

Abstract
Complement fixation to surface-conjugated ligands plays a critical role in determining the fate of targeted colloidal particles after intravenous injection. In the present study, we examined the immunogenicity of targeted microbubbles with various surface architectures and ligand surface densities using a flow cytometry technique. Targeted microbubbles were generated using a post-labeling technique with a physiological targeting ligand, cyclic arginine-glycine-asparagine (RGD), attached to the distal end of the poly(ethylene glycol) (PEG) moieties on the microbubble surface. Microbubbles were incubated in human serum, washed and then mixed with fluorescent antibodies specific for various serum components. We found that complement C3/C3b was the main human serum factor to bind in vitro to the microbubble surface, compared to IgG or albumin. We also investigated the effect of PEG brush architecture on C3/C3b fixation to the microbubble surface. RGD peptide was able to trigger a complement immune response, and complement C3/C3b fixation depended on microbubble size and RGD peptide surface density. When the targeting ligand was attached to shorter PEG chains that were shielded by a PEG overbrush layer (buried-ligand architecture), significantly less complement activation was observed when compared to the more traditional exposed-ligand motif. The extent of this protective role by the PEG chains depended on the overbrush length. Taken together, our results confirm that the buried-ligand architecture may significantly reduce ligand-mediated immunogenicity. More generally, this study illustrates the use of flow cytometry and microbubbles to analyze the surface interactions between complex biological media and surface-engineered biomaterials.
1. Introduction
In recent years, molecularly targeted contrast-enhanced ultrasound has received increasing attention as a diagnostic imaging modality that allows the detection and evaluation of endothelial biomarkers associated with vascular events underlying specific pathologies [1–7]. For such applications, targeted contrast agents are injected intravenously into the bloodstream, where they accumulate at targeted sites along the vascular endothelium. When imaged with ultrasound [8], these bound contrast agents provide an acoustic signal and therefore allow the measurement of specific endothelial receptor expressions that are upregulated. Ultrasound molecular imaging has thus been applied to the assessment of tumor angiogenesis [9–11], thrombosis [12, 13], atherosclerosis [14] and inflammation [15, 16].
Ultrasound contrast agents are typically gas-filled colloidal particles (microbubbles) with diameters less than 10 μm. The surface comprises amphiphilic phospholipids self-assembled to form a lipid monolayer shell. Microbubbles can provide sensitive acoustic responses when detected using ultrasound because of their compressible gas cores [7, 17]. Similar to the design of long-circulating liposomes, poly(ethylene glycol) (PEG) chains, or PEG chain derivatives, can be incorporated into the shell of microbubbles in order to form a steric barrier against coalescence and adsorption of macromolecules, such as antibodies, to the microbubble surface [18, 19]. These agents, owing to their small sizes, can pass through the pulmonary vasculature [20] and have been shown to exhibit contrast persistence longer than 10 min in vivo [21].
When administered intravenously, microbubbles or other conventional colloidal particles are rapidly removed from the bloodstream by the mononuclear phagocyte system (MPS) [22]. The MPS protects the systemic circulation by distinguishing foreign and endogenous substances, and the fast clearance of foreign particles is mediated through endocytosis with recognition of specific cell surface receptors, such as complement receptor 1 (CR1) and Fc receptor [23, 24]. Endocytosis is classified into three categories: receptor-mediated endocytosis (RME), pinocytosis and phagocytosis [25]. Depending on the size of the particle, it can be eliminated from the system either through RME and/or pinocytosis (for small compounds) or phagocytosis (for large particles such as microbubbles). Evidence of microbubble phagocytosis has been demonstrated both in vitro [26] and in vivo [27, 28]. Although not required, phagocytosis is often triggered by specific receptor recognition, and such ligand-receptor interactions typically exist between the cellular receptor specific for the proteins bound to the colloidal particles rather than for the particles themselves. Thus, serum protein adsorption is extremely important in determining particle uptake by phagocytes and predicting the fate of colloidal particles after administration. Immunoglobulin G (IgG) and complement components are known as major opsonins for the uptake of large particles, such as bacteria, viruses, and remnants of dead cells. In particular, complement activation plays a critical role in the recognition of biocolloids by the immune system [29].
The complement system, consisting of over 30 soluble plasma and cell-surface bound proteins, is an important effector arm of innate immunity [24]. There are three pathways to activate the complement system: the classical pathway, the lectin pathway and the alternative pathway. The classical pathway is triggered by the binding of complement component C1q to immune-complexes on the antigen surfaces; the lectin pathway is triggered by the binding of mannose-binding lectin to arrays of carbohydrates on foreign microorganisms; and the alternative pathway is triggered by the binding of spontaneously activated complement component C3 in plasma to the surface of foreign particles. All three pathways converge to the formation of C3 convertases, which cleave C3 into C3b and C3a for further opsonization and mediation of inflammation in the complement cascade. One key site for the activation of the complement system is the foreign particle surface. Regardless of the activation pathway, the main effectors of the complement system (such as C3 convertases and C3b) need to bind to the surface of the particle in order to initiate the phagocytic process. Therefore, the accessibility of the complement component proteins to the foreign particle surface, or rather to the specific recognition sites on the surface, is important in determining complement activation.
Targeted microbubbles are created by attaching a targeting ligand, such as a polysaccharide, monoclonal antibody or peptide, specific for the desired endothelial biomarker, onto the shell. One of the most widely studied targeting ligands is cyclic-arginine-glycine-asparagine (RGD), which has been shown to bind to an overexpressed angiogenic biomarker, αvβ3 integrin, with high affinity and specificity [30, 31]. There are many surface functionalization strategies for conjugating targeting ligands to the surface of microbubbles, most notably through attaching specific ligands to the distal end of tethered PEG chains. However, targeting ligands typically present nucleophilic groups (e.g., hydroxyl and amino) that could trigger the alternative pathway of complement activation and decrease the microbubble circulation persistence. Clinical reports have shown data indicating that long circulating PEGylated liposomes, with similar surface structures to microbubbles, could trigger acute hypersensitivity reaction in sensitive individuals [29, 32]. These reactions are classified as complement activation-related pseudoallergy (CARPA) due to their common trigger mechanism: complement activation [32, 33]. Despite the common belief that water-soluble PEG chains on the surface of Stealth® liposomes can prevent immune recognition by reducing protein adsorption and vesicle aggregation, recent studies have suggested that PEGylated liposomes are capable of triggering the complement system in human serum and fixing opsonic complement proteins [29, 34–36]. It was reported that complement activation was detectable as quickly as 10 min after injection of the marketed liposomal drug Doxil (Centocor Ortho Biotech Inc.; Horsham, PA) [32], which is on the same time scale as microbubble persistence in vivo. Therefore, it is important to design targeted microbubbles with a surface architecture that minimizes complement recognition by minimizing C3/C3b fixation in order to reduce CARPA and prevent premature microbubble clearance from the circulatory system. At the same time, avoidance of complement fixation can keep the ligand pristine and therefore allow it to retain specificity to the target receptor.
