

Abstract
The initiation and progression of cardiovascular diseases involve extensive arterial wall matrix protein degradation. Proteases are essential to these pathological events. Recent discoveries suggest that proteases do more than catabolize matrix proteins. During the pathogenesis of atherosclerosis, abdominal aortic aneuryms, and associated complications, cysteinyl cathepsins and mast cell tryptases and chymases participate importantly in vascular cell apoptosis, foam cell formation, matrix protein gene expression, and pro-enzyme, latent cytokine, chemokine, and growth factor activation. Experimental animal disease models have been invaluable in examining each of these protease functions. Deficiency and pharmacological inhibition of cathepsins or mast cell proteases have allowed their in vivo evaluation in the setting of pathological conditions. Recent discoveries of highly selective and potent inhibitors of cathepsins, chymase, and tryptase, and their applications in vascular diseases in animal models and non-vascular diseases in human trials, have led to the hypothesis that selective inhibition of cathepsins, chymases, and tryptase will benefit patients suffering from cardiovascular diseases. This review highlights recent discoveries from in vitro cell-based studies to experimental animal cardiovascular disease models, from protease knockout mice to treatments with recently developed selective and potent protease inhibitors, and from patients with cathepsin-associated non-vascular diseases to those affected by cardiovascular complications.
Keywords: cathepsin, mast cell, chymase, tryptase, atherosclerosis, abdominal aortic aneurysm, protease inhibitor
1. Introduction
Cysteinyl cathepsins are papain family members of the cysteine protease superfamily. In humans, 11 members have been identified — cathepsins B, C, F, H, K, L, O, S, V, W, and X — all of which share a conserved active three-dimensional pocket formed with cysteine, histidine, and asparagine residues (Mohamed & Sloane, 2006; Turk et al., 2001). Cathepsins are synthesized as preproenzymes. Procathepsin is formed after removal of the prepeptide during the passage to the endoplasmic reticulum. Subsequently, the active cathepsins can be produced after proteolytic removal of the propeptide in the acidic compartments of the late endosomes or lysosomes. Cysteinyl cathepsins typically localize in the lysosomes and endosomes to degrade intracellular and endocytosed “unwanted” proteins. Most cathepsins, such as cathepsins B, F, H, K, L, and V, are optimally active in acidic environments and become inactive at neutral pH. Cathepsin S (CatS), however, remains partially active at neutral pH (Shi et al., 1992).
Under physiological circumstances, cathepsins usually localize in lysosomes, but lysosomal permeabilization due to exogenous oxidants (reactive oxygen species) may induce lysosomal leakage, leading to the release of cathepsins into the cytoplasm (Chwieralski et al., 2006; Ichinose et al., 2006). Cathepsins have been found in cell culture medium and in the circulation under physiological conditions, but increased after cells were induced with inflammatory cytokines or in patients with inflammatory diseases (Sukhova et al., 1998; Liu et al., 2006a; Taleb et al., 2005; Yang et al., 2007), through mechanisms that are not fully understood. Increased extracellular cathepsins may participate in extracellular matrix (ECM) protein remodeling (Tu et al., 2008; Cavallo- Medved et al., 2009; Barascuk et al., 2010).
Cathepsins both inside and outside of cells play both pathological and physiological roles. Besides degrading endocytosed and endogenous proteins, cathepsins have been implicated in antigen processing and presentation (Riese et al., 1996; Shi et al., 1999a) and involved in insulin receptor turnover (Yang et al., 2007; Yang et al., 2008) within the endosome and lysosome compartments. Outside of cells, cathepsins degrade elastin (Shi et al., 1992), collagen (Kitamoto et al., 2007), fibronectin (Yang et al., 2007), and laminin (Wang et al., 2006), and may also degrade other untested ECM. Extracellular cathepsins may use their remaining activity under neutral pH to interact with ECM or by cell–matrix direct contact, which forms acidic milieu for optimal cathepsin ECM degradation (Reddy et al., 1995; Punturieri et al., 2000). All such intracellular and extracellular activities have been implicated in the pathogenesis of autoimmune diseases, cancer, bone metabolism, metabolic diseases, and cardiovascular diseases (Sukhova et al., 2003; Maehr et al., 2005; Yang et al., 2005; Wang et al., 2006; Lutgens et al., 2006; Yang et al., 2007; Kitamoto et al., 2007; Yang et al., 2008; Salminen-Mankonen et al., 2007). Detailed information concerning cysteinyl cathepsin structures, protease-substrate binding specificity, activity regulation, general pathological functions in different dieases, and pharmacological inhibition with small molecule inhibitors has been fully reviewed elsewhere (McGrath, 1999; Turk & Guncar, 2002; Roberts, 2005; Vasiljeva et al., 2007).
Mast cell proteases include mainly tryptase, chymase, and carboxypeptidase A (CPA), although these cells are also rich in cysteinyl cathepsins and matrix metalloproteinases (MMPs) (Mallen-St Clair et al., 2006; Di Girolamo et al., 2006). While both tryptase and chymase belong to the serine protease family, CPA is a zinc- dependent metalloproteinase. Gunnar Pejler and colleagues summarized these mast cell proteases — including chromosome localization, transcription regulation, evolution, pro-enzyme activation, three-dimensional structure, substrate specificity, physiological and pathological roles, and protease inhibitor applications (Pejler et al., 2007). Chymase can generate angiotensin-II (Ang-II) from angiotensin-I (Ang-I) (Sanker et al., 1997; Caughey et al., 2000; Dell'Italia & Husain, 2002), fibronectin from ECM (Lazaar et al., 2002), TGF-β from ECM (Taipale et al., 1995), collagen for fibril formation from type I procollagen (Kofford et al., 1997), and active MMPs from zymogens (Fang et al., 1996; Tchougounova et al., 2005); and activates IL1β, IL18, endothelin-1, and endothelin-2 (Mizutani et al., 1991; Omoto et al., 2006; Kido et al., 1998). Chymase also degrades lipoproteins, thereby promoting macrophage foam cell formation (Lee-Ruechert & Kovanen, 2006). Tryptase activates pro-MMPs (Kaminska et al., 1999) and chemokines (Pang et al., 2006), and degrades lipoproteins (Lee et al., 2002) and fibronectin (Schwartz et al., 1985). These mast cell proteases have also been implicated in collagen synthesis and tissue fibrosis (Abe et al., 1998; Jones et al., 2004; Satomura et al., 2003), angiogenesis (Blair et al., 1997; Russo et al., 2005), immunoglobulin molecule synthesis (Yoshikawa et al., 2001), and associated cardiac disorders (Tsunemi et al., 2002; Hamada et al., 1999; Ortlepp et al., 2001; Hoshino et al., 2003; Takai et al., 2003).
In this review, we will summarize briefly recent discoveries in cysteinyl cathepsins and mast cell chymase and tryptase in cardiovascular diseases, mainly in atherosclerosis and abdominal aortic aneurysms (AAA), including protease functions and drug developments against these proteases.
2. Cathepsins in atherosclerosis
Arterial wall remodeling, including elastinolysis and collagenolysis, is critical in atherogenesis (Lijnen, 2002; Katsuda & Kaji, 2003). Many cathepsins, such as cathepsins S, L, K, B, and V (Guinec et al., 1993; Chapman et al., 1997; Yasuda et al., 2004), are potent elastases and/or collageases. In atherosclerotic lesions, inflammatory cell adhesion on the endothelium either in large vessels or the vasa vasrum, and cross-transmigration through the subendothelium basement membrane, are essential to atherosclerosis neointima formation. These inflammatory cells, including monocytes/macrophages, lymphocytes, mast cells, and neotrophils, all express high levels of cathepsins (Shi et al., 1992; Kitamoto et al., 2007; Sun et al., 2007a; 2009; Blomgran et al., 2007), as well as inflammatory cytokines that can trigger vascular cell cathepsin expression (Sukhova et al., 1998; Shi et al., 1999b). Inflammatory cells and activated vascular cells then release cathepsins or other proteases to degrade extracellular collagen and elastin in the arterial wall, thus promoting atherogenesis.
