

Abstract
Proteomic identification of protein interactions with membrane associated molecules in their native membrane environment pose a challenge because of technical problems of membrane handling. We investigate the possibility of employing membrane nanodiscs for harboring the membrane associated molecule to tackle the challenges. Nanodiscs are stable, homogenous pieces of membrane with a discoidal shape. They are stabilized by an encircling amphipatic protein with an engineered epitope tag. In the present study we employ the epitope tag of the nanodiscs for detection and co-immunoprecipitation of interaction partners of the glycolipid ganglioside GM1 harbored by nanodiscs. Highly specific binding activity for nanodisc-GM1 immobilized on sensorchips was observed by surface plasmon resonance in culture media from enterotoxigenic Escherischia coli. To isolate the interaction partner(s) from enterotoxigenic Escherischia coli, GM1-nanodiscs were employed for co-immunoprecipitation. The B subunit of heat labile enterotoxin was identified as a specific interaction partner by mass spectrometry, thus demonstrating that nanodisc technology is useful for highly specific detection and identification of interaction partners to specific lipids embedded in a membrane bilayer.
The ability of biomolecules to interact with surrounding substances is essential for their function. Whereas robust methods for identification and characterization of such interactions exist for water soluble biomolecules the study of membrane associated molecules is much more challenging in an aqueous environment. Integral membrane proteins and other molecules embedded in biological phospholipid membranes change their functional and structural properties and aggregate when the membrane is removed because of their hydrophobic nature. Thus membrane proteins are less studied than soluble proteins, even though genes that encode integral membrane proteins constitute 25% of the human genome and are primary targets for many drugs. Our aim is to develop methods to isolate and identify proteins that associate with membrane embedded molecules. For this purpose the molecule of interest is often immobilized on solid supports, e.g. sensorchips or affinity media. To study a given biomolecular interaction it is often of importance that the membrane embedded molecule resides in a phospholipid membrane under homogeneous and controlled conditions. Systematic analysis requires the biomolecules of interest to be isolated from additional molecules that are present in the native membrane, yet still to be embedded in a phospholipid bilayer to keep them in a nearly native state.
To study physiologically relevant interactions the biomolecule under study should be maintained under aqueous and nondenaturing conditions. Thus, the membrane must be solubilized for isolation of membrane associated molecules. Under aqueous conditions solubilization of membranes is obtained by addition of detergent above the critical micellar concentration, thus incorporating the phospholipids and other molecules from the membrane in mixed micelles. However, even nonionic detergents that are often considered nondenaturing can change the properties of membrane embedded biomolecules by stripping off the membrane thus exposing hydrophobic areas of the molecule. The new exposed areas can lead to unspecific binding caused by hydrophobic forces.
After purification, membrane associated molecules can be reconstituted in phospholipid membranes as part of synthetic vesicles. However, this approach has disadvantages because vesicles are prone to aggregation because of curvature of the membrane, which exposes hydrophobic sites because of cracks in the hydrophilic surface. This may also be detrimental to reconstituted integral membrane proteins (1–3).
Alternatively, the membrane embedded molecule can be kept in solution while still surrounded by a stable phospolipid membrane as a nanolipid particle termed a nanodisc. Nanodiscs are composed of a discoidal phospholipid bilayer surrounded by two belts of amphipatic helical proteins termed membrane scaffold protein (MSP)1 that stabilize and solubilize the membrane disc by shielding its hydrophobic edge from exposure to the aqueous surrounding (see Fig. 1). Homogenous nanodiscs self-assemble from a mixture of lipids, MSP and detergent when the detergent is removed by dialysis or adsorption to detergent binding beads. Once solubilized as part of a nanodisc the membrane embedded molecule can be studied for identification and characterization of protein interactions using the same methods as those used for soluble proteins, thereby facilitating the study of hydrophobic membrane associated moleculessignificantly. The use of nanodiscs also permits the study of membrane proteins in a well-defined, soluble, uniform, and stable environment, thus offering an improvement over the use of liposomes as a model membrane system employed in studies of molecular interactions with membranes and membrane proteins (4).
Fig. 1.

Strategy for nanodisc assisted detection and co-immunoprecipitation for isolation of interaction partners to membrane embedded molecules: Antibodies that recognize the epitope tag of nanodiscs are immobilized on sensorchip surfaces (A) or affinity beads (B, left). Potential binding activity can be monitored by surface plasmon resonance (SPR) by first capturing nanodiscs and then introducing a flow of a mixture, e.g. a cell lysate or culture medium, containing interaction partners of the nanodisc embedded molecule (A, right). Alternatively the binding components of the mixture can be isolated by incubating the nanodiscs with the mixture and isolating the nanodiscs and associated proteins with the antibody decorated affinity beads (B).
Known molecular interactions have been studied with nanodisc technology in solution using spectroscopic methods, native electrophoresis (5), gel permeation chromatography (5), and enzyme linked immunosorbent assays, electron microscopy (6, 7). An epitope tag engineered into the MSP facilitates anchoring of the nanodiscs to various solid supports, such as sensorchips for kinetic interaction analysis by surface plasmon resonance (SPR) (8, 9) and specialized target plates for mass spectrometric analysis of interaction partners by matrix assisted laser desorption-ionization time-of-flight MS (MALDI-TOF MS) (10). Nanodiscs have also been attached to glass substrates for microfluidic fluorescence analyses of interactions (11), microcantilevers for detection of toxin-glycolipid interaction (12), silver nanoparticles for localized SPR analysis of drug-protein interactions (13), and polymeric beads for drug screening by NMR (14). However, to our best knowledge a nanodisc based method for proteomic identification of interaction partners of membrane embedded molecules does not exist.