Our laboratory recently introduced a microbubble construct for use with ultrasound radiation force (USRF) to allow triggered and specific adhesion with reduced immunogenicity. This stealth microbubble design, the buried-ligand architecture (BLA), involves bimodal PEG polymer chains on the surface. The targeting ligand is attached to the shorter PEG chains, while the longer PEG overbrush serves as a shield to inhibit ligand exposure and reduce the accessibility to opsonins. The purpose of the present study was to experimentally investigate in greater depth the targeted microbubble immunogenicity in vitro between various microbubble surface architectures.
2. Materials and Method
2.1 Materials
All phospholipids were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL), including 1,2-distearyol-sn-glycero-3-phosphocholine (DSPC), 1,2-distearyol-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)2000] (DSPE-PEG2000), 1,2-distearyolsn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)2000] (DSPE-PEG2000-M), 1,2-distearyol-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)3000] (DSPE-PEG3000) and 1,2-distearyol-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)5000] (DSPE-PEG5000). All phospholipids were dissolved in chloroform (Sigma-Aldrich; St. Louis, MO) and stored in the freezer at −20 °C. The perfluorobutane gas (PFB, 99 wt% purity) used for microbubble generation was purchased from FluoroMed, L.P. (Round Rock, TX). The RGD peptide (cyclo [Arg-Gly-Asp-D-Phe-Cys], 99.9% purity) was purchased from Peptides International (Louisville, KY) and was dissolved in 3 vol% degassed acetic acid (Sigma-Aldrich). The dissolved RGD peptide was aliquoted into 50-μL volume and stored in nitrogen at −20 °C. The L-cysteine was purchased from Sigma-Aldrich and was dissolved in 18 MΩ-cm filtered deionized water (Direct-Q Millipore; Billerica, MA). The L-cysteine solution was prepared on each day immediately before use to ensure reactivity.
Human complement-preserved serum was purchased from Valley Biomedical (catalog no. HC1004; Winchester, VA). Serum was thawed once to aliquot into 1-mL eppendorf tubes and stored at −80 °C. Anti-human IgG-FITC antibody (catalog no. F4512) was purchased from Sigma-Aldrich. Both anti-human albumin-FITC antibody (catalog no. CLFAG2140) and anti-human C3/C3b-FITC antibody (catalog no. CL2103F) were purchased from Cedarlane (Burlington, NC). All antibody solutions were stored at 4 °C.
2.2 Microbubble generation and RGD peptide conjugation
The compositions of microbubble samples for all the experiments performed are listed in Table 1. Microbubbles were generated as described elsewhere [37]. Briefly, the indicated amounts of each phospholipid species were mixed, and the chloroform was evaporated. The dried lipid film was hydrated with phosphate buffered saline (PBS) mixture (90 vol% PBS:10 vol% 1,2-propanediol:10 vol% glycerol; Sigma-Aldrich) to a final lipid/surfactant concentration of 1 mg/mL. Fully dispersed lipid suspension was then transferred to a 3-mL serum vial and sealed for headspace PFB gas exchange. Microbubbles were formed by shaking with a VialMix (ImaRx Therapeutics; Tucson, AZ) for 45 s. The generated microbubbles were then diluted to 10-mL suspension with PBS, and washed 3 times by centrifugation flotation in a bucket-rotor centrifuge (Model 5804, Eppendorf; Westbury, NY) at 250G for 5 min. The microbubble cake was then diluted in 5 mM EDTA (pH 6.5) for subsequent experiments.
Table 1.
Microbubble compositions.
| Phospholipid Composition (mol %) | |||||
|---|---|---|---|---|---|
| Samples | DSPC | DSPE-PEG2000 | DSPE-PEG2000-M | DSPE-PEG3000 | DSPE-PEG5000 |
| P2K Control | 90 | 10 | - | - | - |
| P5K Control | 90 | - | - | - | 10 |
| ELA 5% | 90 | 5 | 5 | - | - |
| ELA 0.5% | 90 | 9.5 | 0.5 | - | - |
| ELA 0.05% | 90 | 9.95 | 0.05 | - | - |
| P2K/P5K Control | 90 | 5 | - | - | 5 |
| BLA 5% | 90 | - | 5 | - | 5 |
| BLA 0.5% | 90 | 4.5 | 0.5 | - | 5 |
| BLA 0.05% | 90 | 4.95 | 0.05 | - | 5 |
| BLA-P3K 5% | 90 | - | 5 | 5 | - |
An Accusizer optical particle counter (NICOMP Particle Sizing System; Santa Barbara, CA) was used to measure the size distribution and particle concentration. The amount of RGD peptide needed was then calculated as previously described [38]. RGD peptide was added to react with maleimide functional groups on the distal end of PEG chains at a molar ratio of 30:1 (RGD:maleimide). The reaction was carried out on a benchtop rotator for 12 hours at 4 °C. To ensure there were no unreacted maleimide groups, L-cysteine was added at a molar ratio of 1000:1 (L-cysteine:maleimide) after RGD peptide conjugation. The sample was incubated on a benchtop rotator for 30 min at room temperature. Unreacted RGD peptide was removed by centrifuging the microbubble suspension at 250G for 4 min. RGD peptide conjugation was confirmed using HPLC and MALDI-TOF (data not shown) as reported elsewhere [39, 40]. The concentrated microbubble cake was then re-suspended in PBS and analyzed by Accusizer. The median fluorescence intensity was measured using an Accuri C6 flow cytometer (Accuri Cytometers Inc.; Ann Arbor, MI). For zeta potential measurement, the washed microbubble cake was re-suspended in pH adjusted PBS solution (pH 7.2) and analyzed using a Malvern Zetasizer Nano-ZS (Malvern Instrument Ltd.; Worcestershire, UK).
2.3 Human complement-preserved serum C3/C3b activity assay
Serum aliquots were randomly chosen from each batch to test for complement component C3/C3b activity at different time points throughout the entire immunogenicity study. C3/C3b activity was measured using an ELISA kit purchased from Assaypro (catalog no. EC2101-1) following the manufacturer's instruction. No serum was re-frozen after ELISA assay to ensure complement activity.
2.4 Microbubble serum stability analysis
1 mL serum was preheated in a water bath at 37 °C using a digital block heater (VWR; West Chester, PA) for at least 20 min. A total of 5×108 RGD-conjugated microbubbles were added to the serum, and the size distribution and microbubble concentration was continuously monitored for 2 hr at 37 °C using both Accusizer and flow cytometer. The same amount of sample (6 μL for Accusizer and 4 μL for flow cytometry) was taken out at different time points for measurement to ensure consistency. Incubated samples were vortexed regularly to prevent microbubble aggregation at the top of the serum. Flow cytometry size isolated gating was used for data analysis as previously described [38].
