

Abstract
The expression level of proteins governing the electrical excitability of and conduction within ventricular myocardium are known to vary as a function of distance through the heart wall. The expression patterns of a subset of these proteins are altered in disease. Precise measurement of such patterns is therefore essential to understanding structure-function relationships within the heart in health and disease. Here, we report a new experimental approach using reverse-phase protein microarrays to map the left ventricular transmural proteome. This approach can yield submillimeter spatial resolution, and when coupled with the method of array microenvironment normalization, reduces nonbiological components of variability to ∼10% of overall study variability. In addition, the experimental design provides sufficient statistical power to detect small, yet potentially biologically significant expression changes on the order of 1.1-fold. The usefulness of this technique is demonstrated by mapping the transmural expression of Serca2a in the left ventricle of 12 canine hearts, each in one of three states: normal, dyssynchronous heart failure, and dyssynchronous heart failure followed by cardiac resynchronization therapy. We confirm the existence of a 40% transmural gradient (epi>endo) of Serca2a, and demonstrate the ability of this technique to yield highly significant transmural expression differences within each individual heart.
The cardiac action potential (AP)1 is a rapid rise and slow decrease of cardiac myocyte trans-membrane electrical potential in response to changes in membrane conductance for specific ions. The resulting electrical wave propagates from the right and left atria to the ventricles, initiating the synchronized release of calcium (Ca2+) and subsequent contraction of the heart muscle. Isolated cardiac ventricular myocytes have been shown to exhibit disparate AP characteristics (shape and duration) as a function of transmural location (depth within the ventricle from surface to interior) (1, 2). These differences in AP properties are now known to result from transmural differences in the expression and/or function of key proteins (ion channels, transporters, and signaling) regulating the shape of the cardiac AP (1–9). Therefore, understanding the process of AP generation and propagation throughout the myocardium requires the measurement of transmural differences in protein expression as an initial step to understanding the extent of differences throughout the entire heart.
Currently, measurements of membrane ion channel and transporter expression are made using Western blot or immunofluorescence methods. Such measurements are plagued by low transmural spatial-resolution, low throughput (∼10–15 samples per gel; one tissue sample per immunofluorescent measurement), and high variability (7, 10–13). One estimate of typical across-gel coefficient of variance for Western blots is ∼25%(14). A power calculation using this coefficient of variance indicates that ∼100 replicates per sample would be needed to achieve a power of 0.80 for identifying 10% shifts in expression. Further, the low statistical power of these assays requires that measurements from multiple experimental preparations be pooled in an effort to improve statistical significance. However, the possibility of considerable interindividual differences of transmural protein expression might limit the ability to identify important patterns of transmural expression differences when pooling data across preparations. The ability to measure protein expression with greater statistical power of detection and increased spatial resolution would make it possible to measure transmural expression differences within individual hearts, capturing biologically relevant information on individual variability.
In previous work, we have shown that reverse-phase protein microarrays (RPMAs) can be used to detect small expression changes with high statistical significance(15). RPMAs immobilize lysates from many samples (∼2500) onto a single nitrocellulose surface via robotic printing (see Fig 1C). The relative amount of target protein within each spotted lysate on the array may be measured simultaneously by incubating the entire array with a well-validated antibody (16, 17). Many arrays can be printed from a single set of sample lysates and each probed with a unique antibody of interest. When RPMAs are coupled with the recently developed array microenvironment normalization (AMN) technique (an approach to both array design and data analysis), technical variability can be reduced by up to 70%, enabling consistent detection of expression changes on the order of 1.1-fold.
Fig. 1.

Experimental overview. A, A depiction of the acquisition of an anterior transmural wedge from a left ventricle (z axis represents the transmural dimension), which is immediately snap frozen. B, An individual transmural wedge being sectioned at 1-mm resolution (dotted line represents the sectioning plane x-y), at −24°C in a cryostat. Sample tissue from each disparate 1-mm region is lysed to create a whole-cell lysate. All 1-mm whole-cell lysates are then robotically printed on a RPMA in unique spots, as demonstrated by the arrows. C, A sypro ruby total protein stain of a single HRSH array is shown (the left side of the array contains samples printed at 600 μg/ml, and the right side at 150 μg/ml). The array is magnified to display the AMN format of RPMA printing. D, The structure of replicates printed on the RPMAs for each 1 mm transmural depth of wedge tissue. In total, sixteen replicates are printed that consist of independent sectioning and lysing replicates (reps) as well as repeated spotting on the array.