2.1. Cathepsin S
CatS was one of the first cathepsins found in human atherosclerotic lesions (Sukhova et al., 1998). While healthy human arteries expressed negligible levels of CatS, early human atherosclerotic lesions (or fatty streaks) showed vivid CatS immunoreactivity in intimal and medial smooth-muscle cells (SMCs). In advanced human atherosclerotic plaques, CatS was localized in macrophages and SMCs of the fibrous cap (Sukhova et al., 1998) and in areas of elastin fragmentation (Rodgers et al., 2006). Endothelial cells (ECs) lining the lumen of the arterial wall and microvessels inside plaques also expressed CatS (Liu et al., 2004). In mouse atherosclerotic lesions, CatS, CatL, and CatB were increased and localized in macrophages or lipid-rich areas (Jormsjo et al., 2002). Deficiency of CatS (Ctss−/−) in atherosclerosis-prone low-density lipoprotein receptor–deficient (Ldlr−/−) mice resulted in significant reductions in atherosclerotic plaque area (60% reduction after 12 weeks of an atherogenic diet), and decreases in the number of elastin breaks and elastinolytic activities (Sukhova et al., 2003). Furthermore, CatS deficiency led to reduced SMC content, collagen content, and fibrous cap thickness. Macrophages from Ctss−/− mice showed reduced transmigration through an artificial aortic wall made with EC monolayer and collagen types I and IV (Sukhova et al., 2003). In a separate study using apolipoprotein E–deficient (Apoe−/−) mice (Rodgers et al., 2006), CatS deficiency also reduced atherosclerotic plaque size by 46% (after 12 weeks of an atherogenic diet) and reduced the number of plaque ruptures (as defined by visible defects in the cap, accompanied by the intrusion of erythrocytes into the region below) by 73%. The numbers of elastin breaks in atherosclerotic lesions were not significantly different, however, between Apoe−/− mice and those mutant for CatS, but total plaque elastin content was reduced by 49% in Ctss−/− Apoe−/− mice. Using bone marrow transplantation technology, de Nooijer and colleagues tested the importance of leukocyte-derived CatS in atherogenesis. Leukocyte CatS deficiency resulted in considerably altered plaque morphology, with smaller necrotic cores, reduced apoptosis, and decreased SMC content and collagen deposition (de Nooijer et al., 2009). These data all point to a protective role of CatS inhibition or deficiency in atherogenesis through decreased degradation of ECM components.
2.2. Cathepsin K
CatK shares many similarities with CatS. They have the same chromosomal localization (Shi et al., 1994; Gelb et al., 1997) and share 56% homologies of the primary amino acid sequences (Shi et al., 1995). Like CatS, CatK expression in normal arteries is hardly detectable, but also like CatS, early human atherosclerotic lesions show CatK expression in the intimal and medial SMCs. In advanced atherosclerotic plaques, CatK also localized in macrophages and in SMCs of the fibrous cap (Sukhova et al., 1998). CatK expression is highest in advanced but stable human atherosclerotic plaques, compared with early atherosclerotic lesions and lesions containing thrombi (Lutgens et al., 2006). Augmented CatK proteolysis in atheromata further linked CatK to vascular remodeling and plaque vulnerability. Protease-activatable near-infrared fluorophore probe for in vivo optical imaging demonstrated preferential localization of enzymatically active CatK to macrophage-rich areas in mice (Jaffer et al., 2007). CatK deficiency (Ctsk−/−) in Apoe−/− mice resulted in a 42% reduction in the atherosclerotic plaque area. Although the total number of plaques was unchanged, there was a relative increase in early lesions and a decrease in advanced lesions in the absence of CatK. As anticipated, lack of potent elastase and collagenase CatK led to increases lesion collagen and decreased elastin breaks, although lesion T lymphocyte content and lipid core area were not affected (Lutgens et al., 2006). CatK deficiency appears to increase lesion stability in brachiocephalic arteries by maintaining the integrity of the tunica media and by decreasing plaque vulnerability to rupture (Samokhin et al., 2008). Therefore, most of these beneficial pathologies obtained from the Ctsk−/− mice associate with CatK elastase and collagenase activies.
2.3. Cathepsin L
Like CatK, CatL is a potent elastase and collagenase. But unlike CatK, which is expressed exclusively in osteoclasts under normal conditions (Shi et al., 1995), CatL is expressed at low levels in almost all tested tissue or cell types. Inflammatory cytokines and pro-angiogenic growth factors induce CatL expression in vascular SMCs and ECs (Liu et al., 2006b). Immunostaining using human CatL antibodies demonstrated increased CatL immunoreactivities in human atheroma. CatL localized mainly to SMCs, ECs, and macrophages in advanced atherosclerotic lesions (Liu et al., 2006b), particularly in areas with necrotic cores and with ruptures of the fibrous cap, and correlated with apoptosis and oxidative stress protein (Li et al., 2009). CatL expression was consistently higher in plaques from symptomatic patients than in those from asymptomatic patients. Monocyte-derived macrophages from atherosclerosis patients contained higher amounts of CatL proteins and were more prone to 7β-hydroxycholesterol-induced apoptosis (Li et al., 2009). Oscillatory and lowered shear stress corresponds to different atheroprone areas, while laminar shear stress corresponds to atheroprotected areas (Cheng et al., 2006; Koskinas et al., 2010; Chatzizisis et al., 2011). Secreted CatL was suggested as a shear dependent matrix protease (Zarins et al., 1983). Inhibition of CatL by a cysteine protease inhibitor or CatL silencing RNA (siRNA) treatment inhibited oscillatory shear stress-induced gelatinase and elastase activity by mouse aortic ECs, but not laminar shear stress-induced activity (Platt et al., 2006). In hyperlipidemic mice and swine, areas with low endothelial shear stress contained significantly larger atherosclerotic lesions than those with increased endothelial shear stress or oscillatory shear stress (Cheng et al., 2006; Koskinas et al., 2010; Chatzizisis et al., 2011). Although whether CatL expression and activities are also higher in low-shear-stress segments than in high-shear-stress or oscillatory-shear-stress segments remains untested, both MMP (MMP-2, MMP-9, and MMP-12) and cathepsin (CatS and CatK) mRNA and enzymatic activities are significantly higher in low-shear-stress segments than in high-shear-stress or oscillatory-shear-stress segments (Cheng et al., 2006; Chatzizisis et al., 2011). These observations suggest that CatL is a shear sensitive protease with potential importance in vascular remodeling and atherosclerosis. Using Ldlr−/− mice, we found that the absence of one or both alleles of the CatL gene reduced atherosclerosis at both 12-week and 26-week time points. Lesion lipid core areas, inflammatory cell content, collagen levels, medial SMC content, and elastin fragmentation all were reduced in a gene dose-dependent manner (Kitamoto et al., 2007). Mechanistically, we demonstrated that this potent elastase and collagenase also was involved in T-cell and peripheral blood mononuclear cell transmigration through collagens or collagens coated with endothelium monolayer (Kitamoto et al., 2007).
2.4. Other cysteinyl cathepsins
A few other cathepsins also may participate in atherosclerosis. CatB mRNA and protein levels were increased in atherosclerotic lesions from Apoe−/− mice, and CatB immunoreactivity was highest in areas next to the lumen or rich in macrophages (Chen et al., 2002). CatF was expressed weakly in normal human arteries, but in human atherosclerotic lesions, it localized in macrophages devoid of intracellular lipid and in SMCs and ECs, but not in T lymphocytes (Oorni et al., 2004). CatV is the most potent elastase among all cysteinyl cathepsins. It accounts for one third of intracellular elastase activities in macrophages (Yasuda et al., 2004). Whether other cells also express CatV in human atherosclerotic lesions remains unknown, but total protein extract preparation immunoblot analysis demonstrated CatV expression in human atheromata (Yasuda et al., 2004). Direct participation of these cysteinyl cathepsins in atherogenesis, however, is still untested in animal models.