2.5 Microbubble antibody binding analysis
A total of 5×108 microbubbles (about 100 μL in volume) were incubated with 900 μL serum on a benchtop rotator for 2 hr at room temperature. The sample was then washed once by centrifugation floatation at 250G for 3 min. The concentrated microbubble cake was re-suspended in 100 μL PBS solution and analyzed by both the Accusizer and flow cytometer. For anti-human C3/C3b, anti-human IgG and anti-human albumin FITC-antibody binding, 5 μg antibody was added. All FITC-antibody binding experiments were carried out in the dark on a benchtop rotator for 1 hr at room temperature. At the end of the incubation period, the microbubble sample was again analyzed by flow cytometry and microscopy without washing. The measured median fluorescence intensity data was analyzed according to microbubble diameter using size-isolated microbubble gating information previously described [38].
2.6 Optical microscopy
Direct visual confirmation of microbubble fluorescence was performed within 24 hrs after FITC antibody binding. Microbubble samples were taken out of the reaction syringe and imaged at room temperature. Still images were taken using an Olympus 1×71 inverted microscope (Olympus; Center Valley, PA). Images in both bright field mode and epifluorescence mode were captured for the same field of view using a high-resolution digital camera (Orca HR, Hamamatsu; Japan) with a 100× oil immersion objective and processed with Simple PCI software (C-Imaging; Cranberry Township, PA). Subsequent image analysis was done using ImageJ 1.4g software (NIH; Washington DC.).
3. Results and discussion
3.1 Targeted microbubble generation
For each set of microbubble components, the vial shaking method produced a milky, white microbubble suspension that was stable over the experimental timeframe. We have previously shown that small ligands with molecular weight < 1 kDa, such as RGD peptides, could diffuse freely through the PEG overbrush and react with functional groups at the distal end of buried PEG chains [38]. Here, HPLC and MALDI-TOF were used to ensure the complete attachment of RGD peptides to the surface of BLA microbubbles using the post-labeling technique (data now shown). Since several factors, such as microbubble size and surface charge, could influence the interactions between microbubbles and serum antibodies, the physicochemical properties of the samples were examined (Table 2). Microbubble samples were matched in concentration after the RGD conjugation and/or washing steps. We measured similar size distributions for all samples, with a dominant peak between 1–2 μm and a secondary peak between 4–5 μm (Figure 2A and 2B). The conjugation of RGD peptide to the surface of microbubbles did not affect either the microbubble size distribution or concentration. The number-weighted mean diameters for all microbubble samples were found to be similar, while the volume-weighted mean diameters ranged between 4.7 – 8.2 μm. Measurement of zeta potential showed that the negative charge of P2K microbubbles tended to increase by the conjugation of RGD peptide at pH 7.2. At the same time, the addition of the PEG overbrush (DSPE-PEG5000) into the microbubble shell tended to neutralize this negative charge.
Table 2.
Microbubble physical properties.
| Initial Concentration (#/mL) | Number-Weighted Mean Diameter (im) | Volume-Weighted Mean Diameter (im) | Zeta Potential (mV) | |
|---|---|---|---|---|
| Samples | (Mean) | (Mean ± SD) | (Mean ± SD) | (Mean ± SD) |
| P2K Control | 5.19E+09 | 1.3 ± 0.0 | 5.4 ± 0.6 | −24.1 ± 1.7 |
| P5K Control | 4.19E+09 | 1.4 ± 0.1 | 6.7 ± 2.0 | −9.5 ± 2.1 |
| ELA 5% | 4.59E+09 | 1.4 ± 0.1 | 8.2 ± 2.1 | −38.9 ± 2.8 |
| ELA 0.5% | 6.10E+09 | 1.4 ± 0.0 | 6.9 ± 0.8 | −31.4 ± 0.9 |
| ELA 0.05% | 5.29E+09 | 1.3 ± 0.0 | 6.8 ± 0.4 | −30.0 ± 1.1 |
| P2K/P5K Control | 5.89E+09 | 1.4 ± 0.1 | 4.8 ± 0.8 | −12.0 ± 0.6 |
| BLA 5% | 4.83E+09 | 1.4 ± 0.1 | 5.2 ± 0.7 | −19.2 ± 0.3 |
| BLA 0.5% | 6.57E+09 | 1.5 ± 0.1 | 6.8 ± 0.6 | −14.6 ± 0.8 |
| BLA 0.05% | 6.78E+09 | 1.4 ± 0.1 | 7.0 ± 3.2 | −12.5 ± 1.7 |
| BLA-P3K 5% | 4.63E+09 | 1.3 ± 0.0 | 4.7 ± 0.3 | −37.6 ± 0.2 |
Figure 2.
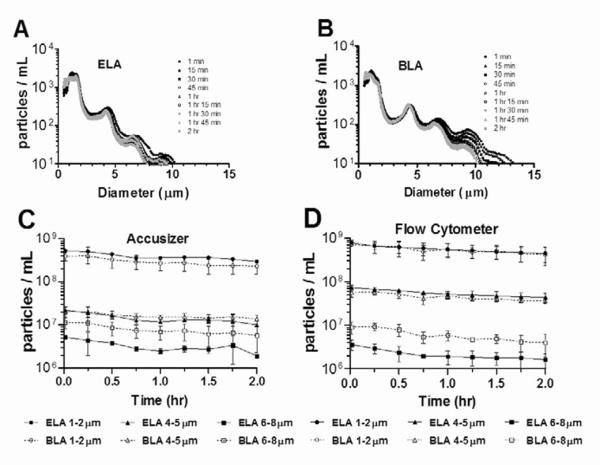
5% RGD peptide labeled microbubble size distribution (A, B) and concentration (C, D) change during human complement-preserved serum incubation at 37 °C. The size distribution was continuously monitored for 2 hrs for both ELA (A) and BLA (B) microbubble samples. With the exception that some smaller microbubbles (diameter <2 μm) showed a decrease in number detected over time, the majority of targeted microbubbles were stable during incubation with no significant change in size. The total concentration, as measured by Accusizer (C) and flow cytometer (D), was plotted against time according to microbubble diameter ranges. Both techniques showed data in good agreement; even though a concentration decrease was observed for both designs at the end of 2-hr incubation time, more than 70% of the targeted microbubbles were stable at 30 min, which was in the same time scale as for a typical ultrasound contrast imaging session.