Here, we demonstrate how RPMAs may be used to measure the transmural expression of a Ca+2 pump located in the sarcoplasmic reticular membrane known as the serca2a pump at millimeter spatial resolution. The expression level of this transporter is known to be an important regulator of AP duration in cardiac myocytes (18). Serca2a expression is also known to vary transmurally, making it a good choice with which to validate the RPMA technique (7, 10).
EXPERIMENTAL PROCEDURES
Detailed methods can be found in the online supplement; here we describe the general methodology in brevity. Fig. 1 displays an overview of the experimental process for reference.
Tissue Collection and Preparation
Left ventricle tissue was collected from mongrel dogs in one of three states: normal, dyssynchronized heart failure (DHF), and DHF with cardiac resynchronization therapy (CRT). All protocols followed US Department of Agriculture and National Institutes of Health guidelines and were approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions. A left bundle-branch ablation by radiofrequency and subsequent right atrial pacing for 6 weeks at 200 bpm was used to induce DHF (8). For CRT, the DHF protocol was stopped after 3 weeks, at which time biventricular pacing was carried out for 3 weeks at the same pacing rate of 200 bpm (19). Left ventricle (LV) tissue was obtained from a total of twelve animals in this study, four from each of the three states (normal, DHF, and CRT).
A wedge of tissue spanning from the epicardial (epi) surface of the LV through to the endocardial (endo) surface is known as a transmural wedge. Wedges midway between the apex and base of the LV at independent anterior and lateral sites were removed from each heart (see Figs. 1A and 1B). Each of the wedges was further split into two ∼10 × 5×15 mm wedges (to be used for sectioning replicates; see Fig. 1D), snap frozen in liquid nitrogen, and embedded in optimal cutting temperature compound (OCT). The embedded transmural wedge of tissue was mounted in a cryostat (Leica CM1850 UV) maintained at –24°C, with the sectioning plane (x-y axes of Fig. 1B) aligned perpendicular to the transmural dimension. For each millimeter of transmural depth sectioned, two 0.5 ml eppendorf tubes (at –24°C) were labeled for their transmural location and each filled with ∼6 mg of tissue. Two tubes per millimeter were collected so that the ensuing lysing procedure could be replicated to account for potential variability (see Fig. 1D). The tissue within each eppendorf tube was then lysed using a whole-cell SDS lysing buffer containing: 2% SDS, 62.5 mm Tris-HCl (pH 6.8), 10% glycerol, 40% TPER, 10% TCEP. At the end of the protocol each tube contained 300 μl of lysate at ∼3 mg/ml total protein concentration. Lysates were stored at –80°C until RPMA fabrication.
Array Printing and Staining
Standard protocols were used for the robotic printing and staining of RPMAs, details can be found in (16, 20, 21). Lysates were diluted in additional SDS buffer to normalize their total protein concentration before deposition on the array at ∼600 and 150 μg/ml (details in supplemental material online). Total protein normalized sample lysates at both concentrations were pipetted into a 384-well plate for robotic deposition in the AMN format(15) (Fig. 1C). The 384-well plates were loaded into an Aushon 2470 arrayer equipped with 185-μm pins (Aushon Biosystems, Billerica, MA), which deposited ∼10 nL of each sample onto glass-backed nitrocellulose slides (Schott, Germany). RPMAs were then stored in desiccant at –20°C until stained.
Because of space limitations on a single array, two types of RPMAs were produced: high resolution single heart (HRSH) arrays, and low resolution multi-heart (LRMH) arrays. The HRSH arrays are called “high resolution” because all of the lysates from each millimeter of sectioned depth were deposited on them, whereas “single heart” means that the deposits only include tissue lysates from a single heart. Independent HRSH arrays were thus fabricated for each of the 12 hearts in the study. In contrast, the LRMH arrays are deemed “low resolution” because they contain a subset of samples from three depths (epi, mid, and endo) of each wedge but are “multiheart” as they include all wedges from all 12 hearts on a single array. The epi, mid, and endo samples for the LRMH arrays were each selected from among the full resolution samples to be within the first 20%, 40–60%, and last 20% of transmural depth, respectively. For both HRSH and LRMH arrays, each sample from a specified millimeter of depth was printed in 16 independent spots on the array with replicate structure shown in Fig. 1D.