2.5. Cathepsins and lipid metabolism
Lipoprotein modification and uptake by atherosclerotic lesion cells, mainly macrophages and SMCs, are important pathologic steps in atherogenesis (Doran et al., 2008; Shashkin et al., 2005). After taking up these lipoproteins and modified lipoproteins, macrophages and SMCs become foam cells filled with lipid droplets. This step can be enhanced, depending on the levels of lipid modification (Shashkin et al., 2005), but proteolysis of lipid components may also facilitate this process. For example, increased uptake and decreased lysosomal degradation of multiple modified low-density lipoproteins (LDLs) causes LDL accumulation in human aortic SMCs, leading to SMC foam cell formation. This process depends partly upon cysteinyl cathepsins (Tertov & Orekhov, 1997).
Cathepsins B, F, K, L, S, and V have been implicated in ApoB-100 proteolytic modification, which enhances extracellular LDL particle aggregation, lipid droplet formation, and LDL retention to human arterial proteoglycans (Oorni et al., 2004). This proteolysis enhanced LDL particle fusion competence to macrophages (Linke et al., 2006). On the other hand, lipids or modified lipoproteins also affect cysteinyl cathepsin cellular expression and localization. When macrophages were exposed to oxidized LDL (ox-LDL) or 7β-hydroxycholesterol, these cells expressed high amounts of CatB and CatL, in addition to forming foam cells. These cathepsins then translocated from lysosomes to cytosol or nuclei. Such relocalization of cathepsins causes macrophage apoptosis (Li & Yuan, 2004); this proposed pathway is consistent with observations of macrophage foam cell formation and apoptosis in human atherosclerotic lesions (Martinet & Kockx, 2001). Cysteinyl cathepsins also participate in the degradation of high-density lipoproteins (HDL), thus reducing macrophage foam cell cholesterol efflux. When recombinant human CatS or CatF was incubated with HDL3, these cathepsins degraded apoA-1 rapidly, leading to complete loss of apoA-1 cholesterol acceptor function, thereby blocking macrophage intracellular cholesterol efflux (Lindstedt et al., 2003). Interestingly, under the same conditions, recombinant human CatK did not act similarly. CatK function in lipid deposition in macrophages also remains unexplained. Lutgens and colleagues showed that CatK deficiency increased macrophage size and consequent lipid deposition (Lutgens et al., 2006). Therefore, different cathepsins show different functions in lipid metabolism, and contribute to atherosclerosis with different mechanisms. The roles of cysteinyl cathepsins in atherosclerosis are summarized in Figure 1.
Figure 1.

Illustration of cysteinyl cathepsin functions in the pathogenesis of vascular diseases, such as atherosclerosis and abdominal aortic aneurysm. Cathepsins can originate from inflammatory cells or from vascular SMCs and ECs under inflammatory conditions.
3. Cathepsins in AAA
The development of AAA involves extensive inflammatory cell infiltration, followed by releasing cytokines, chemokines, and proteases. Increased levels of these inflammatory molecules result in further inflammatory cell recruitment, thereby inducing vascular cell inflammation and apoptosis, catabolizing the vasculature ECM proteins (e.g., elastin and collagen), and promoting cell migration, angiogenesis, and apoptosis. These infiltrates mainly include macrophages, neutrophils, CD4+ T cells, NK T cells, and MCs, which are recruited from the lumen or the vasa vasorum to the adventitia and media of human or animal AAA (Koch et al., 1990; Chan et al., 2005; Duftner et al., 2005; Sun et al., 2007b). As in atherosclerotic lesions, these inflammatory infiltrates release cathepsins, and cause further destructive elastinolysis, inflammatory cell recruitment, vascular cell apoptosis, and angiogenesis. The roles of cysteinyl cathepsins in AAA are largely the same as those proposed in atherogenesis (Figure 1), but different cathepsins may play different roles in AAA.
We reported enhanced CatS and CatK expression in human AAA lesions with concomitant deficiency of their endogenous inhibitor, cystatin C (Shi et al., 1999b). Serum cystatin C levels are significantly lower in AAA patients than in control subjects (Shi et al., 1999b; Lindholt et al., 2001). CatB, CatC, and CatL expression and activity also were increased in the aneurysm wall and thrombus of human aortic aneurysms when compared with normal arteries (Gacko et al., 1999; Gacko & Glowinski, 1998). Protein levels of CatL were also increased in human AAA and atheroma (Liu et al., 2006b), suggesting that these cysteinyl cathepsins participate in aneurysm formation (Abdul-Hussien et al., 2007; Liu et al., 2006b). Several cathepsins — including cathepsins S, C, K, and L — recently have been tested in AAA formation in mouse experimental models.
3.1. Cathepsin S
The role of CatS in AAA has been tested in abdominal aortic elastase perfusion-induced experimental AAA (Thompson RW et al., unpublished data) and in Ang-II minipump perfusion-induced experimental AAA (Qin Y et al., unpublished data). Although detailed discoveries are pending, absence of CatS prevented AAA formation in both models. In the elastase perfusion-induced model, at 14 days post-perfusion, abdominal aortic dilation was significantly reduced in Ctss−/− mice compared with wild-type (WT) control mice. Similar observations were obtained in Apoe−/−Ctss−/− mice. Compared with Apoe−/− control mice, Apoe−/−Ctss−/− mice developed much smaller AAAs at 28 days after Ang-II perfusion.
3.2. Cathepsin C
CatC, also called dipeptidyl peptidase I (DPPI), helps to activate neutrophil-derived neutrophil elastase, CatG, and proteinase 3 (Adkison et al., 2002). DPPI-deficient (DPPI−/−) mice were resistant to elastase perfusion-induced AAA (Pagano et al., 2007). At 14 days post-perfusion, WT mice showed transmural inflammatory cell infiltration and pronounced medial elastin laminae destruction. Such inflammatory responses were significantly reduced in DPPI−/− mice. Mechanistically, DPPI controls neutrophil infiltration and neutrophil chemokine CXCL2 production. Reconstitution of DPPI−/− mice with WT neutrophils, but not those from DPPI−/− mice, fully restored aortic dilation. Both neutrophils and CXCL2 reached a peak at 3 days post-perfusion in WT mice. At the same time point, however, neutrophil levels decreased by 60% and aortic wall CXCL2 levels remained at baseline in DPPI−/− mice. Antibody depletion of neutrophils or chemokine CXCL2 reduced aortic dilation significantly, but why DPPI affects neutrophil recruitment and how DPPI-mediated activation of neutrophil proteases controls neutrophil chemokine CXCL2 levels in the arterial wall remain unknown.