3.2 Microbubble serum stability
The stability of microbubbles with 5% RGD peptide during incubation in human serum at physiological temperature was investigated. Figure 2A and 2B shows the size distribution change for ELA and BLA microbubbles, respectively, during the 2-hr incubation as measured by the Accusizer. For both surface architectures, smaller microbubbles with diameter less than 2 μm showed a decrease in number detected over time, while larger microbubbles showed no significant size change. The total microbubble concentration change was monitored using both the Accusizer and the flow cytometer (Figure 2C and 2D). Both detection methods showed results that were in good agreement: a decrease in microbubble concentration was observed for both surface architectures at all size ranges at the end of 2 hrs, with the highest decrease being 59% and 53% for ELA and BLA 6–8 μm microbubbles, respectively. However, the concentration decrease for all microbubble samples after 30 min incubation ranged from only 11% to 30%. Based on these results, we concluded that targeted microbubbles, regardless of surface architecture, were stable in human serum during incubation at physiological temperature within the time scale for a typical ultrasound contrast imaging session (~30 min) [41].
3.3 Human serum factor binding to microbubbles
It has been reported that the concentration of serum strongly affects activation of the complement system [42]. Therefore, undiluted complement-preserved human serum was used for all experiments. To ensure the validity of our immunogenicity data, complement activity of the serum samples was continuously monitored by measuring complement component C3/C3b activity of randomly chosen serum aliquots throughout the study. Figure 3 shows the quantified C3/C3b activity as measured by ELISA assay for all 38 samples. The measured C3/C3b activity was 30 ± 16 μg/mL of serum (mean ± SD). The human serum samples from different batches were statistically identical in terms of complement C3/C3b activity, and the aliquots were stable for the duration of the experiments. By showing consistent complement activity, we ensured that any measured C3/C3b binding difference in the immunogenicity study was due to the difference in complement activation by various microbubble samples, not due to batch-to-batch variability in serum C3/C3b activity.
Figure 3.
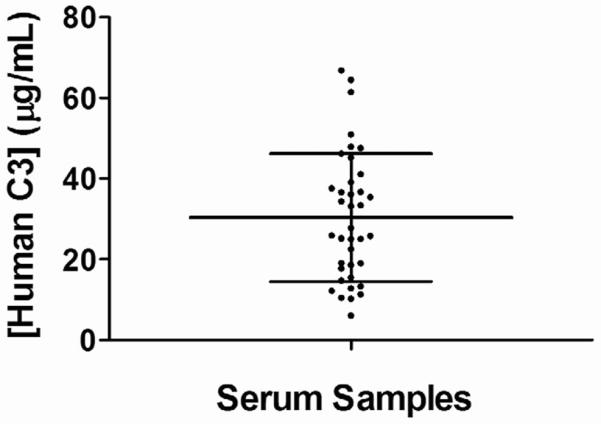
ELISA results of complement component C3/C3b activity for human complement-preserved serum aliquots. Serum aliquots were randomly chosen to be tested throughout the immunogenicity experiments. The average of measured C3/C3b activity was 30 ± 16 μg/mL of serum (mean ± SD). The human serum samples from different batches were statistically identical in terms of complement C3/C3b activity.
Next, we investigated the binding of human complement component C3/C3b, IgG and albumin to targeted microbubbles with 5% RGD conjugated to the surface. Sufficient incubation time (2 hr) was given to allow the full exposure of microbubbles to the serum environment. Detection of fluorescent antibodies by flow cytometry allowed an assessment of serum factor binding to the microbubble shells. Figure 4 shows the median fluorescence intensity (MFI) values for 1–2 μm ELA and BLA microbubbles after incubation with human serum and FITC-antibodies. The 1–2 μm size range was chosen because these microbubbles were the most abundant in all the samples and could correctly represent the MFI trend for the entire population. Measurements made before and after serum incubation showed minimal autofluorescence owing to binding of serum proteins to the microbubbles. All three serum factors were detected on the targeted microbubble samples. However, increases in MFI for IgG and albumin were small (<10%) compared to C3/C3b binding to ELA and BLA microbubbles (3.8-fold and 1.3-fold increase, respectively). We therefore concluded that complement C3/C3b was the main opsonin involved with the recognition of targeted microbubbles by the immune system.
Figure 4.
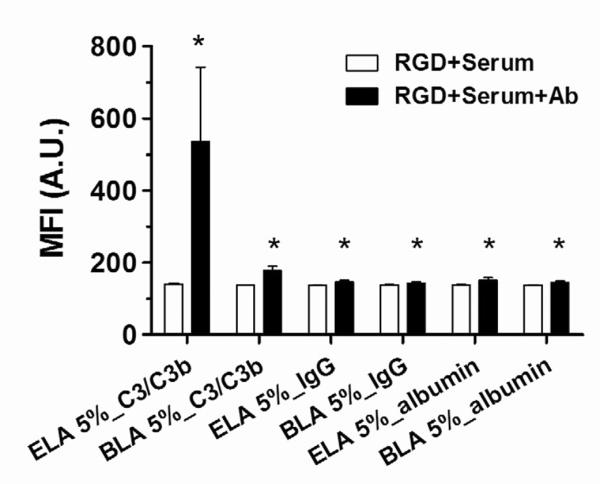
Human serum factor binding to 5% RGD labeled 1–2 μm ELA and BLA microbubbles. The median fluorescence intensity (MFI) was measured after RGD peptide conjugation, after 2 hrs human serum incubation and after 1 hr anti-human serum factor FITC-antibodies incubation. All three serum factors were observed to bind to both targeted microbubbles. However, only complement C3/C3b showed significant MFI increases; while IgG and albumin showed much less binding. “*” denotes a significant increase vs. the corresponding “RGD+Serum” measurement (p<0.05).
Epi-fluorescence microscopy images provided direct visual confirmation of FITC-antibody binding to the surface of targeted microbubbles. Only anti-human C3/C3b FITC-antibody labeled targeted ELA microbubbles were visible under epi-fluorescence mode. Figure 5 shows both the bright field and epi-fluorescence images for the same field of view of these polydisperse microbubbles. All microbubbles appeared to be stable during observation. No visible changes, such as collapse, aggregate formation or vesiculation were observed for these microbubbles over the observation time period (typically around 10–15 min). Almost all microbubbles visible under the bright field mode were also seen under the epifluorescence mode, indicating FITC-antibody binding to the surface. There was no preferential binding due to microbubble size. However, non-uniform FITC-antibody attachment was observed (see enlarged images), indicating heterogeneous binding of complement C3/C3b.
Figure 5.

Microscopic images of 5% RGD labeled ELA microbubbles after C3/C3b binding in both bright field mode (A) and epifluorescence mode (B). Both images show the same field of view. Enlarged images indicate microstructural features of non-uniform C3/C3b binding. Scale bars correspond to 10 μm.
Using streptavidin-FITC as model ligand, we previously showed that the PEG overbrush did not completely eliminate macromolecule conjugation to the surface of microbubbles, suggesting the existence of two distributions of PEG-lipid conjugates on the microbubble surface [38]. In the present study, a small amount of C3/C3b binding to BLA microbubbles was observed, lending further support to our suggestion that such non-uniform distributions of lipid molecules in the microbubble shell (e.g., domains and folds) may yield incomplete shielding of the targeting ligands. The heterogeneous fluorescence observed on the shell was another indication of the existence of such microstructural features (Figure 5).