A total protein (sypro ruby) stain and an antibody stain were then carried out as described in Anderson et al. (15) for each RPMA. For primary antibody staining, arrays were incubated with mouse anti-Serca2a (monoclonal MA3–910, Affinity Bioreagents, Golden, CO) at room temperature, for 30 min. As a negative control, additional arrays were incubated with antibody diluent only and used to demonstrate the specificity of the process to the primary antibody.
On-array Controls
Every fabricated array included a standard panel of control samples that were used to ensure robust and reproducible results. All controls were generated in large quantities at the beginning of the study and were stored at –80°C for use as needed. The on-array controls include: the AMN control (Fig. 1), total protein controls, and specificity controls. Below is a brief description of the composition and purpose of each.
The AMN control is used for the normalization of all samples on every array. As such, the selection of an appropriate AMN control is essential. In our previous work, we found the performance of AMN to be exemplary when the AMN control had a similar relative amount of target protein to the samples of interest (15). To ensure this for all target proteins, we chose to use lysate pooled from three normal LVs using the described lysing technique (see online supplemental material for additional details).
Total protein controls were used to account for the fact that the amount of target protein within each spot on an RPMA will vary proportionally with its deposited total protein amount. The feature of interest in RPMA work is the amount of target protein per unit total protein, which is invariant to the original lysate total protein concentration. Total protein controls were created by serial twofold dilutions of the AMN control lysate. This yielded samples of identical target protein per unit total protein, but unique total protein concentrations ranging from 1 mg/ml down to 62.5 μg/ml.
RPMAs are a single-blot assay (sample protein is not separated before staining i.e. electrophoresis), thus primary antibody specificity is always checked via Western blotting to ensure the antibody is only binding to the molecular weight of interest (single band in image) (17). However, it is preferable to also control for specificity on the arrays themselves to ensure the signal is specific during the array staining. Using a recently developed fractionation protocol, we created three lysates (from the same tissue as the AMN control) enriched for cytosolic, myofilament, and membrane protein respectively (22). Depending on the antibody target, the fractions can be used for high and low controls. For instance, if the RPMA antibody stain targets an ion channel, the membrane fraction serves as a high-expression control, whereas the cytosolic and myofilament serve as low controls.
Array Signal Analysis
Imaging of the emitted RPMA fluorescent signal subsequent to antibody staining and/or sypro ruby staining, was carried out using the Vidar RevolutionTM 4550 laser scanner (Vidar Systems, Herndon, VA) at 10 μm resolution. Array tif images (both antibody and total protein stains) were uploaded into MicroVigene (Vigene Tech Inc., Carlisle, MA) for signal quantification. The background-subtracted mean intensity of each individual spot was output in a “.txt” file for analysis in R (23). Data taken directly from the “.txt” file with no additional processing is referred to as the “primary intensity” if a primary antibody was used, “negative intensity” if the primary was omitted, and “sypro ruby intensity” if it is the result of a sypro ruby stain. The primary intensity data is then total protein normalized (TPN) and finally AMN transformed to yield the final data for investigation.
TPN Transformation
The TPN transformation is the operation that removes the total protein dependence within the primary intensity to yield the feature of interest: the amount of target protein per unit total protein. In the TPN transformation, the total protein controls are used to fit a linear total protein dependence function (primary intensity versus sypro ruby intensity, see Fig. 2A), and the predicted intensity due to total protein dependence is then subtracted (on a log2-log2 scale) from the primary intensity (Fig. 2B) (see supplemental material).
Fig. 2.

Total protein normalization. A, The primary intensity plotted versus the sypro ruby intensity. The red line is the fit total protein dependence using the total protein controls, slope = 1.34. B, A plot of the TPN adjusted intensity versus the same sypro ruby intensity, total protein control slope = –0.0017.