3.3. Cathepsin K
CatK function has been tested in Ang-II perfusion-induced experimental AAA (Bai et al., 2010) and aortic elastase perfusion-induced AAA (Shi, unpublished data). In Ang-II–induced AAA, the absence of CatK in Apoe−/− mice did not affect AAA development. Aneurysm area, numbers of ruptures, and aortic wall elastin breaks were not affected by CatK deficiency. Unexpectedly, AAA lesions from Ctsk−/− mice contained significantly more CD45+ leukocytes and Mac-3+ macrophages, and enhanced CatS and CatC expression. Peripheral blood from Ctsk−/− mice also contained more Ly6G+ granulocytes and CD4+CD25+ T cells than those of the control mice after Ang-II perfusion (Bai et al., 2010). Although the investigators did not examine by what mechanisms CatK deficiency led to increased inflammatory cells and other cathepsin expression in AAA lesions or in peripheral blood, these compensatory changes may explain insignificant AAA lesion sizes between Apoe−/−Ctsk+/+ and Apoe−/−Ctsk−/− mice. In elastase perfusion-induced experimental AAA, however, absence of CatK prevented AAA formation in mice. At 14 days post-perfusion, all WT mice developed AAA, but no Ctsk− /− mice did. Aortic lesions from Ctsk−/− mice contained significantly fewer CD4+ T cells, Ki67+ proliferating cells, and apoptotic cells, and less medial SMC loss than those from control mice. We demonstrated that CatK contributed to lesion T-cell proliferation, medial SMC apoptosis, and elastin fragmentation. In vitro study did not reveal any significant effect of CatK in macrophage proliferation and transmigration, or in angiogenesis. Indeed, lesion total macrophage content and CD3+ microvessel contents were no different between WT and Ctsk−/− mice. One important but unexplained finding is that CatK deficiency reduced activities of CatL, MMP-2, and MMP-9 in SMCs, but not in ECs, and how CatK affects T-cell proliferation and SMC apoptosis remains unknown. In contrast to Apoe−/−Ctsk+/+ and Apoe−/−Ctsk−/− mice, Ang-II infusion significantly increases blood total Ly6G+ neutrophils and Ly6G+CD11b+ activated neutrophils in Ctsk+/+ mice, while showing no effects on Ctsk−/− mice. Blood CD11b+ cells (e.g., monocytes, macrophages, and NK T cells) were significantly fewer in Ctsk−/− mice than in Ctsk+/+ mice, and Ang-II treatment did not affect these populations in blood (Shi, unpublished data). Therefore, altered peripheral inflammatory cell profiles in Apoe−/− mice after Ang-II infusion may have confounded the AAA progression, thus obscuring the contribution of CatK (Bai et al., 2010).
3.4. Cathepsin L
CatL function in AAA has been tested in aortic elastase perfusion-induced experimental AAA (Shi, unpublished data). Similar to our findings in Ctsk−/− mice, Ctsl−/− mice were protected fully from elastase perfusion-induced AAA. At 14 days post-perfusion, none of the Ctsl−/− mice developed AAA. Aortic lesions from Ctsl−/− mice contained significantly fewer macrophages and CD4+ T cells, and reduced chemokine monocyte chemotactic protein-1 (MCP-1). Medial elastin degradation was also significantly impaired in Ctsl−/− mice compared with WT mice. As discussed above, CD4+ T cells and peripheral blood mononuclear cells from Ctsl−/− mice showed impaired transmigration through collagen or EC monolayer-coated collagen matrix (Kitamoto et al., 2007). CatL deficiency therefore affected AAA lesion chemokine expression and inflammatory cell infiltration differently than that in Ctsk−/− mice. CatL deficiency also reduced lesion microvessel growth and EC total cathepsin and MMP expression and activities. Although lesion cell proliferation was reduced in Ctsl−/− mice, cell apoptosis and medial SMC loss did not differ significantly between WT and Ctsl−/− mice. Therefore, both CatK and CatL contributed to elastase perfusion-induced mouse experimental AAA, but through different mechanisms.
4. Cathepsin inhibitors in cardiovascular diseases
Analysis of mice or humans carrying mutations in different cathepsin genes has shown that individual cathepsins play unique physiological roles in cardiac, brain, and skin development (Reinheckel et al., 2001), and in specialized processes such as bone resorption (Saftig et al., 1998; Gelb et al., 1996), periodontal disease (Toomes et al., 1999), and antigen presentation (Villadangos et al., 1999). An increase in the expression or activity of certain cathepsins has been implicated convincingly in several human diseases, such as osteoporosis (Zaidi et al., 2001; Potts et al., 2004), rheumatoid arthritis (Yasuda et al., 2005), cancers (Joyce et al., 2004), metabolic diseases (Yang et al., 2007; 2008), and cardiovascular diseases (Liu et al., 2004). In normal cells, cysteinyl cathepsins are usually located in lysosomes, but during cancer progression they may move to the cell surface, where they can be secreted into the extracellular milieu. This change in cellular localization has important implications for the design and therapeutic efficacy of cathepsin inhibitors. Cathepsin inhibitors may target extracellular or cell surface cathepsins, but not intracellular cathepsins, thus preserving essential cathepsin physiological activities inside the cells and thereby minimizing inhibitor toxicity (Palermo & Joyce, 2008). Cathepsin inhibitors have developed from non-selective to highly selective inhibitors for each individual enzyme and application.
4.1. Endogenous cathepsin inhibitors
The most common endogenous inhibitors of cysteinyl cathepsins are cystatins (Dubin, 2005). This family of inhibitors can be grouped into three different types based on their primary structure, body distribution, and functions. Type I cystatins include cystatins A and B (also called stefins A and B), the smallest cystatins, which do not contain disulfide bonds or carbohydrate side chains and reside mainly inside cells. Type II cystatins — including cystatin C, D, E, F, G, S, SN, and SA — contain two disulfide bonds and are released to the circulation to target extracellular cathepsins. Type III cystatins include kininogens, which have the highest molecular weight and reside in plasma. Cystatin C, with the highest inhibiting properties to cathepsins L and S, is the best-studied cystatin in cardiovascular diseases, followed by cathepsins B and H (Hall et al., 1998).
We showed that cystatin C is deficient in human atherosclerotic and AAA lesions (Shi et al., 1999b). In atherosclerosis-prone Apoe−/− mice, the absence of cystatin C increased lesion cathepsin activities, tunica media elastin degradation, and lesion SMC and collagen contents; decreased media size; and caused more dilated thoracic and abdominal aortae compared with control Apoe−/− mice — although lesion size, intimal macrophage content, and lipid core size were not different between these mice after 12 weeks of an atherogenic diet (Sukhova et al., 2005). SMCs from cystatin C–deficient mice were more proliferative, expressed more cathepsin activities, and degraded more extracellular elastins and collagens. After consuming an atherogenic diet for 25 weeks, however, cystatin C–deficient Apoe−/− mice developed significantly larger subvalvular atherosclerotic lesions than did control Apoe−/− mice (Bengtsson et al., 2005). Lesions from cystatin C–deficient mice contained more macrophages and less collagen than those from WT mice. Bone marrow transplantation demonstrated that cystatin C from non-hematopoietic cells is critical to atherogenesis. We recently have shown that cystatin C deficiency also enhanced Ang-II–induced AAA in Apoe−/− mice (Schulte et al., 2010). Lesion inflammatory cell content, apoptotic SMC numbers, angiogenesis, and cathepsin activities were all significantly increased in the absence of this common cathepsin inhibitor.
Other endogenous proteins inhibit cathepsins. For example, hurpin (also called serpinB13) is a serine protease inhibitor, but specifically inhibits intracellular CatL. In several skin diseases, such as psoriasis, squamous-cell carcinoma, or basal-cell carcinoma, hurpin and CatL both increased and localized to the outmost layers of the granular and upper spinous layers (Bylaite et al., 2006). In normal skin, hurpin protects keratinocytes from UV light-induced apoptosis (Welss et al., 2003). Other serpins, such as B3 and B4, are squamous-cell carcinoma antigens and can inhibit cathepsins L, K, and S (Higgins et al., 2010), but studies of these inhibitors in cardiovascular diseases are limited.