The glycosphingolipid ganglioside GM1 (II3NeuAcGgOse4Cer) is embedded in the epithelia of intestines. The glycomoieties protrude into the intestinal lumen and are thus able to interact with soluble proteins. One known interaction partner is the heat labile toxin (LT), a protein which is secreted from enterotoxigenic Escherichia coli (ETEC). It is an AB5 type of toxin, of which the pentameric B subunit interacts with gangliosides and the A subunit is an ADP ribosylase. It is structurally and functionally homologous to the cholera toxin which was used previously in purified form to develop nanodisc based kinetic interaction analysis (8). Both cholera toxin and LT interact with the glycosyl moiety of GM1, after which they are internalized in the host cell. Inside the host cell, the A subunit modifies the regulatory G-protein Gs-R, thereby causing constitutive cAMP production, which in turn affects the regulation of ion channels. The consequence is fluid loss from the small intestine (15).
In this study we develop nanodiscs-based methodology for detection of binding activity in a complex biological mixture that interacts with defined membrane embedded molecules. Subsequent isolation by a novel nanodisc-based immunoprecipitation procedure further enables the identification of specific binding partners. Here we identify interaction partners of the glycosphingolipid GM1 in a matrix of phosphatidylcholine lipids. However, we envision that the nanodisc co-immunoprecipitation method will be especially powerful for transmembrane proteins that are especially prone to structural rearrangements in the absence of membrane.
EXPERIMENTAL PROCEDURES
Growth of E. coli and Isolation of Culture Medium
The LT-expressing ETEC strain H10407 (16) and the nonLT-expressing reference strain MG1655 were cultured statically overnight in LB broth supplemented with 150 mm NaCl. The culture medium was separated from the bacterial cells by centrifugation at 4000 × g and subsequent clearing by filtration through a 0.0.2 μm filter (Nalgene). Each filtrate was concentrated from 250 ml to 5 ml using a pressurized amicon stirred ultrafiltration cell (Millipore, Billerica MA) with a Millipore regenerated cellulose filter with a nominal molecular weight cut-off of 5000 and a diameter of 76 mm (Millipore, Billerica MA).
Nanodisc Preparation
MSPs and nanodiscs with 4% GM1 (Sigma-Aldrich) in 96% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids, Alabaster, AL) (referred to as GM1 discs) or with 100% POPC (referred to as POPC discs) were produced as described in (8), (17). In brief, dried lipids of the specified molar composition were resolubilized in nanodisc assembly buffer (100 mm NaCl, 20 mm Tris-HCl, 1 mm EDTA, pH 7.4) supplied with 25 mm sodium cholate. The 6xHis-tagged MSP variant MSP1T2 (17) or the Flag-tagged MSP variant MSPFC (8) was then added to obtain a final 1:60 MSP:lipid ratio. The assembly process was initiated by removal of cholate with biobeads SM-2 and continued for three hours at 4 °C. The discs were purified to free the preparation from minor amounts of aggregated lipids and MSP by gel filtration on a Superdex 200 10/300 GL gel filtration column (GE Healthcare Biosciences, Uppsala, Sweden) with nanodisc assembly buffer as eluent at a flow rate of 400 μl/min.
Surface Plasmon Resonance Analysis
Nanodisc binding was analyzed by surface plasmon resonance using a Biacore 3000 instrument (Biacore AB, Uppsala, Sweden). HEPES-buffered saline, which contained 10 mm HEPES, pH 7.4, 150 mm NaCl, 3.33 mm EDTA, was used as running buffer. All buffers were filtered (0.22 μm) and thoroughly degassed. All analyses were performed at 25 °C.
Anti-tetra-His antibody (Qiagen, Hilden, Germany) was immobilized covalently on CM5 sensorchips (GE Healthcare Biosciences, Uppsala, Sweden) by amine coupling with 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride and N-hydroxysuccinimide (Pierce, Rockford, IL) according to the guidelines of Biacore AB. For sensorchip capture, 6xHis-tagged discs were injected at a flow rate of 5 μl/min until 1000 RU had been captured (6–8 min) on a flow cell. GM1 nanodiscs were injected over a sample flow cell and control nanodiscs were injected over the reference flow cell. Subsequently, concentrated culture media or commercially available LT (Sigma-Aldrich) was injected for 4 min at a flow rate of 20 μl/min. To assess the stably bound material, the flow was changed to buffer without analyte. Flow cells were regenerated by injection of regeneration solutions as stated in the figure legends followed by a 1-min pulse of 50 mm Glycine-HCl, pH 2.2, which strips off discs and potentially associated proteins, leaving the antibody ready for another round of disc capture, analyte binding, and regeneration.
Data Analysis
Signal from the reference flow cell was subtracted from the sample flow cell to remove bulk responses caused by different refractive indexes of sample and running buffer. The SPR response immediately before sample injection was defined as baseline value by adjusting the curves to zero on the y (SPR response) axis by subtraction of the SPR-response at that time from all data-points. For quantitation in the regeneration experiments, the baseline-subtracted SPR responses were read 15 s before injection of regeneration solution (Rbound) and 20 s after injection of regeneration solution had ended and the flow had been changed back to running buffer (Rregenerated). The ratios Rregenerated/Rbound are reported. The data were plotted and analyzed using BIAevaluation software 4.1 (Biacore AB, Uppsala, Sweden).
Co-immunoprecipitation
Co-Immunoprecipitation from the concentrated culture medium was carried out with protein G Dynabeads (Invitrogen, Carlsbad, CA) essentially as described by the manufacturer. To prepare the beads for immunoprecipitation, 40 μl bead slurry was washed two times with 200 μl phosphate buffered saline containing 0.005% P20 (PBSP). Then the anti-FLAG antibody (Sigma-Aldrich) was captured on the beads by resuspending the beads in 40 μl PBSP containing 6 μl anti-FLAG antibody and washing once in 200 μl PBSP to remove unbound antibody. Subsequently, the beads were washed twice in 200 μl phosphate buffered saline without P20 to wash out P20 detergent that might interfere with the nanodisc structure.