3.4 Human serum complement C3/C3b binding to control microbubbles
First, we tested control microbubbles without RGD peptide for immunogenicity after human serum incubation (Figure 6). Three different surface architectures were tested. Significantly lower MFI values were detected for P2K/P5K control than those for P2K control in all microbubble size classes. P5K control microbubbles showed the lowest MFI among all three control samples. For the same methoxy DSPE-PEG surface coverage, the MFI for P5K control 4–5 μm and 6–8 μm microbubbles was only 36% and 31%, respectively, of those for their corresponding P2K control groups. These data suggest that some complement C3/C3b may be binding to the underlying phospholipid as well, and that the longer PEG reduces this effect [43].
Figure 6.
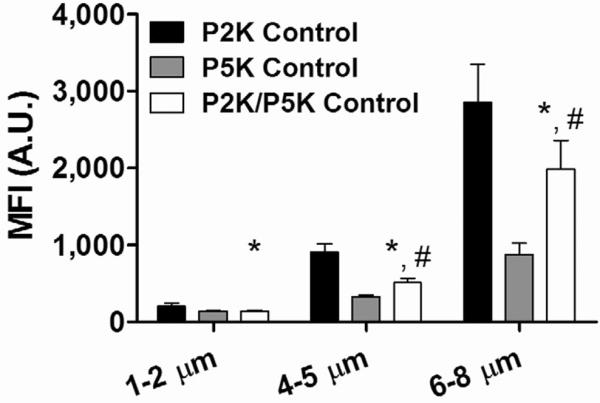
Human complement C3/C3b binding to control microbubbles. P2K/P5K control microbubbles showed significant lower C3/C3b binding than P2K control in all microbubble size ranges. P5K control microbubbles showed the lowest amount of C3/C3b binding, suggesting a thicker and denser protective layer was formed by the DSPE-PEG5000 chains than either DSPE-PEG2000 or DSPE-PEG2000/5000 mixture. “*” denotes a significant difference vs. the corresponding P2K control, and “#” denotes a significant difference vs. the corresponding P5K control (p<0.05).
It is commonly accepted that water-soluble, nonionic PEG can protect colloidal particles, such as microbubbles and liposomes, from aggregation and macromolecule adsorption due to the steric hindrance effect of the polymer brush; each PEG chain forms an impermeable “cloud” over the surface because of its large excluded volume, which inhibits most macromolecules from diffusing into the brush layer. Here, we showed evidence to further support this conclusion: the incorporation of DSPE-PEG5000 into the microbubble shell forced the PEG chains to extend further away from the surface than either the DSPE-PEG2000 alone or the DSPE-PEG2000/5000 mixture, therefore forming a thicker and denser protective layer against complement protein adsorption.
For a given architecture, we observed an increase in complement C3/C3b fixation with microbubble size (Figure 6). The increase in MFI was found to be proportional to microbubble surface area, indicating that the surface density of C3/C3b was independent of microbubble diameter. This result was consistent across all microbubble samples, including those with RGD targeting ligands (see below). In addition to the higher total complement fixation, the larger microbubbles could also render them more susceptible to splenic clearance, possibly resulting in shorter circulation persistence. Interestingly, our group previously found that the echoes from untargeted 6–8 μm microbubbles persisted much longer than their 1–2 μm or 4–5 μm counterparts, suggesting other mechanisms, such as gas core dissolution, may be at play [21].
3.5 Effect of RGD ligand surface density
Next, we tested complement fixation on targeted microbubbles. Figure 7 shows the dependence of complement C3/C3b binding on RGD peptide surface density. Targeted ELA microbubbles showed more C3/C3b binding than targeted BLA microbubbles for all RGD peptide surface coverages. For targeted ELA microbubbles, as the amount of conjugated RGD peptide increased, the binding of C3/C3b increased accordingly, indicating a correlated immune response. Complement binding was linearly dependent on microbubble surface area, and the slope (change of MFI per μm2) increased as the RGD surface density increased. However, this trend was not seen for targeted BLA microbubbles. For each size class, the MFI did not increase significantly even when the amount of conjugated RGD peptide was increased by two orders of magnitude. These results agreed with our previous findings that the buried-ligand architecture protects the targeting ligands by inhibiting the adsorption of macromolecules to the microbubble surface [38]. Since the extent of opsonization dictates the degree of complement activation, we conclude that BLA microbubbles triggered less immune recognition than their ELA counterparts.
Figure 7.
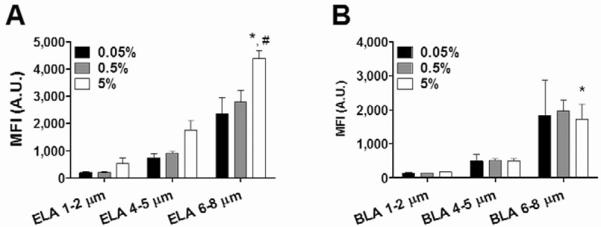
RGD surface coverage and size dependence of complement C3/C3b binding to targeted ELA (A) and BLA (B) microbubbles. For ELA microbubbles, higher RGD surface coverage led to more complement C3/C3b binding. But for BLA microbubbles, the PEG overbrush successfully protected the RGD peptide; no significant increase of MFI values was detected when the RGD conjugation amount was increased by two orders of magnitude. For both surface architectures, large targeted microbubbles (6–8 μm) showed significantly higher C3/C3b binding than smaller ones (1–2 μm and 4–5 μm), possibly due to the higher amount of RGD peptide presented on the surface. “*” denotes a significant increase vs. the corresponding 4–5 μm sample with 5% RGD, and “#” denotes a significant increase vs. the corresponding 6–8 μm sample with 0.5% RGD (p<0.05).
To further investigate the role of targeting ligand presentation on human complement C3/C3b binding, MFI values were compared for targeted and control microbubbles with the same surface PEG brush layer configurations (Figure 8). The 1–2 μm size range was used to represent the entire population of microbubbles. The exposed-ligand architecture led to a significant increase in complement activity compared to the P2K control (3.8-fold increase for ELA 5% vs. 1.5-fold increase for P2K control). We suggest that this increase in C3/C3b binding was due to the presence of RGD peptide on the surface, which interacts with complement proteins in serum. C3 molecules contain unstable thioester bonds upon cleavage of C3a from C3b [24]. RGD peptides contain such nucleophilic groups (e.g., the carbonyl group on Asp and the amino group on Arg), which could trigger the immobilization of C3/C3b molecules on the ELA microbubble surface and activate the alternative pathway. The addition of RGD peptide to the surface of BLA microbubbles similarly led to a significant increase in C3/C3b binding. However, when compared to ELA microbubbles, the increase was much lower (1.3-fold increase for BLA 5% vs. no increase for P5K control). Such a small difference in MFI suggested that the buried-ligand architecture indeed partially inhibited the binding of C3/C3b to the microbubble surface and decreased the immunogenicity of targeted microbubbles.