4.2. Non-selective cathepsin inhibitors
One of the first broad-spectrum cysteinyl cathepsin inhibitors studied was E64 — a covalent, irreversible, epoxysuccinyl-based inhibitor originally isolated from Aspergillus japonicus. Extracts from human atheroma showed a twofold increase in elastolytic activity compared with normal arteries, which could be inhibited up to 40% by a cysteine protease inhibitor (E64) (Sukhova et al., 1998). The ethyl ester of E64 (E64d) (Figure 2A) was tested in a clinical trial of Japanese patients with muscular dystrophy, but the trial was stopped at phase III due to suboptimal performance (Otto & Schirmeister, 1997). JPM was originally named JPM-565, after Joseph P. Meara from the University of Wisconsin–Madison (Shi et al., 1992). It is structurally similar to E64. Its ethyl ester (Figure 2B) derivative has been used recently to block tumor growth in several studies (Joyce et al., 2004; Bell-McGuinn et al., 2007; Schurigt et al., 2008). No information is available, however, regarding these non-selective inhibitors in treating cardiovascular diseases.
Figure 2.

Chemical structures of the broad cathepsin inhibitors E64d (A) and JPM-ethyl ester (B), and of the CatS-selective inhibitors K11777 (C) and LHVS (D).
4.3. Selective cathepsin inhibitors
4.3.1. Cathepsin S inhibitors
Several CatS-selective inhibitors have entered phase I trials for rheumatoid arthritis and psoriasis, and clinical trials for other autoimmune conditions, such as multiple sclerosis and lupus, may follow (Palermo & Joyce, 2008). Development of CatS-selective inhibitors for experimental and clinical applications began after the first demonstration of CatS function in invariant chain processing and antigen presentation (Riese et al., 1996, 1998; Shi et al., 1999a). Following non-selective inhibitors, epoxysuccinyl-based E64 or JPM, vinyl sulfone derivatives were among the earliest generation of CatS irreversible inhibitors, such as K11777 (N-methyl-piperazine-Phe-homoPhe-vinylsulfone-phenyl) (Figure 2C) and LHVS (morpholinurea-leucine-homophenylalanine-vinyl phenyl sulfone) (Figure 2D). K11777 has shown considerable potency in animal models of the parasitic disease schistosomiasis (Abdulla et al., 2007). The vinyl sulfone scaffold was used to derive LHVS, which can perturb antigen presentation in vivo (Riese et al., 1998) and reverse neuropathic painina in a rat model of nerve injury (Clark et al., 2007; Irie et al., 2008). To our knowledge, however, no CatS-selective inhibitors have been used to treat cardiovascular diseases in humans or animal experimental models, and experiments in this field are limited to cell cultures. We showed that cytokine-stimulated SMC elastolytic activity was inhibited more than 80% by LHVS or E64 (Sukhova et al., 1998). Another study showed that SMC invasion through ECM is cathepsin-dependent (Cheng et al., 2006). In the presence of low-dose LHVS (5 ng/ml), presumably inhibiting CatS only (Riese et al., 1998), SMC invasion into an elastin gel was reduced by 90%, and SMC invasion into a collagen gel was reduced by 30%. Potent and selective CatS inhibitors therefore hold future therapeutic potential for vascular diseases (Wiener et al., 2010).
4.3.2. Cathepsin K inhibitors
Since the discovery of essential role of CatK in bone resorption in humans (Gelb et al., 1996) and in mice (Saftig et al., 1998), it has become an attractive target for bone development. For example, the novel selective CatK inhibitor OST-4077 (furan-2-carboxylic acid (1-{1-(4-fluoro-2-(2-oxo-pyrrolidin-1-yl)-phenyl)-3-oxo-piperidin-4-ylcarb amoyl}-cyclohexyl)-amide) potentially could treat diseases characterized by excessive bone loss, including osteoporosis (Kim et al., 2006). To date, clinical trials have studied the effects of several CatK-selective inhibitors on osteoporosis, skeletal disorders, and osteoarthritis (Abbenante & Fairlie, 2005; Yasuda et al., 2005; Lewiecki, 2009). Table 1 lists selected clinical trials of CatK inhibitors. A phase II study of the CatK inhibitor balicatib (AAE581) for the treatment of osteoporosis was reported initially in 2003, and the trial was performed between 2006 and 2008. Preliminary results showed that the compound was tolerated well, inhibited bone resorption, and improved bone formation. In 2002, results were reported for a phase I trial of relacatib (SB-462795) for the treatment of postmenopausal osteoporosis and osteoarthritis; relacatib inhibited bone resorption in monkeys (Kumar et al., 2007; Yamashita et al., 2006). Odanacatib is probably the most promising CatK inhibitor to date. A phase II trial showed increased bone mineral density and reduced bone turnover markers in postmenopausal women. Phase III results are expected shortly (Lewiecki, 2009). Although CatK has been proven important to both atherosclerosis (Lutgens et al., 2006) and AAA (Shi, unpublished data), no CatK inhibitor data are available in vascular diseases in either humans or animals.
Table 1.
Cathepsin K inhibitors in clinical trials.
| Drug name | Company | Clinical trial (year[s]) | Function | Reference |
|---|---|---|---|---|
| Odanacatib | Merck & Co., Inc | Phase III (2009) Phase II (2008) |
Post-menopausal osteoporosis Breast cancer bone metastasis |
Lewiecki, 2009 cme.medscape.com/viewarticle/727651_6 |
| MIV-701 | MedivirAB | Initial trial (2007) | Osteoporosis | www.mskreport.com/articles.cfm?articleID=1219 |
| Balicatib | Novartis | Phase II (2006–2009) | Osteoporosis | clinicaltrials.gov/ct2/show/NCT00371670 |
| Relacatib | GlaxoSmith Kline | Phase I (2006–2009) | Osteoporosis, osteoarthritis | clinicaltrials.gov/ct2/show/NCT00411190 |
4.3.3. Cathepsin L inhibitors
Several different classes of CatL-selective inhibitors have been developed. Some have low potency and poor selectivity, and a few show high potency and target selectivity. The epoxysuccinate derivatives CLIK (cathepsin L inhibitor Katunuma) series compounds 195 and 148 (Katunuma et al., 1999), for example, have been used in both in vitro cell culture studies and in vivo animal models (Yang et al., 2007; Yamada et al., 2010), but these compounds have relatively low potency and selectivity (Table 2). In contrast, the azepanone-based CatL inhibitors are much more potent and selective, although these compounds have been used only in cell culture or cytochemical studies (James et al., 2001; Marquis et al., 2005). Whether these compounds acted on CatL irreversibly, like E64 or JPM (Grubb et al., 1990), or reversibly, is unknown. The field is currently focused on the development of reversible cathepsin inhibitors (Wiener et al., 2010). Few peptide-based CatL inhibitors were found reversibly binding to CatL with high potency (Ki = 19~45 nM) and selectivity (64~333-fold), compared with CatL’s inhibitions to CatB or CatK (Table 2) (Chowdhury et al., 2002; Shenoy et al., 2009). The most recent CatL-selective and reversible inhibitors are thiocarbazate derivatives (Table 2). Compound SID 26681509 demonstrated high potency (IC50: 1.0 nM) and bound to CatL in a slow-binding and slowly reversible manner. It has 7-fold to 50-fold higher selectivity to cathepsins V, S, B, and K (Shah et al., 2008). None of these reversible inhibitors, however, has been tested in animal disease models. We anticipate that these reversible compounds may prove invaluable in treating humans or animals with vascular diseases in the near future.
Table 2.
