, especially for chronic liver diseases and cirrhosis, and its sensitivity shows only 60 to 80% at cutoff value of 20 μg/L in serum (37). As it is urgent to find another noninvasive, reliable method for detecting HCC as early as possible, the application of high-throughput screening technologies like proteomics is becoming the trend in biomarker discovery.
In this study, the developed method was applied in the differential glycoprotein screening between normal people and HCC patients, the three validated differential glycoproteins have already been found in association with the generation and development of human cancer. The IGF signaling pathway is regulated by a six-member family of IGF binding proteins (IGFBP-1 to 6). Among them, IGFBP-3 has the highest affinity for IGF-1 and the most abundant IGFBP family member in the circulation. IGFs are synthesized mainly in the liver and also in peripheral tissues (including the prostate). IGFBP-3 could sequester the ligand, preventing IGF-1-induced IGF-R1 autophosphorylation and signaling. Furthermore, IGFBP-3 has been shown to exhibit effects on proliferation, migration, and apoptosis that are independent of its involvement in IGF signaling (38–39). IGFBP-3 was found to be inversely associated with benign prostate hyperplasia risk, although findings were statistically significant only for men with severe symptoms (40). Epidemiologic studies have also suggested that high circulating levels of IGF-I and low levels of IGFBP-3 are associated with increased risk of premenopausal breast cancer (41–42) and lung cancer (43). In liver cancer, only one report showed that in 12 human HCC, nine showed reduced expression of the IGFBP-3 (44).
Galectin-3-binding protein (lectin, galactoside-binding soluble, 3-binding protein), also named Mac-2BP or S90K, is a large oligomeric glycoprotein first identified as a tumor-associated antigen in breast cancer (45). Mac-2BP binds to multiple proteins and molecules mediating cell-matrix and cell-cell adhesions such as collagens, fibronectin and nidogen, which are critical during tumor cell invasion and migration (46).In immune system. Mac-2BP stimulates activity of natural killer cells and lymphokine-activated killer cells partly mediated through the induction of IL-2. In noninflammatory cells, Mac-2BP was identified as an IL-6 stimulatory factor in BMSC and a critical mediator in bone invasion in metastatic neuroblastoma (47). Mac-2BP is elevated and correlates with prognosis in patients with a variety of different cancers, including breast, nasopharyngeal, and lung cancer (48–50). More recently, in defining a molecular signature of potential prognostic value of Ewing's sarcoma, Mac-2BP was identified and confirmed as the most important predictor of event-free survival and overall survival. Forced expression of Mac-2BP showed reduced ability to form colonies and to migrate as well as higher cellular aggregation. In addition, cells with higher expression of Mac-2BP displayed stronger adhesion to vitronectin but reduced adhesion to laminin and collagen IV, the 2 major components of basal lamina, implying that through favoring cell-cell aggregation and inhibiting cell adhesion to basal lamina, Mac-2BP maybe keep EWS cells in fluid blood circulation, where they are more prone to die, and prevent EWS cell adhesion to endothelium and extravasation (51). In liver diseases, serum 90K/Mac-2BP was found as an independent predictor of disease severity during HCV infection (52). In another report, serum Mac-2BP was evaluated in 11 chronic active hepatitis, 48 liver cirrhosis and 36 hepatocellular carcinoma. At a cut-off point of 14 μg/ml (100% of specificity in 50 controls), increasing positivity was observed from chronic active hepatitisto cirrhosis and then to HCC (27%, 50 and 78%, respectively) (53).
Thrombospondin-1 (TSP-1) is a large matricellular glycoprotein secreted by many cell types. It is a component of the extracellular matrix during active and subacute processes. TSP-1 has various functions in different human diseases like hemostasis, angiogenesis, tumor cell biology, wound healing, and arterial remodeling (54). It has been found that TSP-1 is an endogenous inhibitor of tumor growth and angiogenesis through direct effects on endothelial cell migration and survival and through indirect effects on growth factor mobilization. TSP-1 that is present in the tumor microenvironment also acts to suppress tumor cell growth through activation of transforming growth factor β in those tumor cells that are responsive to TGF-β (55). TSP-1 has been shown to be highly expressed in human malignant tissues and present in higher than normal levels in the plasma of cancer patients (56), like beast, lung, and colorectal carcinoma (57), which is in contrast with our result, suggesting a possible unique figure of hepatocellular carcinoma.