To co-immunoprecipitate GM1 interacting proteins 40 μl FLAG-tagged GM1 nanodiscs or control nanodiscs were added to 300 μl filtered and concentrated culture medium and incubated 25 min at room temperature with end-over mixing. The nanodisc-interaction partner complexes were then captured on the washed beads by addition of the nanodisc-culture medium mixture to the anti-FLAG antibody/protein G Dynabeads and incubation for 15 min at room temperature. The supernatant was aspirated and the beads were washed three times with 200 μl phosphate buffered saline. After resuspension of the beads in the last washing solution, the slurry was split in two 100 μl portions by transfer to new microcentrifuge tubes. The beads of one tube were eluted twice by 5-min incubations with 10 μl 3xFLAG peptide at a concentration of 125 ng/μl, whereas the beads of the other tube were eluted two times by 5-min incubations with 5 mm 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC) (Avanti Polar Lipids).
SDS-PAGE, Protein Detection and In Gel-digestion
Proteins were solubilized in Laemmli-buffer and separated by SDS-PAGE on 4–20% Nupage gels with the MES buffer system (Invitrogen, Carlsbad, CA) according to the manufacturer's recommendation. To visualize the proteins, the gel was stained with silver nitrate according to (18).
The distinct bands were cut out of the gel and digested with sequencing grade porcine modified trypsin (Promega, Madison, WI) after reduction and alkylation in low binding 0.5 ml microcentrifuge tubes (Sorenson Bioscience, Salt Lake City, Utah) according to (18).
Mass Spectrometric Analysis
Peptides from the in-gel digest were applied to a ca 2 mm long self-packed reversed phase microcolumn of POROS R2–20 (Applied Biosystems, Foster City, CA) in eppendorf gel-loader-tips (Eppendorf, Hamburg, Germany) according to (19) for desalting and up-concentration. The peptides were eluted onto a stainless steel-target with less than half a microliter of 70% acetonitrile, 0.1% trifluoroacetic acid, 30% water and 10 mg/ml α-cyano hydroxy cinnamic acid as matrix. Peptide mass fingerprints were recorded by MALDI-TOF MS on a Bruker ultraflex (Bruker, Bremen, Germany) or a 4800 plus MALDI TOF/TOF analyzer (Applied Biosystems) in the positive ion mode with delayed extraction. MS spectra were recorded with an acceleration voltage of 20 kV. Intense peptides that did not have mass values that coincided with trypsin autodigestion products or tryptic keratin peaks were subjected to MS/MS by the 4800 MALDI TOF/TOF. Collision-induced dissociation was performed at collision energy of 1 kV with an indicated collision gas pressure of 32 ∼1 × 10−6 Torr.
The spectra were annotated and analyzed using Data Explorer v. 4.5 (Applied Biosystems) without smoothing or noise reduction. The MS/MS data from each gel band was combined into a single mass list using an in-house developed script (Jakob Bunkenborg, University of Southern Denmark, Denmark).
Protein Identification
Peptide mass values of individual bands, as well as the MS/MS spectra were searched against proteins of the National Center for Biotechnology Information nonredundant database, release 20101030. Protein sequences from E. coli (158,800 sequences) were searched first and the unidentified proteins were then searched against human and murine sequences because proteins of the nanodisc co-immunoprecipitation system origin from these sources. The peptide MS and MS/MS tolerances were set to 100 ppm and 0.6 Da, respectively. The MASCOT search engine located on an in-house server (version 2.2.06, Matrix Science, London, UK)) was used for searching and scoring the identified proteins. The search parameters were as follows: Specificity of protease digestion was set to trypsin with one missed trypsin site allowed. Oxidation of methionine was set as variable modification for all Mascot searches, and carbamidomethyl on cysteine was set as a fixed modification. Individual peptides with a MASCOT score >35 (probability value of p < 0.05) were accepted as identified. Using these parameters a false discovery rate of 0.00% was obtained as judged by MASCOT's decoy database searching.
RESULTS
Detection of Specific Binding Activity to GM1-nanodiscs in Cell Culture Medium
Previous studies have demonstrated that SPR can be used for kinetic analysis of binding between antibody-captured nanodiscs that harbor the glycosphingolipid ganglioside GM1 and cholera toxin subunit B (8). In the present study, we perform SPR analyses to determine if the sensitivity and specificity of nanodiscs is suitable for interaction studies with binding partners in complex mixtures. In the Biacore system that we used for SPR measurements, a continuous flow of running buffer or analyte is directed over a sensor chip to which a ligand is attached. The optical SPR phenomenon measures the refractive index in the vicinity of the sensorchip surface. The refractive index change accompanied with injection of a binding analyte is composed of the increment in refractive index caused by the up-concentration of analyte in the vicinity of the sensor chip caused by binding to the immobilized ligand plus the change in refractive index caused by mismatch between the refractive index of the analyte solution and running buffer. The latter bulk refractive index change can be corrected for by subtracting the SPR signal from a reference flow cell that does not contain a ligand for the analyte. The reference flow cell is also used to estimate potential nonspecific binding. The binding-dependent refractive index change correlates linearly with the mass of analyte bound to the sensorchip (20, 21). Thus we can use SPR to detect binding activity in complex mixtures to GM1 harbored by nanodiscs immobilized on SPR sensorchips (see Fig. 1A for outline).