Cathepsin L-selective inhibitors.
| Name and structure (Compound ID) |
Inhibitory potency and selectivity (Reference) |
||
|---|---|---|---|
| Epoxysuccinate derivatives | |||
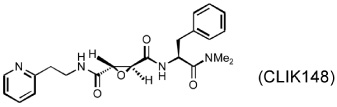 |
% of inhibition at 1 ×106 M | ||
| CatL: CatS: CatB, CatK, CatC: |
100% 30% 0% |
||
| (Katunuma et al., 1999) | |||
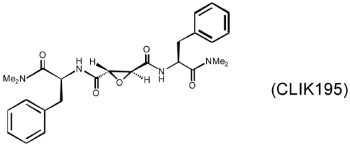 |
% of inhibition at 1 × 106 M | ||
| CatL: CatS: CatB, CatK, CatC: |
100% 25% 0% |
||
| (Katunuma et al., 1999) | |||
| Azepanone-based derivatives | |||
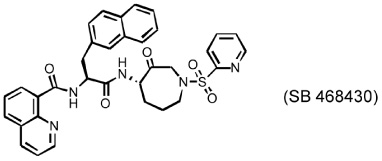 |
CatL: Ki = 0.0099 nM CatK: Ki = 503 nM |
||
| (James et al, 2001) | |||
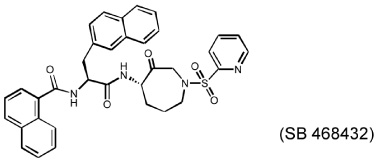 |
CatL: Ki = 0.033 nM; Ki,app = 0.43 nM CatK: Ki = 420 nM; Ki,app > 10,000 nM CatS: Ki,app = 15.6 nM CatB: Ki,app = 150 nM |
||
| (James et al., 2001) | |||
| Peptide-based derivatives | |||
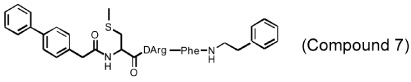 |
CatL: Ki = 19 nM CatK: Ki = 5.9 µM CatB: Ki = 4.1 µM |
||
| (Chowdhury et al., 2002; Shenoy et al., 2009) | |||
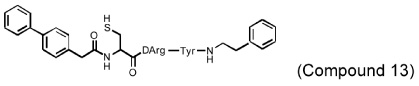 |
CatL: Ki = 45 nM CatK: Ki = 2.9 µM CatB: Ki = 15 µM |
||
| (Chowdhury et al., 2002; Shenoy et al., 2009) | |||
| Thiocarbazate derivative | |||
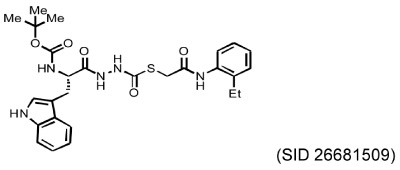 |
IC50 at 90 min | Selectivity index | |
| CatL: 50 nM CatV: 576 nM CatS: 721 nM CatB: 1,562 nM CatK: 2,512 nM |
1 7–11 7–14 31–46 40–50 |
||
| (Shah et al., 2008) | |||
5. Mast cell chymase and tryptase in cardiovascular diseases
Mast cells were first implicated in atherosclerosis more than half a century ago (Constantinides, 1953; Cairns & Constantinides, 1954). Petri Konaven, Ken Lindstedt, and colleagues have contributed most of our current knowledge of these inflammatory cells in atherosclerosis during the last 25 years (Lindstedt & Konaven, 2004). Limited knowledge is available, however, regarding mast cells in AAA. Although mast cells were first detected in human cerebral aneurysms 30 years ago (Faleiro et al., 1981), the first demonstration of mast cells in AAA was rather recent (Ihara et al., 1999). Using mast cell–deficient KitW-sh/W-sh mice, atherosclerosis-prone Ldlr−/− mice, and aortic elastase perfusion–induced experimental AAA, we demonstrated that mast cells participate in atherosclerosis and AAA by releasing pro-inflammatory cytokines, chemokines, and proteases to induce inflammatory cell recruitment, cell apoptosis, angiogenesis, and matrix protein remodeling (Sun et al., 2007a; 2007b). Pharmacological stabilization of mast cells reduced atherosclerosis in Apoe−/− mice (Bot et al., 2007) and reduced elastase perfusion–induced AAA (Sun et al., 2007b). Mast cell chymase and tryptase are unique to mast cells and have been directly and indirectly proven to participate in atherosclerosis and AAA.
5.1. Chymases and tryptases in atherosclerosis
Mast cells contain their own unique neutral serine proteases, chymase, and tryptase. Mast cell chymase is one of the important enzymes in generating Ang-II from Ang-I (Akasu et al., 1998; Caughey et al., 2000; Miyazaki & Takai, 2001). High levels of Ang-II forming activity and chymase expression have been demonstrated in human atherosclerotic lesions (Ihara et al., 1999). Using chymase-deficient (Sun et al., 2009) and tryptase-deficient (Shi, unpublished data) mice, we proved that both proteases promote aortic SMC apoptosis. Activated mast cells release TNF-α and chymase to inactivate the focal adhesion kinase–mediated cell survival signaling in EC, thereby inducing EC apoptosis (Heikkila et al., 2008). Thus, by inducing EC and SMC apoptosis, mast cell proteases may contribute to plaque erosion and complications of atherosclerosis. As discussed, chymase- and tryptase-mediated bioactivation of pro-enzyme and latent cytokines also appears important to atherogenesis. Several studies, including ours (Xiang et al., 2011), have established an association of blood tryptase levels with atherosclerotic plaque instability (Deliargyris et al., 2005; Filipiak et al., 2003). Patients with acute myocardial infarction (MI) or unstable angina pectoris have significantly higher serum tryptase levels than those without significant coronary heart disease or with stable angina pectoris. Serum chymase levels also were higher in patients with acute MI or unstable angina pectoris than in patients with stable angina pectoris or those without significant coronary heart disease (Xiang et al., 2011). In vitro, treatment of human coronary arteries intraluminally with recombinant tryptase or chymase induced endothelial damage, characterized by disruption of EC adhesion followed by retraction and desquamation (Mayranpaa et al., 2006). In mice, both chymases and tryptases control vascular cell atherosclerosis-pertinent cathepsin expression (Sun et al., 2009).
5.2. Chymases and tryptases in AAA
Immune cells in AAA — including macrophages, lymphocytes, neutrophils, and mast cells — produce cytokines and proteases to promote inflammatory reactions, extracellular matrix degradation, and neovascularization (Rizas et al., 2009). We found that mast cells use their inflammatory cytokines to induce vascular cell protease expression, thereby enhancing angiogenesis, vascular cell apoptosis, and ECM degradation (Sun et al., 2007b). Mayranpaa and colleagues investigated the relationship between mast cells and inflammation, neovascularization, and intraluminal thrombus in human AAA, and their results support a direct participation of mast cells in the pathogenesis of AAA — particularly regarding neovascularization of the aortic wall (Mayranpaa et al., 2009).
In a small-population AAA follow-up study, we demonstrated significant correlations of serum chymase (Sun et al., 2009) and tryptase levels (Shi, unpublished data) with AAA annual expansion rate. More interesting discoveries include that high serum tryptase levels significantly increased the risks of later surgical repair or mortality in this population. Both mast cell proteases were expressed highly in the media and adventitia of human AAA, but were negligible in normal aortas. In an elastase perfusion– induced mouse AAA model, mice lacking connective tissue chymase mMCP-4 (Sun et al., 2009) or tryptase mMCP-6 (Shi, unpublished data) developed significantly smaller AAA lesions than did WT mice. Mechanistically, we found that chymase contributed to microvessel growth, vascular SMC apoptosis, and vascular cell cysteinyl cathepsin expression and activities. Compared with mast cells from WT mice, those from chymase mMCP-4–deficient mice showed significantly impaired induction of microvessel sprouting in an aortic ring assay, aortic SMC apoptosis, and cathepsin expression and activities in EC and SMC (Sun et al., 2009). In contrast, tryptase contributed to SMC apoptosis, vascular EC and SMC cathepsin expression, and monocyte transmigration, but showed no effect on microvessel growth (Shi, unpublished data). Figure 3 summarizes our current understanding of mast cell chymase and tryptase functions in atherogenesis and AAA formation.
Figure 3.

Illustration of mast cell protease functions in the pathogenesis of atherosclerosis and abdominal aortic aneurysm.