AMN Transformation
Subsequent to the TPN transformation, AMN is employed to remove the technical variability as described in Anderson et al. (Equation 1)(15). This step is performed on data in the linear scale.
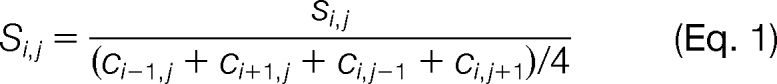 |
Where indices i,j are the array row and column positions respectively, s is the TPN sample signal on a linear scale, and c is the TPN transformed AMN control signal (see Fig. 1C). The full protocol for processing RPMA data was automated by creating R scripts (23).
Nested ANOVA
To demonstrate the ability of the TPN and AMN processing steps to reduce nonbiological variability, nested ANOVA was employed. Nested ANOVA is a common mathematical process for tracking sources of variability (24). Fig. 3 shows the nested group structure of all samples and replicates within the normal hearts only for ease of viewing and will be discussed in detail in results. The percent of overall study variability associated with each group was calculated using the sum of squares across each successive grouping versus that within each group. The remaining variability unaccounted for by any of the groups is called “within.” The groups of state, heart, location, and depth are sources of biological variability. “State” refers to Normal, DHF, or CRT; “heart” is a number identifier for each independent heart; “location” refers to the anterior or lateral LV location of the transmural wedge; and “depth” is the transmural depth of each wedge lysate. The groups of section replicate, lysate replicate, and within are all nonbiological sources of variability.
Fig. 3.

Nested ANOVA displaying the four normal hearts assayed using this technique. All replicates from all samples are displayed, grouped according to their origin as seen on the bottom of the graph. Unprocessed Serca2a primary intensity is on top, TPN adjusted intensity in the middle, and the final TPN and AMN adjusted intensity is shown on the bottom.
RESULTS
The first step of RPMA analysis is to ensure the quality of the data. The specificity of the antibody directed against the membrane protein Serca2a is verified in Fig. 2B by the expression difference of eightfold in the membrane fraction versus cytosolic and myofilament fractions. In addition, a Western blot performed showed only the band of interest for serca2a (see supplemental Fig. 1). The linear dynamic range is confirmed, because the total protein controls extend above and below the range of the samples themselves and exhibit linear behavior (Fig. 2A). Further, the ability to detect the decrease in expression in the cytosolic and myofilament fraction, and the increase in that of the membrane fraction confirms that the assay is capable of detecting both increases and decreases in sample expression per unit protein.
Fig. 2A shows that before the TPN transformation was carried out there was significant dependence on the total protein deposited within each spot that needed to be addressed. The study samples exhibited a Pearson correlation of 0.67 between primary intensity and sypro ruby intensity, while displaying a fit slope of 1.34 for the total protein controls. Fig. 2B demonstrates that TPN minimizes the potential effect of differences in spotted total protein among the samples. These data show that the correlation of the study samples' TPN transformed primary intensity versus sypro ruby intensity was reduced to –0.19, whereas the fit slope of the total protein controls fell to 0.0017, indicating minimal remaining dependence on the deposited total protein amount.
Nested ANOVA analysis was used to track sources of variability at each step of the analysis: primary intensity, TPN transformed intensity, and TPN and AMN transformed intensity. Results of nested ANOVA analysis for the Serca2a antibody stain are shown in Table I. In Fig. 3, individual data points from all normal samples, including all replicates, are scatter plot by their discussed nested ANOVA structure (only normal samples shown because of space). The nonbiological variability in the data is shown to diminish as the TPN and AMN transformations are performed. Before the TPN and AMN transformations, only 25% of all variability in the study is attributable to biological sources. The TPN transformation alone increases that to nearly 50%. After both TPN and AMN, the biological groupings of the samples account for 90% of the variability in the study, with 75% of all variability associated with the transmural depth of the lysate. A negative control stain was also analyzed by nested ANOVA and 85% of variability remained nonbiological after TPN and AMN, indicating the analysis process is working properly (Table I). This result was expected, because we have shown previously that TPN and AMN remove ∼70% of technical variability (15). Reducing technical variability to this extent makes it possible to accurately assess sample-specific expression patterns, patterns that are crucially important to understanding the inherent animal to animal differences in protein expression in the normal, DHF, and CRT states.