The binding to GM1 of components in media filtrates from two E. coli strains was compared: (1) An ETEC strain (H10407) that expresses heat labile enterotoxin (LT) (16), which is a homolog of CT, and 2) and a nonpathogenic laboratory strain (MG1655) that does not contain the LT genes. Thus the H10407 culture medium is expected to display increased binding compared with the culture medium of MG1655 because of the presence of LT protein in H10407 only. The media concentrates were injected over two flow cells: One flow cell coated with antibody-captured GM1/POPC nanodiscs (8% GM1 in 92% POPC) and one flow cell coated with antibody-captured POPC nanodiscs. The former is expected to bind LT, whereas the latter is not. Thus, POPC nanodiscs can be used for estimation of nonspecific binding and serve as a reference for subtraction of bulk refractive index caused by different composition of the culture medium from the running buffer of the SPR system. From the reference-subtracted SPR sensorgrams in Fig. 2A it is seen that a specific binding activity of 275 response units (RU) to 1000 RU captured GM1-nanodiscs was observed after H10407 medium had been injected for 4 min, whereas injection of MG1655 medium to the same amount of captured nanodiscs led to specific binding of only 20 RU as seen from Fig. 2B. Thus SPR studies with nanodiscs could clearly distinguish between media from the different E. coli strains, presumably because of the presence of LT in H10407 only. To estimate the amount of nonspecific binding, the nonreference-subtracted sensorgrams were inspected for binding to the flow cell that contained POPC nanodiscs. Only 10 RU binding activity was observed in sensorgrams for these control nanodiscs. This amounts to only 4% of the binding activity to GM1-containing nanodiscs (data not shown). Thus nanodisc technology appears to be well suited for detection and capture of interaction partners in complex mixtures.
Fig. 2.
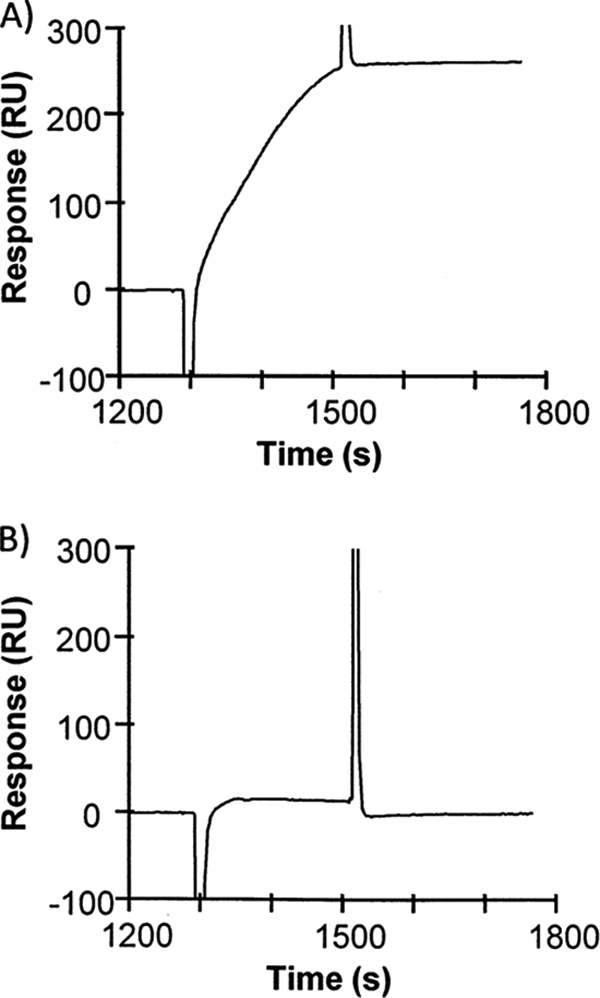
SPR analysis of binding activity in culture media of two E. coli strains. Media cleared after growth of enterotoxigenic E. coli H10407 (A) and the nonpathogenic E. coli MG1655 (B) were injected over ganglioside GM1 nanodiscs captured on antibody coated sensorchips. The presented sensorgrams are produced by reference subtraction of the signal of binding to 900 RU GM1-free (control) nanodiscs from the signal of binding to 940 RU GM1 nanodiscs.
Test of Elution Conditions by Surface Plasmon Resonance
The epitope-tagged proteins can often be eluted from their respective antibodies with low pH buffer. This is also the case for anti-tetra-His antibody as seen by the dissociation of nanodiscs from the SPR sensorchip after a 1-min injection of glycine at pH 2.2 (8). Low pH destabilizes the nanodisc and often leads to aggregation and precipitation of molecules embedded in membranes. Furthermore, low pH presumably also releases potential nonspecific binding components from the antibodies and agarose beads. Therefore we tested the possibility of using mild nondenaturing detergents for solubilization of the nanodiscs with the purpose of releasing inserted phospholipids and GM1 from the MSP. 6xHis tagged nanodiscs were captured by antibody-coated sensorchips for 360 s, and after a continuous buffer flow for 386 s, a 60 s pulse of detergent or buffer was injected. The drop in RU caused by detergent injection reflects a drop in refractive index, which corresponds to a drop in mass bound to the sensorchip (Fig. 3A). The detergents Triton X-100, cholate, n-octyl glucopyranoside (OGP), DHPC, and Tween-20 were tested at approximately twice their critical micellar concentrations (see the legend for Fig. 3 for actual concentrations). All the tested detergents except Tween 20 released mass, presumably lipids, from the flowcell sensorchip on which nanodiscs were immobilized as summarized in Fig. 3B. Strikingly, cholate, DHPC, and Triton X-100 all released 80% of the mass. It seems reasonable to speculate that the released material is lipid and that at least one MSP remains bound to the immobilized antibody after injection with detergent because the used detergents are considered nondenaturing. The assumption is supported by the following stoichiometric calculations: According to references (22–24) the refractive index of lipid bilayers is in the same range as the refractive index of proteins. A nanodisc contains 2 MSPs and ca. 120 POPC molecules (25). One MSP has a molecular weight of 25,000 Da and a POPC molecule 760 Da, i.e. the lipid component constitutes 120 x 760 Da = 91,200 Da and the nanodisc has a total molecular weight of 141200. Therefore, 1 MSP (25,000 Da) represents 18 mass percent of a total nanodisc (141,200 Da), which corresponds nicely to the observed retained mass of 20%. OGP released essentially all of the captured mass from the sensorchip, which means that it released both lipids and MSP from the sensorchip.