6. Chymase and tryptase inhibitors in cardiovascular diseases
Although both chymases and tryptases are essential to mast cell biology and to the pathogenesis of atherosclerosis, AAA, and associated complications, most current studies focus on the development of chymase inhibitors in experimental cardiovascular disease in animals. Tryptase inhibitors receive much less attention, but this does not mean that tryptase is less important than chymase. We demonstrated that mMCP-6–null mice are resistant to elastase perfusion–induced AAA (Shi, unpublished observation). In THP-1– derived macrophages, the tryptase inhibitor APC-366 blocked ox-LDL–induced foam cell formation. Mechanistically, APC-366 inhibited tryptase activities known to reduce nuclear receptor LXRa (regulates lipid homeostasis), ATP-binding cassette transporters A1 and G1 (ABCA1, ABCG1; involved in the cholesterol efflux pathway), and sterol regulatory element binding protein-1c (SREBP-1c) (regulates gene for de novo lipogenesis) (Yeong et al., 2010).
6.1. Chymase inhibitors in atherosclerosis
At least three chymase inhibitors have been tested in atherosclerosis and related conditions. In a dog vein graft disease experimental model (Takai et al., 2001), each animal underwent right common carotid artery bypass grafting with the ipsilaterial external jugular vein. Grafting increased vascular cell proliferation, chymase activity, stem cell factor activation, mast cell accumulation, and intima thickening. When dogs received chymase inhibitor NK3201 orally (Table 3), however, all such phenotypes were significantly suppressed. The same group performed a similar experiment using a balloon catheter–induced carotid artery injury model to test the role of NK3201 in intimal hyperplasia (Takai et al., 2003). Both chymase activity and carotid artery intimal thickening were significantly reduced in dogs receiving the treatment. These experiments suggest that chymase enhances vascular cell proliferation.
Table 3.
Chymase inhibitors in cardiovascular studies.
| Structure (Compound ID) |
Application [Animal type] (Reference) |
Major finding |
|---|---|---|
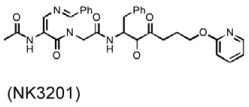 |
Vein graft disease [dog] (Takai et al., 2001) |
Reduced mast cell accumulation Reduced intimal thickening Reduced vascular cell proliferation |
| Balloon injury intimal thickness [dog] (Takai et al., 2003) |
Reduced intimal thickening | |
| Abdominal aortic aneurysm [hamster, dog, Apoe−/− mouse] (Tsunemi et al., 2004; Furubayashi et al., 2007; Inoue et al., 2009) |
Reduced lesion inflammatory cells Reduced MMP9 activity Reduced luminal area & AAA size |
|
| Myocardial infarction [hamster, rat] (Jin et al., 2003; Kanemitsu et al., 2006, 2008) |
Reduced mortality Improved fractional shortening Reduced collagen-I, III expression Reduced fibrotic area |
|
 |
Atherosclerosis [hamster] (Arakawa & Urata, 2000, Uehara et al., 2002) |
Suppressed aortic lipid deposition |
 |
Atherosclerosis [Apoe−/− mouse] (Bot et al., 2010) |
Reduced atheroma plaque size Reduced lesion necrotic core size Enhanced lesion collagen content Impaired intraplaque haemorrhage |
 |
Myocardial infarction [hamster] (Jin et al., 2002) |
Improved hemodynamics Reduced mortality |
 |
Myocardial infarction [hamster] (Hoshino et al., 2003) |
Reduced microvascular leakage Reduced cardiac hypertrophy Reduced end-diastolic LV pressure Increased survival rate |
 |
Myocardial infarction [dog] (Jin et al., 2004) |
Decreased plasma Ang-ll level Suppressed ventricular arrhythmias |
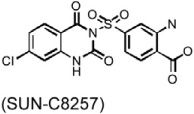
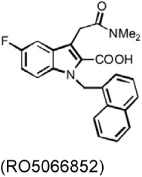
In patients with atherosclerosis, serum total or LDL cholesterol levels correlated with arterial chymase-dependent Ang-II forming activity. Hamsters consuming a high-cholesterol diet developed lipid deposition in the aortic cusp. Plasma total or LDL cholesterol levels also correlated with adventitial Ang-II immunoreactivity. Oral administration of the chymase inhibitor SUN-C8257 completely suppressed aortic lipid deposition and Ang-II forming activity (Arakawa & Urata, 2000; Uehara et al., 2002) (Table 3). Similar observations were made in atherosclerosis-prone Apoe−/− mice (Bot et al., 2010). A newly developed chymase inhibitor, RO5066852, reduced chymase and tryptase expression and activity, reduced spontaneous atherosclerosis, prevented local mast cell activation–induced acceleration of plaque progression, enhanced lesion collagen content and reduced lesion necrotic core size, and completely normalized the increased frequency and size of intraplaque hemorrhages after acute perivascular collar placement-induced carotid plaque (Table 3).
6.2. Chymase inhibitors in MI
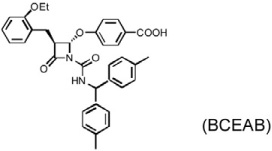
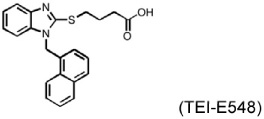
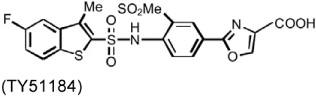
MI is a common complication of atherosclerosis, although experimental MI has different mechanisms of pathogenesis from that in humans. Several chymase inhibitors — including BCEAB, NK3201, TEI-E548, and TY51184 — have been used to examine the role of chymase in left coronary artery ligation-induced MI in hamsters, dogs, and rats (Table 3). In hamsters, orally active inhibitor BCEAB treatment starting 3 days before surgery yielded suppression of cardiac chymase activity, improvement of hemodynamics (increase of maximal negative and positive rates of pressure development [+dP/dt and – dP/dt] and left ventricular systolic pressure [LVSP]), and reduction of mortality (Jin et al., 2002). NK3201 caused similar results in this model, reducing mortality and improving fractional shortening (Jin et al., 2003). In the same hamster experimental model, TEI-E548 completely inhibited chymase-induced microvascular leakage, increased survival rate, and attenuated cardiac hypertrophy and end-diastolic left ventricular pressure. Plasma Ang-II levels also were suppressed after chymase inhibition (Hoshino et al., 2003). In MI in dogs, chymase inhibitor TY51184 decreased plasma Ang-II levels and suppressed total numbers of ventricular arrhythmias by more than 80% (Jin et al., 2004). In MI in rats, Kanemitsu et al. demonstrated that chymase inhibition with NK3201 inhibited TGF-β1 expression, thereby reducing collagen type-I and type-III levels. Rats receiving NK3201 had significantly reduced fibrotic areas. Consequently, these rats showed significantly reduced left-ventricular end-diastolic pressure, increased maximal end-systolic pressure–volume relationship, and decreased time constant of left ventricular relaxation (Kanemitsu et al., 2006; 2008).
6.3. Chymase inhibitors in AAA
As discussed earlier, CatC participates in AAA formation by activating neutrophil proteases and thereby controlling neutrophil chemokine CXCL2 production and neutrophil recruitment (Pagano et al., 2007). CatC also activates inactive chymase in mast cell granules (Wolters et al., 2001), thereby enhancing mast cell Ang-II production, which ultimately induces vascular wall MMP-9, a well-known protease involved in AAA pathogenesis (Pyo et al., 2000). There are many more chymase functions in AAA beyond MMP-9 expression (Sun et al., 2009). Chymase inhibitor NK3201 has been used in three experimental AAA models (Table 3). In aortic elastase perfusion–induced experimental AAA in hamsters and dogs, NK3201 reduced lesion Ang-II forming activity, MMP activity, mast cell and neutrophil accumulation, and lesion luminal areas (Tsunemi et al., 2004; Furubayashi et al., 2007). The same compound yielded similar observations in Ang-II perfusion–induced AAA in Apoe−/− mice (Inoue et al., 2009). NK3201 substantially reduced AAA severity, luminal area, lesion monocyte/macrophage content, and MMP-9 activity in mice.