Figure 8.
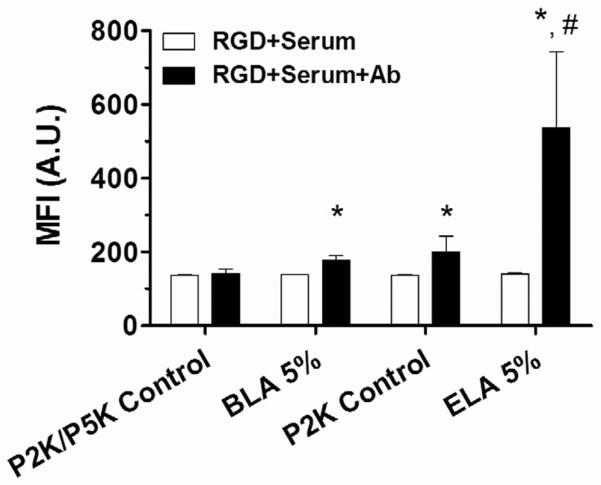
Flow cytometric analysis of complement C3/C3b binding to 5% RGD labeled 1–2 μm ELA and BLA microbubbles. Significant binding occurred to RGD labeled ELA microbubbles in comparison with P2K control, suggesting the targeting ligand was immunogenic. BLA microbubbles also showed C3/C3b binding, indicating partial complement protein fixation. However, the increase of MFI value for targeted ELA microbubbles was much higher than that for BLA microbubbles, supporting our hypothesis that the buried-ligand architecture could indeed successfully shield the RGD peptide from complement recognition. “*” denotes a significant increase vs. the corresponding “RGD+Serum” measurement, and “#” denotes a significant increase vs. BLA 5% (p<0.05).
3.6 Effect of PEG overbrush height
To further illustrate the protective role of PEG chains, we compared the MFI for 5% conjugated RGD peptides for microbubbles with different overbrush lengths (Figure 9). In addition to the ELA and BLA-P5K design, a different bimodal brush layer using DSPE-PEG3000 to form a shorter PEG overbrush was tested for complement C3/C3b binding. We hypothesized that the shorter PEG overbrush would result in an intermediate inhibition of C3/C3b binding. Indeed, for all three size ranges, BLA-P3K microbubbles showed a measured MFI value that fell between the values detected for ELA and BLA-P5K designs when the same amount of RGD peptide was conjugated to the surface. For 6–8 μm microbubbles, there was no significant difference in C3/C3b binding between targeted BLA-P3K and BLA-P5K microbubbles.
Figure 9.
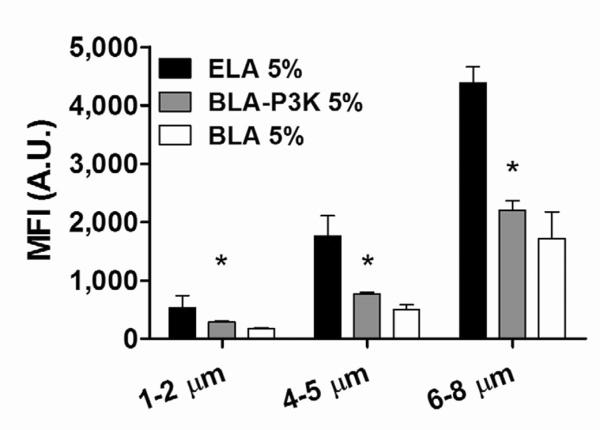
The effect of PEG overbrush length on complement C3/C3b fixation. For targeted microbubbles, BLA-P3K showed an intermediate MFI value, supporting the hypothesis that DSPE-PEG3000 chains formed an intermediate brush layer to protect the targeting ligand from the complement system. “*” denotes a significant difference vs. the corresponding ELA 5% (p<0.05).
The buried-ligand architecture did not completely inhibit the binding of complement C3/C3b to the targeted microbubble surface, presumably due to the phase separation of the phospholipid species in the lipid monolayer coating the microbubble shell. It is also possible that the transient excursion of the PEG polymer chains could bring the RGD ligands past the overbrush and result in complement recognition. However, when compared to targeted ELA microbubbles with the same amount of RGD conjugated to the surface, the amount of C3/C3b binding for BLA microbubbles was significantly reduced (~ −52% and ~ −68% for BLA-P3K and BLA-P5K, respectively, for 5% RGD peptide). It is presumably the combined effect of the PEG overbrush shielding the RGD peptide and inhibiting C3/C3b fixation on the microbubble surface that resulted the reduced complement activation. Therefore, we concluded that the buried-ligand architecture successfully protects RGD peptides on the surface of microbubbles from complement recognition, and targeted BLA microbubbles are significantly less immunogenic than ELA microbubbles in vitro.
4. Conclusion
Complement activation-related pseudoallergy (CARPA) is an important adverse effect that has been observed clinically with non-targeted ultrasound contrast agents [44–46]. Increased incidence of CARPA arising from immunogenic groups on targeted microbubbles is therefore a significant safety concern for the viability of ultrasound molecular imaging. In this study, the immunogenicity of the targeted microbubbles with various surface architectures and ligand surface densities was investigated. We generated RGD-bearing microbubbles with either the exposed- or buried-ligand architecture (ELA or BLA) using a post-labeling technique, and showed that they were stable during incubation in human complement-preserved serum at physiological conditions for at least 2 hrs with minimal change in size distribution or concentration. Three serum factors (albumin, IgG and complement C3/C3b) were tested for microbubble binding, and only C3/C3b binding was detected using our system. Even though RGD peptide is a physiological ligand, we showed that it could still trigger an immune response when conjugated to the surface of microbubbles. However, the PEG overbrush on the surface of BLA microbubbles partially inhibited the recognition of RGD peptide by the complement system and, therefore, reduced the ligand-mediated immunogenicity. When the surface coverage of RGD peptide was increased by two orders of magnitude, C3/C3b binding was increased significantly for ELA microbubbles, while it stayed relatively constant for BLA microbubbles. In addition, we showed evidence of the protective role of the PEG overbrush on complement C3/C3b fixation by showing that P5K control microbubbles had the lowest C3/C3b binding and that BLA-P3K microbubbles could still partially inhibit complement binding even with a shorter PEG overbrush layer. Our results show that the BLA strategy offers a means of targeting microbubbles without increasing complement activation and suggest that further studies are warranted to investigate the in vivo molecular imaging capabilities of such microbubbles.
Figure 1.

Cartoon illustration showing the dimensions of a microbubble with either an exposed-or buried-ligand architecture (ELA or BLA). The PEG chain length was estimated using self-consistent field (SCF) theory [45] using values of 0.44 nm2 for the average projected area per lipid molecule [46] and 0.35 nm for PEG monomer length. The lipid monolayer thickness was estimated to be ~3 nm based on the persistence length of the stearoyl chains [41].