Fig. 3.
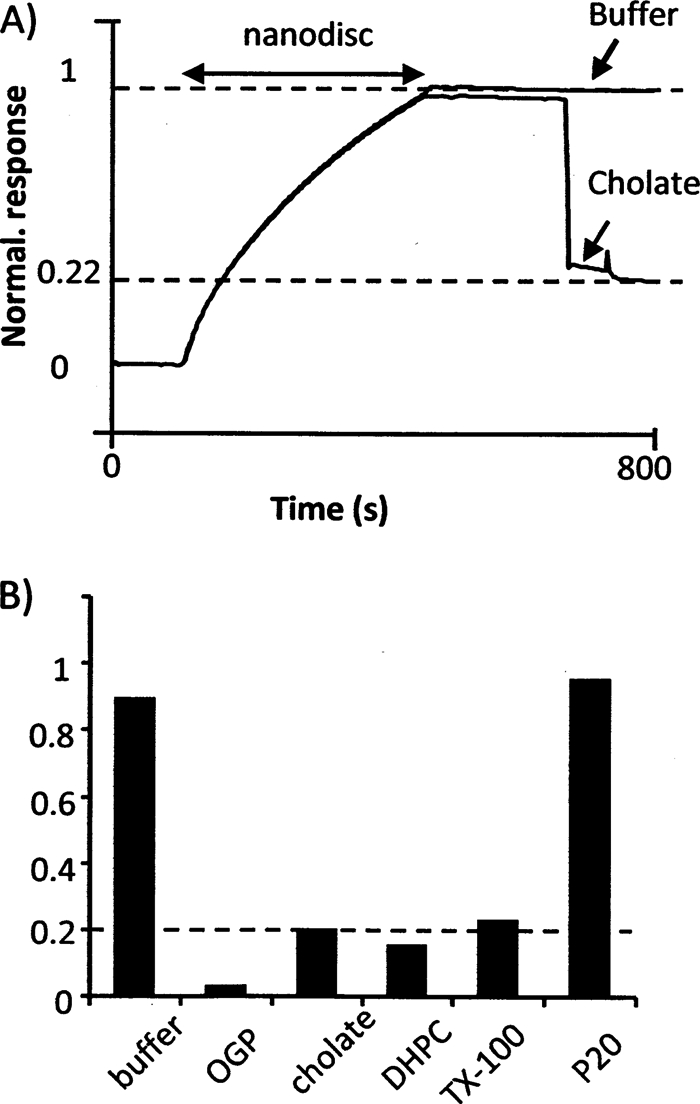
SPR analysis of detergent based elution from nanodiscs captured by antibody. 6xHis-tagged Nanodiscs were injected over immobilized anti-tetra-His antibodies. Then the flow was changed to buffer without nanodiscs and subsequently an injection of detergent or buffer without detergent was injected (A). The remaining signal normalized to after detergent injection is reported in (B). The concentrations of detergent were as follows: Octyl glucopyranoside (OGP) 50 mm; cholate 25 mm; DHPC 5 mm; Triton X-100 (TX-100) 1 mm; P20 (tween 20) 0.1 mm.
Choice of Tag for Co-affinity Purification of GM1-nanodisc Binding Proteins
For affinity purification and subsequent identification of interaction partners the tagged nanodiscs must be captured effectively and specifically. In addition, the specific binding proteins should be released from or eluted along with the captured nanodiscs under sufficiently specific conditions so that nontagged proteins that bind to the tag-capturing agent (e.g. antibody and affinity beads) are not eluted. In a previous study, we have tested different methods for nanodisc capture via affinity-tags (8). We tested two affinity tags: a 6xHis-tag and a C-terminal FLAG epitope that consists of the sequence DYKDDDDK. 6xHis-tags can be captured by Ni2+-immobilized metal-ion affinity chromatography (Ni2+-IMAC) and eluted with imidazole, EDTA, or low pH. Alternatively 6xHis-tags can be captured by antibodies directed against consecutive histidine residues and eluted with low pH buffer. Capture of 6xHis-tags by anti-tetra-His antibodies was stable as determined by SPR, and because of this kinetically stable interaction between the tag and antibody, tagged nanodiscs cannot be eluted competitively after capture.
The FLAG-tag is captured by anti-FLAG antibody and can be eluted either with a peptide that consists of three consecutive FLAG epitope sequences or by low pH. In our previous study we concluded that FLAG-tagged nanodiscs captured by anti-FLAG antibody were unsuitable for kinetic SPR studies because they leak from the surface during the course of the experiment (8). However, they are well-known to be suitable for immunoprecipitation, and commercial kits that include peptides for competitive elution exist and have been used in several applications. Thus we chose FLAG tagged nanodiscs for co-immunoprecipitation of GM1 interacting proteins.
Co-immunoprecipitation From Nanodisc Coated Magnetic Beads
Proteins captured on SPR sensorchips can be eluted for mass spectrometric identification (26). However, the maximum amount that can be captured and eluted in an SPR/MS experiment is typically a few nanograms, making robust mass spectrometric identification challenging. In our view it is often more practical to scale up the affinity purification experiment by using affinity beads that have much larger capacity for capture than sensorchips, even in small volumes. The information on sensitivity and specificity of the conditions attained in the SPR experiments can be implemented in the affinity bead-based protocols.