In summary, an effective approach in the serum glycoprotein quantification was developed and applied in the biomarker discovery for HCC diagnosis. Thirty-eight glycoproteins with abundance changes were identified, including AFP, the clinical marker for HCC. The concentrations changes of three glycoproteins, i.e. insulin-like growth factor-binding protein 3, galectin-3-binding protein, and thrombospondin-1, were confirmed by ELISA with another samples collection and demonstrated good specificity for the discrimination between HCC patients and healthy people. However, it should be noted that the aim of this study was the establishment of the method for high-throughput screening of differential glycoproteins in human serum and the developed method has been proved to be accurate and reliable. The clinical applicability of these proteins could be examined by further validation with samples from liver disease like cirrhosis and early stages of HCC. As this effective approach was developed, it can also be applied in the biomarker discovery of other diseases.
* Financial supports from the National Natural Sciences Foundation of China (20735004, 30921006, 90713032, 06MA154) and the Creative Research Group Project by NSFC (No.21021004), the China State Key Basic Research Program Grant (2007CB914102), the China High Technology Research Program Grant (2006AA02A309, 2006AA02A249), National Key Special Program on Infection diseases (2008ZX10002-017, 2008ZX10002-020), the Analytical Method Innovation Program of MOST (2009IM031800, 2010IM030500), the Knowledge Innovation program of CAS (KJCX2.YW.HO9, KSCX2-YW-R-079) and the Knowledge Innovation program of DICP to H. F. Zou are gratefully acknowledged.
1 The abbreviations used are:
- LC-MS
- Liquid chromatography mass spectrometry
- LC-MS/MS
- Liquid chromatography tandem mass spectrometry
- PNGase F
- Peptide N Glycosidase F
- AFP
- Alpha-fetoprotein
- HCC
- Hepatocellular carcinoma
- S90K
- or Mac-2 BP, Galectin-3 binding protein
- IGFBP-3
- Insulin Growth Factor Binding Protein 3
- TSP-1
- Thrombospondin-1.
REFERENCES
- 1. Ong S. E., Mann M. (2005) Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 [DOI] [PubMed] [Google Scholar]
- 2. Gygi S. P., Rist B., Gerber S. A., Turecek F., Gelb M. H., Aebersold R. (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotech. 17, 994–999 [DOI] [PubMed] [Google Scholar]
- 3. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 4. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 5. Bondarenko P. V., Chelius D., Shaler T. A. (2002) Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed-phase liquid chromatography-tandem mass spectrometry. Anal. Chem. 74, 4741–4749 [DOI] [PubMed] [Google Scholar]
- 6. Liu H., Sadygov R. G., Yates J. R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 7. Block T. M., Comunale M. A., Lowman M., Steel L. F., Romano P. R., Fimmel C., Tennant B. C., London W. T., Evans A. A., Blumberg B. S., Dwek R. A., Mattu T. S., Mehta A. S. (2005) Use of targeted glycoproteomics to identify serum glycoproteins that correlate with liver cancer in woodchucks and humans. Proc. Natl. Acad. Sci. U.S.A. 102, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anderson N. L., Anderson N. G. (2002) The human plasma proteome - History, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 9. Gahmberg C. G., Tolvanen M. (1996) Why mammalian cell surface proteins are glycoproteins. Trends Biochem. Sci. 21, 308–311 [PubMed] [Google Scholar]
- 10. Yang Z., Hancock W. S. (2004) Approach to the comprehensive analysis of glycoproteins isolated from human serum using a multi-lectin affinity column. J. Chromatogr. A 1053, 79–88 [PubMed] [Google Scholar]