Acknowledgements
We thank Dr. Shashank Sirsi for insightful suggestions, and the Materials Research Science and Engineering Center (MRSEC) at Columbia University for access to the Zetasizer Nano-ZS. This research was supported by NIH RO1 EB 009066.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Schutt EG, Klein DH, Mattrey RM, Riess JG. Injectable microbubbles as contrast agents for diagnostic ultrasound imaging: the key role of perfluorochemicals. Angew Chem Int Ed. 2003;42:3218–35. doi: 10.1002/anie.200200550. [DOI] [PubMed] [Google Scholar]
- [2].Quaia E. Microbubble ultrasound contrast agents: an update. Eur Radiol. 2007;17:1995–2008. doi: 10.1007/s00330-007-0623-0. [DOI] [PubMed] [Google Scholar]
- [3].Ferrara K, Pollard R, Borden M. Ultrasound microbubble contrast agents: fundamentals and application to gene and drug delivery. Annu Rev Biomed Eng. 2007;9:415–47. doi: 10.1146/annurev.bioeng.8.061505.095852. [DOI] [PubMed] [Google Scholar]
- [4].Stride E, Saffari N. Microbubble ultrasound contrast agents: a review. Proc Inst Mech Eng H. 2003;217:429–47. doi: 10.1243/09544110360729072. [DOI] [PubMed] [Google Scholar]
- [5].Dijkmans PA, Juffermans LJ, Musters RJ, van Wamel A, ten Cate FJ, van Gilst W, et al. Microbubbles and ultrasound: from diagnosis to therapy. Eur J Echocardiogr. 2004;5:245–56. doi: 10.1016/j.euje.2004.02.001. [DOI] [PubMed] [Google Scholar]
- [6].Lindner JR. Microbubbles in medical imaging: current applications and future directions. Nat Rev Drug Discov. 2004;3:527–32. doi: 10.1038/nrd1417. [DOI] [PubMed] [Google Scholar]
- [7].Sirsi S, Borden M. Microbubble compositions, properties and biomedical applications. Bubble Sci, Eng & Tech. 2009;1:3–17. doi: 10.1179/175889709X446507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lanza GM, Wickline SA. Targeted ultrasonic contrast agents for molecular imaging and therapy. Prog Cardiovasc Dis. 2001;44:13–31. doi: 10.1053/pcad.2001.26440. [DOI] [PubMed] [Google Scholar]
- [9].Ning SC, Tian JQ, Marshall DJ, Knox SJ. Anti-alpha v integrin monoclonal antibody intetumumab enhances the efficacy of radiation therapy and reduces metastasis of human cancer xenografts in nude rats. Cancer Res. 2010;70:7591–9. doi: 10.1158/0008-5472.CAN-10-1639. [DOI] [PubMed] [Google Scholar]
- [10].Anderson CR, Rychak JJ, Backer M, Backer J, Ley K, Klibanov AL. scVEGF microbubble ultrasound contrast agents: a novel probe for ultrasound molecular imaging of tumor angiogenesis. Invest Radiol. 2010;45:579–85. doi: 10.1097/RLI.0b013e3181efd581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tardy I, Pochon S, Theraulaz M, Emmel P, Passantino L, Tranquart F, et al. Ultrasound molecular imaging of VEGFR2 in a rat prostate tumor model using BR55. Invest Radiol. 2010;45:573–8. doi: 10.1097/RLI.0b013e3181ee8b83. [DOI] [PubMed] [Google Scholar]
- [12].Wang YX, Zhou J, Zhang YR, Wang XM, Chen JA. Delivery of TFPI-2 using SonoVue and adenovirus results in the suppression of thrombosis and arterial re-stenosis. Exp Biol Med. 2010;235:1072–81. doi: 10.1258/ebm.2010.010046. [DOI] [PubMed] [Google Scholar]
- [13].Rossi S, Ghittoni G, Ravetta V, Viera FT, Rosa L, Serassi M, et al. Contrast-enhanced ultrasonography and spiral computed tomography in the detection and characterization of portal vein thrombosis complicating hepatocellular carcinoma. Eur Radiol. 2008;18:1749–56. doi: 10.1007/s00330-008-0931-z. [DOI] [PubMed] [Google Scholar]
- [14].Kaufmann BA, Sanders JM, Davis C, Xie A, Aldred P, Sarembock IJ, et al. Molecular imaging of inflammation in atherosclerosis with targeted ultrasound detection of vascular cell adhesion molecule-1. Circulation. 2007;116:276–84. doi: 10.1161/CIRCULATIONAHA.106.684738. [DOI] [PubMed] [Google Scholar]
- [15].Lindner JR, Coggins MP, Kaul S, Klibanov AL, Brandenburger GH, Ley K. Microbubble persistence in the microcirculation during ischemia/reperfusion and inflammation is caused by integrin- and complement-mediated adherence to activated leukocytes. Circulation. 2000;101:668–75. doi: 10.1161/01.cir.101.6.668. [DOI] [PubMed] [Google Scholar]
- [16].Kaufmann BA, Carr CL, Belcik JT, Xie A, Yue Q, Chadderdon S, et al. Molecular imaging of the initial inflammatory response in atherosclerosis: implications for early detection of disease. Arterioscler Thromb Vasc Biol. 2010;30:54–U132. doi: 10.1161/ATVBAHA.109.196386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Borden MA, Dayton PA. Targeted Ultrasound Contrast Agents. In: Pomper MG, Gelovani JG, editors. Molecular Imaging in Oncology. Informa Healthcare USA, Inc.; New York: 2008. p. 329. [Google Scholar]
- [18].Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Letters. 1990;268:235–7. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- [19].Klibanov AL. Targeted delivery of gas-filled microspheres, contrast agents for ultrasound imaging. Adv Drug Deliv Rev. 1999;37:139–57. doi: 10.1016/s0169-409x(98)00104-5. [DOI] [PubMed] [Google Scholar]
- [20].Lindner JR, Song J, Jayaweera AR, Sklenar J, Kaul S. Microvascular rheology of Definity microbubbles after intra-arterial and intravenous administration. J Am Soc Echocardiogr. 2002;15:396–403. doi: 10.1067/mje.2002.117290. [DOI] [PubMed] [Google Scholar]
- [21].Sirsi S, Feshitan J, Kwan J, Homma S, Borden M. Effect of microbubble size on fundamental mode high frequency ultrasound imaging in mice. Ultrasound Med Biol. 2010;36:935–48. doi: 10.1016/j.ultrasmedbio.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mosqueira VCF, Legrand P, Gulik A, Bourdon O, Gref R, Labarre D, et al. Relationship between complement activation, cellular uptake and surface physicochemical aspects of novel PEG-modified nanocapsules. Biomaterials. 2001;22:2967–79. doi: 10.1016/s0142-9612(01)00043-6. [DOI] [PubMed] [Google Scholar]