The SPR experiments had demonstrated that antibody-captured nanodiscs bound interaction partners specifically. Thus, we determined to test co-immunoprecipitation for isolation of GM1 interaction partners by (1) incubation of nanodiscs with the same H10407 culture medium that was used in the SPR experiments, (2) capture of the newly formed nanodisc-interaction partner complexes by beads coated with anti-FLAG antibody, (3) aspirating the nonbound proteins, (4) washing off liquid in and between the beads as well as loosely bound material, and (5) competitive elution or solubilization with gentle detergents of the nanodiscs. In our first attempts we employed an agarose bead-based commercially available kit for immunoprecipitation of FLAG-tagged proteins. The agarose beads are precoated with anti-FLAG antibody. Unexpectedly, experiments of competitive elution with FLAG peptides from agarose beads with- or without the addition of GM1/POPC nanodiscs or POPC nanodiscs resulted in elution of the same subset of proteins in specific as well as control experiments, suggesting that these were interacting with either the FLAG antibody or the agarose beads. In the SPR experiments only negligible binding to antibodies was observed, suggesting that the agarose beads were the source of nonspecific interaction (data not shown).
Next, we tested whether magnetic beads coated with Protein G that bind antibodies displayed lower nonspecific binding. The procedure for binding and elution was the same as for the agarose beads, except that FLAG antibody was captured by Protein G before initiation of the experiment. The captured proteins were eluted with either FLAG peptides or DHPC at the concentration tested in the SPR experiments. Eluted proteins as well as unbound material released during washing steps were separated by SDS-PAGE and visualized by silver-staining (Fig. 4). Clearly, most proteins of the culture medium did not bind the affinity beads and were readily washed off. When comparing the proteins eluted from GM1/POPC discs (Fig. 4A) with proteins eluted from POPC control discs without GM1 (Fig. 4B), it is seen that one 12 kDa protein (marked 1*) is released specifically from the GM1/POPC discs, both by competitive elution with FLAG peptides and by solubilization of nanodisc contents with DHPC. Two proteins, labeled 2 and 3, are released from both GM1 and control discs by competitive elution, and three proteins, labeled 3, 4, and 5, are released from both GM1 and control discs by solubilization of discs contents with DHPC. The GM1 nonspecific protein, labeled 3, is eluted in the same amounts from elution with FLAG peptides as from elution with DHPC (Fig. 4). In summary, the specifically interacting GM1 protein is released in same quantities by both methods.
Fig. 4.

Co-immunoprecipitation of proteins that bind to GM1 containing nanodiscs (A) and nanodiscs without GM1 (B). Fifteen microliters of the flow through, washes and eluents of 3xFLAG peptide incubation or DHPC incubation were subjected to SDS-PAGE as indicated. An aliquot of the FLAG M2 antibody that was used for immunoprecipitation is shown in (C) for comparison. The gel bands of the eluents are indicated with numbers that are also used in the text and Table I.
Mass Spectrometric Identification of the Proteins Eluted From the Nanodisc Co-immunoprecipitation Procedure
To identify the eluted proteins, the silver stained bands were cut out of the gel and subjected to in gel digestion with trypsin. The peptide mass fingerprints were recorded by MALDI TOF and searched against NCBInr. To further validate the hits from the peptide mass search, selected peptides were subjected to fragmentation by MALDI TOF-TOF and the resulting fragment ions were searched. The protein in the GM1 specific band 1* was identified to be LTB, subunit B of the heat labile toxin as illustrated by the peptide mass fingerprint and MS/MS experiment (Fig. 5). The protein in band 2, which was eluted by competitive elution with FLAG, but not with DHPC, could not be identified. The proteins in band 4 and 5 which were eluted predominantly by DHPC could not be identified by searching the E. coli sequence database. Because of their size and the obvious fact that antibodies were used for immunoprecipitation, we suspected that these two bands were the small and large chains of the anti FLAG antibody. Upon searching the mouse sequence database with MS/MS data band 5 was identified as Ig gamma-1 chain C region, thus verifying that this band was derived from the antibody. The protein in band 3 which was eluted in equal amounts by DHPC and FLAG peptide could not be identified by searching the H10407 protein database. However, it migrates as MSP, and its identity as MSP was indeed confirmed by searching the MALDI TOF MS data against the MSP1T2 sequence and by identifying the protein as apolipoprotein A-I, which MSP1T2 is derived from, in the human protein database. The results of the protein identifications are summarized in Table I and detailed in a protein identification table (supplemental Table S1).
Fig. 5.

Identification of the protein that binds specifically to GM1 nanodiscs. The protein of band 1 in Fig. 4A was digested with trypsin after reduction and alkylation. A, shows the peptide mass map with peaks that are annotated to LTB indicated Signal denoted “T” correspond to commonly observed trypsin peptides. Signals denoted “K” correspond to commonly observed keratin peptides. Of the 40 searched peptides 6 were assigned to LTB, 3 to trypsin, and 6 to keratin. B, shows the annotated MALDI MS/MS spectrum of the peptide ion 1553.65.
Table I. Summary of proteins identified by MS/MS from the immunoprecipitation experiments. A detailed table of each protein identification is present as Supplementary Table S1. N.D., a protein could not be identified from the spectra.
| Band no.a | Protein name | Accession no.b | MASCOT scorec | No. peptidesd |
|---|---|---|---|---|
| 1* | Heat labile enterotoxin subunit B | gi 212285888 | 416 | 5 |
| 2 | N.D. | – | – | – |
| 3 | Apolipoprotein A1 | gi 178775. human | 143 | 3 |
| 4 | N.D. | – | – | – |
| 5 | Antibody heavy chain | gi 300244464. mouse | 147 | 2 |
a According to figure 4.
b Organism is given when the protein was not identified as an E. coli protein.
c Sum of MASCOT peptide scores derived from significant MS/MS spectra.
d Number of peptides identified by MS/MS spectra.