Table I. Variance components expressed as percent of total variability from nested ANOVA performed on RPMA Serca2a expression (negative control stain components).
| Variance components | Intensity | TPN adjusted | TPN and AMN adjusted | |||
|---|---|---|---|---|---|---|
| State (Norm,DHF,CRT) | 0.0 | (0.0) | 0.0 | (0.0) | 0.0 | (0.0) |
| Heart | 16.2 | (6.9) | 8.2 | (6.8) | 14 | (11.9) |
| Location (Ant,Lat) | 0.0 | (0.6) | 0.4 | (2.0) | 0.5 | (0.3) |
| Depth (Epi,Mid,Endo) | 9.4 | (0.0) | 38.9 | (0.0) | 75.2 | (2.6) |
| Section replicate | 17.5 | (28.3) | 1.6 | (0.0) | 3.1 | (3.3) |
| Lysate replicate | 19.4 | (50.8) | 0.0 | (24.0) | 1.8 | (15.8) |
| Within (unaccounted) | 37.6 | (13.3) | 50.9 | (67.3) | 5.4 | (66.0) |
| Biological | 25.6 | (7.5) | 47.5 | (8.8) | 89.7 | (14.8) |
| Nonbiological | 74.5 | (92.4) | 52.5 | (91.3) | 10.3 | (85.1) |
After the quality of the data had been assured (Figs. 2 and 3), we analyzed the RPMAs (both LRMH and HRSH) for biologically relevant information. An advantage of the LRMH RPMAs is that all 12 hearts used in this study may be analyzed using a single array, eliminating across-array variability. Fig. 4 shows the Serca2a expression from a single LRMH array (lateral wedge only) in panel A, as well as the same samples' negative control stain in panel B. The Kruskal-Wallis nonparametric test was used to test for the presence of significant changes in expression across heart states as well as transmural depths. Within all three states there is a significant dependence (p < 0.01) on the transmural depth, whereas there is no dependence on heart state. The negative stain showed no significant differences, as expected, indicating there is no detected bias in the data.
Fig. 4.

LRMH Serca2a staining results (A) relative to a negative stain (B) (lateral wedge only). The 16-replicate mean and standard deviation are shown for each depth of each heart, grouped by N-normal (green), d-DHF (black), and C-CRT (red). Four shapes within each state are used to represent each unique heart (filled square, filled triangle, open triangle, and open circle). The within-state means of the four hearts at each depth are shown by a horizontal line. A, shows the result of staining a LRMH array with a SERCA2a antibody, B, is an identical array that was used as a negative control (stained with secondary antibody only).
Each of the hearts shown in Fig. 4 was also profiled for Serca2a expression using the HRSH RPMAs. Fig 5A displays results from a Serca2a stain of HRSH RPMA composed of a single DHF heart. The high-resolution data shows a very clear, smooth transmural gradient in Serca2a expression, with no gradient in the negative control (Fig. 5B). The within-depth coefficient of variation (CV), which includes the section and lysing replicates as well as any remaining technical variability, is 8 ± 2%. Using this CV, a power calculation (assuming normal distribution of data) indicates that 16 replicates per sample (as printed) enables the detection of a 1.1-fold shift in expression among spatially independent samples with greater than 80% power. To verify this estimate, all 55 unique combinations of the 11 transmural depths from the anterior wedge shown in Fig. 5A were tested for statistically significant differences. Shifts greater than 1.1-fold were detected as statistically significant (p < 0.05) 41 out of 42 times by a student's t test whereas shifts smaller than 1.1-fold were not detected as significant in all 13 occurrences, verifying that the power estimate is approximately correct. Using Tukey's honest significance test, 23 out of 24 wedges exhibited a significant epi > endo transmural gradient in Serca2a expression. Each was fit by linear regression to determine the slope and the data was combined as there were no significant differences detected among states or locations (lateral and anterior). For all wedges, there is a 38 ± 15% epi > endo transmural gradient similar to that shown in Fig. 5A (see supplemental Fig. 2).
Fig. 5.