- [23].Harashima H, Sakata K, Funato K, Kiwada H. Enhanced hepatic-uptake of liposomes through complement activation depending on the size of liposomes. Pharm Res. 1994;11:402–6. doi: 10.1023/a:1018965121222. [DOI] [PubMed] [Google Scholar]
- [24].Murphy K, Travers P, Walport M. Janeway's Immunobiology. 7 ed. Garland Science, Taylor & Francis Group, LLC; New York: 2008. [Google Scholar]
- [25].Lodish H, Berk A, Kaiser C, Krieger M, Scott MP, Bretscher A, et al. Molecular Cell Biology. 6 ed. W. H. Freeman and Company; New York: 2008. [Google Scholar]
- [26].Yanagisawa K, Moriyasu F, Miyahara T, Yuki M, Iijima H. Phagocytosis of ultrasound contrast agent microbubbles by Kupffer cells. Ultrasound Med Biol. 2007;33:318–25. doi: 10.1016/j.ultrasmedbio.2006.08.008. [DOI] [PubMed] [Google Scholar]
- [27].Quaia E, Blomley MJK, Patel S, Harvey CJ, Padhani A, Price P, et al. Initial observations on the effect of irradiation on the liver-specific uptake of Levovist. Eur J Radiol. 2002;41:192–9. doi: 10.1016/s0720-048x(01)00458-2. [DOI] [PubMed] [Google Scholar]
- [28].Lim AKP, Patel N, Eckersley RJ, Taylor-Robinson SD, Cosgrove DO, Blomley MJK. Evidence for spleen-specific uptake of a microbubble contrast agent: a quantitative study in healthy volunteers. Radiology. 2004;231:785–8. doi: 10.1148/radiol.2313030544. [DOI] [PubMed] [Google Scholar]
- [29].Szebeni J. Complement activation-related pseudoallergy caused by amphiphilic drug carriers: the role of lipoproteins. Curr Drug Deliv. 2005;2:7. doi: 10.2174/156720105774370212. [DOI] [PubMed] [Google Scholar]
- [30].D'Andrea LD, Del Gatto A, Pedone C, Benedetti E. Peptide-based molecules in angiogenesis. Chem Biol Drug Des. 2006;67:115–26. doi: 10.1111/j.1747-0285.2006.00356.x. [DOI] [PubMed] [Google Scholar]
- [31].Miller JC, Pien HH, Sahani D, Sorensen AG, Thrall JH. Imaging angiogenesis: applications and potential for drug development. J Natl Cancer Inst. 2005;97:172–87. doi: 10.1093/jnci/dji023. [DOI] [PubMed] [Google Scholar]
- [32].Szebeni J. Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology. 2005;216:106–21. doi: 10.1016/j.tox.2005.07.023. [DOI] [PubMed] [Google Scholar]
- [33].Moghimi SM, Andersen AJ, Hashemi SH, Lettiero B, Ahmadvand D, Hunter AC, et al. Complement activation cascade triggered by PEG-PL engineered nanomedicines and carbon nanotubes: the challenges ahead. J Control Release. 2010;146:175–81. doi: 10.1016/j.jconrel.2010.04.003. [DOI] [PubMed] [Google Scholar]
- [34].Moghimi SM, Hamad I. Liposome-mediated triggering of complement cascade. J Liposome Res. 2008;18:195–209. doi: 10.1080/08982100802309552. [DOI] [PubMed] [Google Scholar]
- [35].Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42:463–78. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- [36].Moghimi SM, Hamad I, Andresen TL, Jorgensen K, Szebeni J. Methylation of the phosphate oxygen moiety of phospholipid-methoxy(polyethylene glycol) conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production. FASEB J. 2006;20:2591–+. doi: 10.1096/fj.06-6186fje. [DOI] [PubMed] [Google Scholar]
- [37].Feshitan JA, Chen CC, Kwan JJ, Borden MA. Microbubble size isolation by differential centrifugation. J Colloid Interface Sci. 2009;329:316–24. doi: 10.1016/j.jcis.2008.09.066. [DOI] [PubMed] [Google Scholar]
- [38].Chen CC, Borden MA. Ligand conjugation to bimodal poly(ethylene glycol) brush layers on microbubbles. Langmuir. 2010;26:13183–94. doi: 10.1021/la101796p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nallamothu R, Wood GC, Pattillo CB, Scott RC, Kiani MF, Moore BM, et al. A tumor vasculature targeted liposome delivery system for combretastatin A4: design, characterization, and in vitro evaluation. AAPS PharmSciTech. 2006;7:10. doi: 10.1208/pt070232. [DOI] [PubMed] [Google Scholar]
- [40].Liu XY, Ruan LM, Mao WW, Wang JQ, Shen YQ, Sui MH. Preparation of RGD-modified long circulating liposome loading matrine, and its in vitro anti-cancer effects. Int J Med Sci. 2010;7:197–208. doi: 10.7150/ijms.7.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Borden MA, Zhang H, Gillies RJ, Dayton PA, Ferrara KW. A stimulus-responsive contrast agent for ultrasound molecular imaging. Biomaterials. 2008;29:597–606. doi: 10.1016/j.biomaterials.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Toda M, Kitazawa T, Hirata I, Hirano Y, Iwata H. Complement activation on surfaces carrying amino groups. Biomaterials. 2008;29:407–17. doi: 10.1016/j.biomaterials.2007.10.005. [DOI] [PubMed] [Google Scholar]
- [43].Moghimi SM, Hamad I, Bunger R, Andresen TL, Jorgensen K, Hunter AC, et al. Activation of the human complement system by cholesterol-rich and PEGylated liposomes - Modulation of cholesterol-rich liposome-mediated complement activation by elevated serum LDL and HDL levels. J Liposome Res. 2006;16:167–74. doi: 10.1080/08982100600848801. [DOI] [PubMed] [Google Scholar]
- [44].Herzog CA. Incidence of adverse events associated with use of perflutren contrast agents for echocardiography. JAMA. 2008;299:2023–5. doi: 10.1001/jama.299.17.2023. [DOI] [PubMed] [Google Scholar]
- [45].Wei K, Mulvagh SL, Carson L, Davidoff R, Gabriel R, Grimm RA, et al. The safety of Definity and Optison for ultrasound image enhancement: a retrospective analysis of 78,383 administered contrast doses. J Am Soc Echocardiogr. 2008;21:1202–6. doi: 10.1016/j.echo.2008.07.019. [DOI] [PubMed] [Google Scholar]
- [46].Main ML. Ultrasound contrast agent safety from anecdote to evidence. JACC. 2009;2:1057–9. doi: 10.1016/j.jcmg.2009.05.008. [DOI] [PubMed] [Google Scholar]