DISCUSSION
We have demonstrated that epitope tagged nanodiscs can be employed for immunoprecipitation experiments with the purpose of identifying interaction partners of membrane embedded molecules. The development of the procedure was aided by SPR experiments that were designed to select capture and elution strategies and to assess the specificity of nanodisc binding. The results of these experiments demonstrated that epitope tagged nanodiscs can be captured by antibodies (Fig. 2 and 4, and reference (8)) and that SPR analysis using immobilized nanodiscs that harbored GM1 could specifically detect binding of a component secreted by pathogenic ETEC, but not by nonpathogenic E. coli.
To identify the proteins that bind specifically to GM1 harbored by nanodiscs we chose FLAG antibody for capture of FLAG-tagged nanodiscs because an established method for gentle competitive elution exists. The method worked well as judged from the silver-stained SDS-PAGE gel of flow-through, washes and eluents: When GM1 nanodiscs in complex with interaction partners were eluted with 3xFLAG peptides only three clear bands were observed. One of the bands (band number 1) was specific for GM1 nanodiscs when compared with SDS-PAGE of control nanodiscs. It was identified as heat labile enterotoxin (LTB). Of the two remaining nonspecific bands, one was identified as MSP (band number 3), whereas the other intense band (band number 2) could not be identified. Because of its nonfocused shape we speculate that it is either a modified protein or not protein at all. SPR experiments had demonstrated that mild detergents could also be used for elution of the interacting proteins, presumably because of release of the lipid contents from the nanodiscs. With this elution procedure four proteins could be identified from four bands on an SDS-PAGE gel. As in the FLAG peptide elution the only specifically eluting protein was identified as LTB. Additionally, MSP was identified. Two proteins were eluted with detergent from both GM1 and control nanodisc resin (bands 4 and 5). These proteins were ascribed to the antibody based on mass spectrometric identification and migration as judged from the SDS-PAGE gel. Based on these observations we deduce that the antibody and with that MSP are released from the beads by mild detergent. The release of MSP appears to contradict our interpretation of the SPR results which implied that the MSP is retained on the sensorchip. However, in the immunoprecipitation experiments the antibody was also released from the protein G beads. This was not the case for the SPR experiments, in which the antibody was immobilized covalently on the sensorchip. Thus the detergent releases the antibody from the protein G magnetic beads and with that the nanodiscs. In summary, the immunoprecipitation method is sufficiently sensitive to identify a specific interaction partner by mass spectrometry. Furthermore it is specific in that only one nonspecifically binding component was detected with FLAG elution and none with mild detergent elution. The rest of the bands could be ascribed to the MSP and antibody. Thus we have developed a highly specific method for detection and identification of interaction partners of membrane embedded molecules.
Most other methods for identification of interaction partners to molecules naturally situated in membranes rely on conditions where the membrane associated molecule is freed from membrane by solubilization with detergent or by deletion of the membrane embedded part of the molecule (e.g. transmembrane regions from integral membrane proteins) (27), which might change the structure of membrane proteins and/or expose sticky parts of the molecule. Alternatively, the interaction partners can be covalently cross-linked, generating a pool of cross-linked protein complexes that can be separated from noncross-linked proteins before identification (28). We expect the nanodisc co-immunoprecipitation method to be generally applicable for targeted identification of interaction partners in many biological systems involving membrane embedded molecules, such as specialized lipids and transmembrane proteins: Antibodies and affinity matrices for immunoprecipitation are general reagents of many protein chemical and molecular biology laboratories. Furthermore, it lends itself to scale-up for identification of interaction partners of lower abundance or for downstream applications that require higher amounts of interacting protein. Also, the elution conditions demonstrated here are sufficiently gentle to leave isolated protein(s) native and thus enabling activity studies after nanodisc immunoprecipitation.
Footnotes
* The work was supported by grants from The Lundbeck Foundation.
 This article contains supplemental Table S1.
This article contains supplemental Table S1.
1 The abbreviations used are:
- MSP
- membrane scaffold protein
- DHPC
- diheptoyl phosphatidyl choline
- ETEC
- enterotoxigenic Eschiricia coli
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholin
- OGP
- n-octyl glucopyranoside
- SPR
- surface plasmon resonance
- MALDI-TOF MS
- matrix assisted laser desorption-ionization time-of-flight MS
- LT
- labile toxin
- RU
- response units.