HRSH RPMA results and correlation to LRMH RPMAs. A, Serca2a expression of a single DHF heart (lateral wedge shown as black open triangle in Fig. 3) at full 1-mm resolution. B, Negative stain transmural expression of an identical HRSH RPMA from the same printing run. C, Scatter plot of serca2a expression for samples that were on the LRMH array and the twelve HRSH arrays (R2 = 0.85).
Also of interest is the consistency of measurements across independent RPMA printing runs. Fig. 5C shows a scatter plot of AMN and TPN adjusted intensity among the subset of samples that were analyzed both in the LRMH and HRSH RPMAs (Pearson correlation of 0.85). This correlation is made possible by the identical AMN control, present on every array, used for normalization. Note that the HRSH RPMA arrays for each independent heart were printed and stained at independent times whereas the LRMH RPMA data was from a single array printing and staining run. The across array CV in the sample means was found to be 5 ± 4%. The 5% across array CV is in addition to the within array CV of 8%. Although AMN allows for reproducible results across arrays, the greatest statistical power will be achieved by designing within-array studies for analysis.
DISCUSSION
RPMAs coupled with use of AMN (Fig. 1) provide a powerful alternative to conventional methods for quantitative measurement of protein expression. The effect of the TPN and AMN transformations on the quality of the data is demonstrated in Figs. 2 and 3 as well as Table I. Nonbiological sources are reduced to contributing ∼10% of the overall variability resulting in a within array CV of 8 ± 2% and sufficient statistical power to consistently detect 1.1-fold shifts in Serca2a expression. This methodology enabled the precise measurement of heart-specific expression patterns (Fig. 5A), therefore making it possible to understand the nature of biological variability across preparations in potential future experiments.
Overall, a significant transmural gradient of 38 ± 15% in Serca2a was measured across all 12 hearts. This is consistent with previous findings of Laurita et al. who uncovered an approximate 40% decrease from epi to endo in Serca2a expression among normal canine hearts using the same antibody in Western blots(7). However, because of statistical power limitations, Laurita et al. could not say whether the mid region was statistically different from the endo region, and the robustness of the gradient within individual hearts was unknown. High resolution Serca2a expression profiles showing a significant transmural gradient for all 12 hearts can be found in the online supplemental Fig. 2. Given the statistical power to now detect biological variability among hearts, the robustness of the Serca2a transmural gradient within 23 of 24 wedges indicates that this gradient is a biologically important phenomenon.
The strength of the RPMA assay lies in its ability to simultaneously measure the amount of an antibody bound to a target protein at over 2500 unique spots on an array. These spots can be customized as the researcher prefers to optimize performance for the study objectives. In our study, we presented two RPMA layouts, both using AMN: LRMH and HRSH RPMAs. The LRMH arrays have the advantage of being able to fit many hearts on a single array for comparison of expression levels across different states at the cost of spatial resolution. The HRSH arrays yield 1 mm transmural spatial-resolution of expression for each heart, but require each heart to be printed on a separate array. The LRMH RPMAs revealed that there was no change in expression of Serca2a among the three unique states, however there was significant transmural dependence of expression (Fig. 4A). The HRSH RPMAs yield high confidence in the presence of a transmural gradient within each individual heart, whereas the AMN experimental design allows for comparison across arrays (R2 = 0.85), albeit with slightly reduced statistical power than comparing within an array because of the additional 5% CV across arrays.
An important advantage for RPMAs versus other technologies is that while sample collection and preparation for printing RPMAs can be time consuming, fabricated arrays can be stored for future use. Results in the online supplemental Fig. 3 show that storage for 5 months followed by repeated antibody probing yielded a Pearson correlation of 0.988 between the mean sample intensity pre and post storage, indicating nearly identical information is extracted at a later time. The AMN experimental design aids in this reproducibility as the AMN controls and study samples are exposed to identical conditions within each array. Thus, as new research hypothesizes arise and antibodies become available, data can be obtained from the same sample set in a matter of days by using previously printed and stored arrays rather than repeating the process of sample collection and processing as is required in Western blot and immunofluorescent work.
One shortcoming of RPMAs, immunochemistry, and any nonsandwich antibody assay, remains their dependence on primary antibodies that are highly specific to their protein of interest. Mass spectrometry is an antibody free assay alternative. However, as with RPMA, many replicates will be needed to control for experimental variability in order to expose the biological variability of interest at high spatial resolution. An experimental replicate and control design similar to that presented here will be a challenge for mass spectrometry. Multiple reaction monitoring based mass spectrometry may offer a possibility for such a design in the future (25).