- 11. Wang Y., Ao X., Vuong H., Konanur M., Miller F. R., Goodison S., Lubman D. M. (2008) Membrane glycoproteins associated with breast tumor cell progression identified by a lectin affinity approach. J. Proteome Res. 7, 4313–4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Plavina T., Wakshull E., Hancock W. S., Hincapie M. (2007) Combination of abundant protein depletion and multi-lectin affinity chromatography (M-LAC) for plasma protein biomarker discovery. J. Proteome Res. 6, 662–671 [DOI] [PubMed] [Google Scholar]
- 13. Gong Y., Li X., Yang B., Ying W., Li D., Zhang Y., Dai S., Cai Y., Wang J., He F., Qian X. (2006) Different immunoaffinity fractionation strategies to characterize the human plasma proteome. J. Proteome Res. 5, 1379–1387 [DOI] [PubMed] [Google Scholar]
- 14. Zhang H., Li X. J., Martin D. B., Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 15. Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 16. Schiess R., Mueller L. N., Schmidt A., Mueller M., Wollscheid B., Aebersold R. (2009) Analysis of Cell Surface Proteome Changes via Label-free, Quantitative Mass Spectrometry. Mol. Cell. Proteomics 8, 624–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 18. Whelan S. A., Lu M., He J. B., Yan W. H., Saxton R. E., Faull K. F., Whitelegge J. P., Chang H. R. (2009) Mass spectrometry (LC-MS/MS) site-mapping of N-glycosylated membrane proteins for breast cancer biomarkers, J Proteome Res. 8, 4151–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou Y., Aebersold R., Zhang H. (2007) Isolation of N-linked glycopeptides from plasma. Anal. Chem. 79, 5826–5837 [DOI] [PubMed] [Google Scholar]
- 20. Wang F., Dong J., Jiang X., Ye M., Zou H. (2007) Capillary trap column with strong cation-exchange monolith for automated shotgun proteome analysis. Anal. Chem. 79, 6599–6606 [DOI] [PubMed] [Google Scholar]
- 21. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data - The protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 22. Chen R., Jiang X., Sun D., Han G., Wang F., Ye M., Wang L., Zou H. (2009) Glycoproteomics Analysis of Human Liver Tissue by Combination of Multiple Enzyme Digestion and Hydrazide Chemistry. J. Proteome Res. 8, 651–661 [DOI] [PubMed] [Google Scholar]
- 23. Sun B., Ranish J. A., Utleg A. G., White J. T., Yan X., Lin B., Hood L. (2007) Shotgun glycopeptide capture approach coupled with mass Spectrometry for comprehensive glycoproteomics. Mol. Cell. Proteomics 6, 141–149 [DOI] [PubMed] [Google Scholar]
- 24. McDonald C. A., Yang J. Y., Marathe V., Yen T. Y., Macher B. A. (2009) Combining Results from Lectin Affinity Chromatography and Glycocapture Approaches Substantially Improves the Coverage of the Glycoproteome. Mol. Cell. Proteomics 8, 287–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang G., Wu W. W., Zeng W., Chou C. L., Shen R. F. (2006) Label-free protein quantification using LC-coupled ion trap or FT mass spectrometry: Reproducibility, linearity, and application with complex proteomes. J. Proteome Res. 5, 1214–1223 [DOI] [PubMed] [Google Scholar]
- 26. Zybailov B., Coleman M. K., Florens L., Washburn M. P. (2005) Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal. Chem. 77, 6218–6224 [DOI] [PubMed] [Google Scholar]
- 27. Burnett W. (1960) An assessment of the value of serum cholinesterase as a liver function test and in the diagnosis of jaundice. Gut 1, 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKeownLongo P. J., Panetti T. S. (1996) Structure and function of vitronectin. Trends Glycosci. Glyc. 8, 327–340 [Google Scholar]
- 29. Inuzuka S., Ueno T., Torimura T., Tamaki S., Sakata R., Sata M., Yoshida H., Tanikawa K. (1992) Vitronectin in liver disorders - Biomedical and immunohistochemical studies. Hepatology 15, 629–636 [DOI] [PubMed] [Google Scholar]