REFERENCES
- 1. Hunte C., von Jagow G., Schägger H. (2003) Membrane protein purification and crystallization a practical guide. Academic Press, Oxford [Google Scholar]
- 2. Torchilin V. P., Weissig V. (2003) Liposomes, A practical approach, second ed., Oxford University Press, Oxford [Google Scholar]
- 3. Selinsky B. S. (2003) Membrane protein protocols. Expression, purification, and characterization, Methods in Molecular Biology, Humana Press, Hohokus, NJ [Google Scholar]
- 4. Borch J., Hamann T. (2009) The nanodisc: a novel tool for membrane protein studies. Biol. Chem. 390, 805–814 [DOI] [PubMed] [Google Scholar]
- 5. Dalal K., Nguyen N., Alami M., Tan J., Moraes T. F., Lee W. C., Maurus R., Sligar S. S., Brayer G. D., Duong F. (2009) Structure, Binding, and Activity of Syd, a SecY-interacting Protein. J. Biol. Chem. 284, 7897–7902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ye F., Hu G., Taylor D., Ratnikov B., Bobkov A. A., McLean M. A., Sligar S. G., Taylor K. A., Ginsberg M. H. (2010) Recreation of the terminal events in physiological integrin activation. J. Cell Biol. 188, 157–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsukamoto H., Sinha A., DeWitt M., Farrens D. L. (2010) Monomeric Rhodopsin Is the Minimal Functional Unit Required for Arrestin Binding. J. Mol. Biol. 399, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borch J., Torta F., Sligar S. G., Roepstorff P. (2008) Nanodiscs for immobilization of lipid bilayers and membrane receptors: Kinetic analysis of cholera toxin binding to a glycolipid receptor. Anal. Chem. 80, 6245–6252 [DOI] [PubMed] [Google Scholar]
- 9. Shaw A. W., Pureza V. S., Sligar S. G., Morrissey J. H. (2007) The local phospholipid environment modulates the activation of blood clotting. J. Biol. Chem. 282, 6556–6563 [DOI] [PubMed] [Google Scholar]
- 10. Marin V. L., Bayburt T. H., Sligar S. G., Mrksich M. (2007) Functional assays of membrane-bound proteins with SAMDI-TOF mass spectrometry. Angew. Chem. 46, 8796–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goluch E. D., Shaw A. W., Sligar S. G., Liu C. (2008) Microfluidic patterning of nanodisc lipid bilayers and multiplexed analysis of protein interaction. Lab. Chip. 8, 1723–1728 [DOI] [PubMed] [Google Scholar]
- 12. Tark S. H., Das A., Sligar S., Dravid V. P. (2010) Nanomechanical detection of cholera toxin using microcantilevers functionalized with ganglioside nanodiscs, Nanotechnology 21, 435502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Das A., Zhao J., Schatz G. C., Sligar S. G., Van Duyne R. P. (2009) Screening of Type I and II Drug Binding to Human Cytochrome P450–3A4 in Nanodiscs by Localized Surface Plasmon Resonance Spectroscopy. Anal. Chem. 81, 3754–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Früh V., Zhou Y. P., Chen D., Loch C., Eiso A. B., Grinkova Y. N., Verheij H., Sligar S. G., Bushweller J. H., Siegal G. (2010) Application of Fragment-Based Drug Discovery to Membrane Proteins: Identification of Ligands of the Integral Membrane Enzyme DsbB. Chem. Biol. 17, 881–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Turner S. M., Scott-Tucker A., Cooper L. M., Henderson I. R. (2006) Weapons of mass destruction: virulence factors of the global killer Enterotoxigenic Escherichia coli. FEMS Microbiol. Lett. 263, 10–20 [DOI] [PubMed] [Google Scholar]
- 16. Evans D. G., Silver R. P., Evans D. J., Jr., Chase D. G., Gorbach S. L. (1975) Plasmid-controlled colonization factor associated with virulence in Escherichia-coli enterotoxigenic for humans. Infect. Immun. 12, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Denisov I. G., Grinkova Y. V., Lazarides A. A., Sligar S. G. (2004) Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 126, 3477–3487 [DOI] [PubMed] [Google Scholar]
- 18. Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 19. Gobom J., Nordhoff E., Mirgorodskaya E., Ekman R., Roepstorff P. (1999) Sample purification and preparation technique based on nano-scale reversed-phase columns for the sensitive analysis of complex peptide mixtures by matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 34, 105–116 [DOI] [PubMed] [Google Scholar]
- 20. Fägerstam L. G., Frostellkarlsson A., Karlsson R., Persson B., Ronnberg I. (1992) Biospecific interaction analysis using surface-plasmon resonance detection applied to kinetic, binding-site and concentration analysis. J. Chromatogr. 597, 397–410 [DOI] [PubMed] [Google Scholar]
- 21. Karlsson R., Löfås S. (1998) Kinetic analysis of the interaction between an analyte in solution and an immobilized protein, In Immobilized biomolecules in analysis, A practical approach. eds. Cass T., Ligler F.S., Oxford University Press, Oxford [Google Scholar]
- 22. Salamon Z., Wang Y., Tollin G., Macleod H. A. (1994) Assembly and molecular-organization of self-assembled lipid bilayers on solid substrates monitored by surface-plasmon resonance spectroscopy. Biochim. Biophys. Acta-Biomembranes 1195, 267–275 [DOI] [PubMed] [Google Scholar]
- 23. Jonsson M. P., Jönsson P., Höök F. (2008) Simultaneous nanoplasmonic and quartz crystal microbalance sensing: analysis of biomolecular conformational changes and quantification of the bound molecular mass, Anal. Chem. 80, 7988–7995 [DOI] [PubMed] [Google Scholar]
- 24. Salamon Z., Tollin G. (2001) Optical anisotropy in lipid bilayer membranes: Coupled plasmon-waveguide resonance measurements of molecular orientation, polarizability, and shape. Biophys. J. 80, 1557–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bayburt T. H., Grinkova Y. V., Sligar S. G. (2002) Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2, 853–856 [Google Scholar]
- 26. Mattei B., Borch J., Roepstorff P. (2004) Biomolecular interaction analysis and MS, Anal. Chem. 76, 18A–25A [Google Scholar]
- 27. Daulat A. M., Maurice P., Jockers R. (2009) Recent methodological advances in the discovery of GPCR-associated protein complexes. Trends Pharmacol. Sci. 30, 72–78 [DOI] [PubMed] [Google Scholar]
- 28. Gubbens J., de Kroon A. I. (2010) Proteome-wide detection of phospholipid-protein interactions in mitochondria by photocrosslinking and click chemistry. Molecular Biosystems 6, 1751–1759 [DOI] [PubMed] [Google Scholar]