The RPMA assay described here provides improved statistical power and spatial resolution in expression measurements compared with Western blot or immunofluorescence methods. Using this assay, it is now possible to maximize the information that can be obtained from each individual heart, enabling the exploration of biological differences from heart to heart. When combined with other functional data (e.g. ECG recordings, pressure-volume loops, cardiac deformation, etc.) obtained from the same animal, use of the RPMA assay as described here may make it possible to relate protein expression to these important measures of cardiac function.
Acknowledgments
We thank Dave Hopkins for his assistance in creating Figure 1, and the laboratory of Dr. Gordon Tomaselli for assistance in collecting samples.
* Support was provided by National Institutes of Health R33HL87345, NO1-HV-28180, PO1 HL081427, P01 HL077180, as well as the College of Life Sciences at George Mason University.
 This article contains supplemental Figs. S1 to S3.
This article contains supplemental Figs. S1 to S3.
Conflict of interest: Authors, except for Raimond Winslow, have affiliation with Theranositics Health.
1 The abbreviations used are:
- AP
- action potential
- AMN
- array microenvironment normalization
- CRT
- cardiac resynchronization therapy
- CV
- coefficient of variation
- DHF
- dyssynchronous heart failure
- endo
- endocardial
- epi
- epicardial
- HRSH
- high resolution single heart
- LRMH
- low resolution multi heart
- LV
- left ventricle
- RPMA
- reverse phase protein microarray
- TPN
- total protein normalization
- TPER
- transmembrane protein extraction
- TCEP
- tris[2-carboxylethy]phosphine.
REFERENCES
- 1. Liu D. W., Gintant G. A., Antzelevitch C. (1993) Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ. Res. 72, 671–687 [DOI] [PubMed] [Google Scholar]
- 2. Antzelevitch C., Sicouri S., Litovsky S. H., Lukas A., Krishnan S. C., Di Diego J. M., Gintant G. A., Liu D. W. (1991) Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ. Res. 69, 1427–1449 [DOI] [PubMed] [Google Scholar]
- 3. Drouin E., Charpentier F., Gauthier C., Laurent K., Le Marec H. (1995) Electrophysiologic characteristics of cells spanning the left ventricular wall of human heart: evidence for presence of M cells. J. Am. Coll. Cardiol. 26, 185–192 [DOI] [PubMed] [Google Scholar]
- 4. Liu D. W., Antzelevitch C. (1995) Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weajer IKs contributes to the longer action potential of the M cell. Circ. Res. 76, 351–365 [DOI] [PubMed] [Google Scholar]
- 5. Watanabe T., Delbridge L. M., Bustamante J. O., McDonald T. F. (1983) Heterogeneity of the action potential in isolated rat ventricular myocytes and tissue. Circ. Res. 52, 280–290 [DOI] [PubMed] [Google Scholar]
- 6. Boukens B. J., Christoffels V. M., Coronel R., Moorman A. F. (2009) Developmental basis for electrophysiological heterogeneity in the ventricular and outflow tract myocardium as a substrate for life-threatening ventricular arrhythmias. Circ. Res. 104, 19–31 [DOI] [PubMed] [Google Scholar]
- 7. Laurita K. R., Katra R., Wible B., Wan X., Koo M. H. (2003) Transmural heterogeneity of calcium handling in canine. Circ. Res. 92, 668–675 [DOI] [PubMed] [Google Scholar]
- 8. Spragg D. D., Leclercq C., Loghmani M., Faris O. P., Tunin R. S., DiSilvestre D., McVeigh E. R., Tomaselli G. F., Kass D. A. (2003) Regional alterations in protein expression in the dyssynchronous failing heart. Circulation 108, 929–932 [DOI] [PubMed] [Google Scholar]