- 30. Edwards S., Lalor P. F., Tuncer C., Adams D. H. (2006) Vitronectin in human hepatic tumours contributes to the recruitment of lymphocytes in an alpha v beta 3-independent manner. Brit. J. Cancer 95, 1545–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Paradis V., Degos F., Dargère D., Pham N., Belghiti J., Degott C., Janeau J. L., Bezeaud A., Delforge D., Cubizolles M., Laurendeau I., Bedossa P. (2005) Identification of a new marker of hepatocellular carcinoma by serum protein profiling of patients with chronic liver diseases. Hepatology 41, 40–47 [DOI] [PubMed] [Google Scholar]
- 32. Podor T. J., Campbell S., Chindemi P., Foulon D. M., Farrell D. H., Walton P. D., Weitz J. I., Peterson C. B. (2002) Incorporation of vitronectin into fibrin clots - Evidence for a binding interaction between vitronectin and gamma A/gamma ' fibrinogen. J. Biol. Chem. 277, 7520–7528 [DOI] [PubMed] [Google Scholar]
- 33. Comunale M. A., Wang M., Hafner J., Krakover J., Rodemich L., Kopenhaver B., Long R. E., Junaidi O., Bisceglie A. M., Block T. M., Mehta A. S. (2009) Identification and Development of Fucosylated Glycoproteins as Biomarkers of Primary Hepatocellular Carcinoma. J. Proteome Res. 8, 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taketa K., Sekiya C., Namiki M., Akamatsu K., Ohta Y., Endo Y., Kosaka K. (1990) Lectin-reactive Profiles of Alpha-fetoprotein Characterizing Hepatocellular-carcinoma and Related Conditions. Gastroenterology 99, 508–518 [DOI] [PubMed] [Google Scholar]
- 35. Bosch F. X., Ribes J., Diaz M., Cleries R. (2004) Primary liver cancer: Worldwide incidence and trends. Gastroenterology 127, S5–S16 [DOI] [PubMed] [Google Scholar]
- 36. Chen D. S., Sung J. L., Sheu J. C., Lai M. Y., How S. W., Hsu H. C., Lee C. S., Wei T. C. (1984) Serum alpha-fetoprotein in the early stage of human hepatocellular carcinoma. Gastroenterology 86, 1404–1409 [PubMed] [Google Scholar]
- 37. Buamah P. K., Gibb I., Bates G., Ward A. M. (1984) Serum alpha-fetoprotein heteogeneity as a means of differentiatting between primary heaptocellular carcinoma and heaptic secondaries. Clin. Chim. Acta 139, 313–316 [DOI] [PubMed] [Google Scholar]
- 38. Liu B., Lee K. W., Anzo M., Zhang B., Zi X., Tao Y., Shiry L., Pollak M., Lin S., Cohen P. (2007) Insulin-like growth factor-binding protein-3 inhibition of prostate cancer growth involves suppression of angiogenesis. Oncogene 26, 1811–1819 [DOI] [PubMed] [Google Scholar]
- 39. Zhu W., Shiojima I., Ito Y., Li Z., Ikeda H., Yoshida M., Naito A. T., Nishi J., Ueno H., Umezawa A., Minamino T., Nagai T., Kikuchi A., Asashima M., Komuro I. (2008) IGFBP-4 is an inhibitor of canonical Wnt signalling required for cardiogenesis. Nature 454, 345–349 [DOI] [PubMed] [Google Scholar]
- 40. Neuhouser M. L., Schenk J., Song Y. J., Tangen C. M., Goodman P. J., Pollak M., Penson D. F., Thompson I. M., Kristal A. R. (2008) Insulin-like growth factor-I, insulin-like growth factor binding protein-3 and risk of benign prostate hyperplasia in the prostate cancer prevention trial. Prostate 68, 1477–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fletcher O., Gibson L., Johnson N., Altmann D. R., Holly J. M. P., Ashworth A., Peto J., Silva I. D. (2005) Polymorphisms and circulating levels in the insulin-like growth factor system and risk of breast cancer: A systematic review. Cancer Epidem. Biomar. 14, 2–19 [PubMed] [Google Scholar]
- 42. Allen N. E., Roddam A. W., Allen D. S., Fentiman I. S., Silva I. D., Peto J., Holly J. M. P., Key T. J. (2005) A prospective study of serum insulin-like growth factor-I (IGF-I), IGF-II, IGF-binding protein-3 and breast cancer risk. Brit. J. Cancer 92, 1283–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. London S. J., Yuan J. M., Travlos G. S., Gao Y. T., Wilson R. E., Ross R. K., Yu M. C. (2002) Insulin-like growth factor I, IGF-binding protein 3, and lung cancer risk in a prospective study of men in China. J. Nat.l Cancer Inst. 94, 749–754 [DOI] [PubMed] [Google Scholar]
- 44. Hanafusa T., Yumoto Y., Nouso K., Nakatsukasa H., Onishi T., Fujikawa T., Taniyama M., Nakamura S., Uemura M., Takuma Y., Yumoto E., Higashi T., Tsuji T. (2002) Reduced expression of insulin-like growth factor binding protein-3 and its promoter hypermethylation in human hepatocellular carcinoma. Cancer Lett. 176, 149–158 [DOI] [PubMed] [Google Scholar]
- 45. Iacobelli S., Arnò E., D'Orazio A., Coletti G. (1986) Detection of antigens recognized by a novel monoclonal-antibody in tissue and serum from patients with breast cancer. Cancer Res. 46, 3005–3010 [PubMed] [Google Scholar]
- 46. Inohara H., Akahani S., Koths K., Raz A. (1996) Interactions between galectin-3 and Mac-2-binding protein mediate cell-cell adhesion. Cancer Res. 56, 4530–4534 [PubMed] [Google Scholar]
- 47. Fukaya Y., Shimada H., Wang L. C., Zandi E., DeClerck Y. A. (2008) Identification of galectin-3-binding protein as a factor secreted by tumor cells that stimulates interleukin-6 expression in the bone marrow stroma. J. Biol. Chem. 283, 18573–18581 [DOI] [PubMed] [Google Scholar]
- 48. Iacobelli S., Sismondi P., Giai M., Degidio M., Tinari N., Amatetti C., Distefano P., Natoli C. (1994) Prognostic value of a novel circulation serum 90K antigen in breast cancer. Brit.J. Cancer 69, 172–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marchetti A., Tinari N., Buttitta F., Chella A., Angeletti C. A., Sacco R., Mucilli F., Ullrich A., Iacobelli S. (2002) Expression of 90K (Mac-2 BP) correlates with distant metastasis and predicts survival in stage I nonsmall cell lung cancer patients. Cancer Res. 62, 2535–2539 [PubMed] [Google Scholar]
- 50. Zeimet A. G., Natoli C., Herold M., Fuchs D., Windbichler G., Daxenbichler G., Iacobelli S., Dapunt O., Marth C. (1996) Circulating immunostimulatory protein 90K and soluble interleukin-2-receptor in human ovarian cancer. Int. J. Cancer 68, 34–38 [DOI] [PubMed] [Google Scholar]
- 51. Zambelli D., Zuntini M., Nardi F., Manara M. C., Serra M., Landuzzi L., Lollini P. L., Ferrari S., Alberghini M., Llombart-Bosch A., Piccolo E., Iacobelli S., Picci P., Scotlandi K. (2010) Biological indicators of prognosis in Ewing's sarcoma: an emerging role for lectin galactoside-binding soluble 3 binding protein (LGALS3BP). Inter. J. Cancer 126, 41–52 [DOI] [PubMed] [Google Scholar]
- 52. Kittl E. M., Hofmann J., Hartmann G., Sebesta C., Beer F., Bauer K., Huber K. R. (2000) Serum protein 90K/Mac-2BP is an independent predictor of disease severity during hepatitis C virus infection. Clin. Chem. Lab. Med. 38, 205–208 [DOI] [PubMed] [Google Scholar]
- 53. Correale M., Giannuzzi V., Iacovazzi P. A., Valenza M. A., Lanzillotta S., Abbate I., Quaranta M., Caruso M. L., Elba S., Manghisi O. G. (1999) Serum 90K/MAC-2BP glycoprotein levels in hepatocellular carcinoma and cirrhosis. Anticancer Res. 19, 3469–3472 [PubMed] [Google Scholar]
- 54. Esemuede N., Lee T., Pierre-Paul D., Sumpio B. E., Gahtan V. (2004) The role of thrombospondin-1 in human disease. J. Surg.Res. 122, 135–142 [DOI] [PubMed] [Google Scholar]
- 55. Murphy-Ullrich J. E., Poczatek M. (2000) Activation of latent TGF-beta by thrombospondin-1: Mechanisms and physiology. Cytokine Growth Factor Rev. 11, 59–69 [DOI] [PubMed] [Google Scholar]
- 56. Tuszynski G. P., Smith M., Rothman V. L., Capuzzi D. M., Joseph R. R., Katz J., Besa E. C., Treat J., Switalska H. I. (1992) Thrombospondin levels in patients with malignancy. Thromb. Haemostasis 67, 607–611 [PubMed] [Google Scholar]
- 57. Yamashita Y., Kurohiji T., Tuszynski G. P., Sakai T., Shirakusa T. (1998) Plasma thrombospondin levelsin patients with colorectal carcinoma. Cancer 82, 632–638 [DOI] [PubMed] [Google Scholar]