- 9. Greenstein J. L., Wu R., Po S., Tomaselli G. F., Winslow R. L. (2000) Role of the calcium-independent transient outward current I(to1) in shaping action potential morphology and duration. Circ. Res. 87, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 10. Prestle J., Dieterich S., Preuss M., Bieligk U., Hasenfuss G. (1999) Heterogeneous transmural gene expression of calcium-handling proteins and natriuretic peptides in the failing human heart. Cardiovasc. Res. 43, 323–331 [DOI] [PubMed] [Google Scholar]
- 11. Xiong W., Tian Y., DiSilvestre D., Tomaselli G. F. (2005) Transmural heterogeneity of Na+-Ca2+ exchange: evidence for differential expression in normal and failing hearts. Circ. Res. 97, 207–209 [DOI] [PubMed] [Google Scholar]
- 12. Akar F. G., Wu R. C., Juang G. J., Tian Y., Burysek M., Disilvestre D., Xiong W., Armoundas A. A., Tomaselli G. F. (2005) Molecular mechanisms underlying K+ current downregulation in canine tachycardia-induced heart failure. Am. J. Physiol. Heart Circ Physiol. 288, H2887–2896 [DOI] [PubMed] [Google Scholar]
- 13. Poelzing S., Akar F. G., Baron E., Rosenbaum D. S. (2004) Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am. J. Physiol. Heart Circ. Physiol. 286, H2001–2009 [DOI] [PubMed] [Google Scholar]
- 14. Lundebye A.-K., Vedel G. R., Krogsdam Christensen A. M., Kristiansen K., Hunter D., Depledge M. H. (1995) Improved quantification of stress proteins by western blotting. Analytica Chimica Acta 311, 109–114 [Google Scholar]
- 15. Anderson T., Wulfkuhle J., Liotta L., Winslow R. L., Petricoin E., 3rd (2009) Improved reproducibility of reverse-phase protein microarrays using array microenvironment normalization. Proteomics 9, 5562–5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spurrier B., Ramalingam S., Nishizuka S. (2008) Reverse-phase protein lysate microarrays for cell signaling analysis. Nat. Protoc. 3, 1796–1808 [DOI] [PubMed] [Google Scholar]
- 17. Paweletz C. P., Charboneau L., Bichsel V. E., Simone N. L., Chen T., Gillespie J. W., Emmert-Buck M. R., Roth M. J., Petricoin E. F., 3rd, Liotta L. A. (2001) Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 20, 1981–1989 [DOI] [PubMed] [Google Scholar]
- 18. Winslow R. L., Rice J., Jafri S., Marbán E., O'Rourke B. (1999) Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ. Res. 84, 571–586 [DOI] [PubMed] [Google Scholar]
- 19. Aiba T., Hesketh G. G., Barth A. S., Liu T., Daya S., Chakir K., Dimaano V. L., Abraham T. P., O'Rourke B., Akar F. G., Kass D. A., Tomaselli G. F. (2009) Electrophysiological consequences of dyssynchronous heart failure and its restoration by resynchronization therapy. Circulation 119, 1220–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petricoin E. F., 3rd, Espina V., Araujo R. P., Midura B., Yeung C., Wan X., Eichler G. S., Johann D. J., Jr, Qualman S., Tsokos M., Krishnan K., Helman L. J., Liotta L. A., 3rd, (2007) Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 67, 3431–3440 [DOI] [PubMed] [Google Scholar]
- 21. Vanmeter A. J., Rodriguez A. S., Bowman E. D., Harris C. C., Deng J., Calvert V. S., Silvestri A., Fredolini C., Chandhoke V., Petricoin E. F., 3rd, Liotta L. A., Espina V. (2008) LCM and protein microarray analysis of human NSCLC: Differential EGFR phosphorylation events associated with mutated EGFR compared to wild type. Mol. Cell Proteomics, 7, 1902–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kane L. A., Neverova I., Van Eyk J. E. (2007) Subfractionation of heart tissue: the “in sequence” myofilament protein extraction of myocardial tissue. Methods Mol. Biol. 357, 87–90 [DOI] [PubMed] [Google Scholar]
- 23. (2005) R Core Development Team, R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: ISBN 3–900051-07–0 URL: www.R-Project.org [Google Scholar]
- 24. Sokal R. R., Rohlf F. J. (1995) Biometry: The principles and practice of statistics in biological research. 3rd ed., W.H. Freeman, New York [Google Scholar]
- 25. Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]