7. Future perspectives
Experimental disease in animals has helped to affirm the role of cysteinyl cathepsins and mast cell–specific proteases in cardiovascular diseases, although many current models do not fully recapitulate human disease. Protease inhibitors are much more potent and selective than they were 10 years ago; highly selective and potent cathepsin and chymase inhibitors (Table 2 and Table 3) were developed in the 21st century. Why there is no clinical trial to test whether these potent and selective chymase inhibitors are effective in humans, however, remains unknown. Drug companies may be waiting for more data from chymase-deficient or tryptase-deficient animals, as these inhibitors may have off-target effects. Direct approvals in protease-deficient animals (Sun et al., 2009) will help to advance the progress of human trials.
We also do not know why no cathepsin inhibitors (Table 2) (Palermo & Joyce, 2008) have been enrolled in human trials against cardiovascular diseases. We believe that this is mainly because most drug companies have been focused on the role of cathepsins in autoimmune and bone diseases, due to several important discoveries in the 20th century (Gelb et al., 1996; Riese et al., 1996; 1998; Shi et al., 1999a). Several programs have been halted because of compound toxicities, likely due to the compound-target irreversible binding and associated side effects. Scientists are learning that reversible cathepsin inhibitors might have been used. We are certain that in the near future, some cathepsin inhibitors will be used in human trials. In addition, broad inhibition of major inflammation-inducible cathepsins, such as CatS, CatK, CatL, and CatC, may not be a bad idea, as long as the dose is adequate. We have learned from animal disease models and histological analysis of human pathological tissue that these cathepsins are either not expressed or expressed at low levels under healthy conditions, whereas increased expression appeared in pathological tissues. Compounds have main targets, but their minor inhibitory activities toward other cathepsins also may be beneficial — a hypothesis that certainly will be tested in the near future.
Acknowledgments
We thank Ms. Sara Karwacki for editorial assistance. This study is partially supported by grants from the National Natural Science Foundation of China (81070251) and Beijing Natural Science Foundation of China (7082020) to YQ, and from the American Heart Association (0840118N to GPS) and the U.S. National Institutes of Health (HL60942, HL67283, and HL81090 to GPS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbenante G, Fairlie DP. Protease inhibitors in the clinic. Med Chem. 2005;1:71–104. doi: 10.2174/1573406053402569. [DOI] [PubMed] [Google Scholar]
- Abdul-Hussien H, Soekhoe RG, Weber E, von der Thusen JH, Kleemann R, Mulder A, van Bockel JH, Hanemaaijer R, Lindeman JH. Collagen degradation in the abdominal aneurysm: a conspiracy of matrix metalloproteinase and cysteine collagenases. Am J Pathol. 2007;170:809–817. doi: 10.2353/ajpath.2007.060522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulla MH, Lim KC, Sajid M, McKerrow JH, Caffrey CR. Schistosomiasis mansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 2007;4:e14. doi: 10.1371/journal.pmed.0040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe M, Kurosawa M, Ishikawa O, Miyachi Y, Kido H. Mast cell tryptase stimulates both human dermal fibroblast proliferation and type I collagen production. Clin Exp Allergy. 1998;28:1509–1517. doi: 10.1046/j.1365-2222.1998.00360.x. [DOI] [PubMed] [Google Scholar]
- Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akasu M, Urata H, Kinoshita A, Sasaguri M, Ideishi M, Arakawa K. Differences in tissue angiotensin II-forming pathways by species and organs in vitro. Hypertension. 1998;32:514–520. doi: 10.1161/01.hyp.32.3.514. [DOI] [PubMed] [Google Scholar]
- Arakawa K, Urata H. Hypothesis regarding the pathophysiological role of alternative pathways of angiotensin II formation in atherosclerosis. Hypertension. 2000;36:638–641. doi: 10.1161/01.hyp.36.4.638. [DOI] [PubMed] [Google Scholar]
- Bai L, Beckers L, Wijnands E, Lutgens SP, Herias MV, Saftig P, Daemen MJ, Cleutjens K, Lutgens E, Biessen EA, Heeneman S. Cathepsin K gene disruption does not affect murine aneurysm formation. Atherosclerosis. 2010;209:96–103. doi: 10.1016/j.atherosclerosis.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Barascuk N, Skjøt-Arkil H, Register TC, Larsen L, Byrjalsen I, Christiansen C, Karsdal MA. Human macrophage foam cells degrade atherosclerotic plaques through cathepsin K mediated processes. BMC Cardiovasc Disord. 2010;10:19. doi: 10.1186/1471-2261-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell-McGuinn KM, Garfall AL, Bogyo M, Hanahan D, Joyce JA. Inhibition of cysteine cathepsin protease activity enhances chemotherapy regimens by decreasing tumor growth and invasiveness in a mouse model of multistage cancer. Cancer Res. 2007;67:7378–7385. doi: 10.1158/0008-5472.CAN-07-0602. [DOI] [PubMed] [Google Scholar]
- Bengtsson E, To F, Hakansson K, Grubb A, Branen L, Nilsson J, Jovinge S. Lack of the cysteine protease inhibitor cystatin C promotes atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2151–2156. doi: 10.1161/01.ATV.0000179600.34086.7d. [DOI] [PubMed] [Google Scholar]
- Blair RJ, Meng H, Marchese MJ, Ren S, Schwartz LB, Tonnesen MG, Gruber BL. Human mast cells stimulate vascular tube formation. Tryptase is a novel, potent angiogenic factor. J Clin Invest. 1997;99:2691–2700. doi: 10.1172/JCI119458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgran R, Zheng L, Stendahl O. Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J Leukoc Biol. 2007;81:1213–1223. doi: 10.1189/jlb.0506359. [DOI] [PubMed] [Google Scholar]
- Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, Biessen EA. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115:2516–2525. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- Bot I, Bot M, van Heiningen SH, van Santbrink PJ, Lankhuizen IM, Hartman P, Gruener S, Hilpert H, van Berkel TJ, Fingerle J, Biessen EA. Mast cell chymase inhibition reduces atherosclerotic plaque progression and improves plaque stability in ApoE-/- mice. Cardiovasc Res. 2011;89:244–252. doi: 10.1093/cvr/cvq260. [DOI] [PubMed] [Google Scholar]
- Bylaite M, Moussali H, Marciukaitiene I, Ruzicka T, Walz M. Expression of cathepsin L and its inhibitor hurpin in inflammatory and neoplastic skin diseases. Exp Dermatol. 2006;15:110–118. doi: 10.1111/j.1600-0625.2005.00389.x. [DOI] [PubMed] [Google Scholar]
- Cairns A, Constantinides P. Mast cells in human atherosclerosis. Science. 1954;120:31–32. doi: 10.1126/science.120.3105.31. [DOI] [PubMed] [Google Scholar]
- Caughey GH, Raymond WW, Wolters PJ. Angiotensin II generation by mast cell alpha- and beta-chymases. Biochim Biophys Acta. 2000;1480:245–257. doi: 10.1016/s0167-4838(00)00076-5. [DOI] [PubMed] [Google Scholar]
- Cavallo-Medved D, Rudy D, Blum G, Bogyo M, Caglic D, Sloane BF. Live-cell imaging demonstrates extracellular matrix degradation in association with active cathepsin B in caveolae of endothelial cells during tube formation. Exp Cell Res. 2009;315:1234–1246. doi: 10.1016/j.yexcr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WL, Pejnovic N, Hamilton H, Liew TV, Popadic D, Poggi A, Khan SM. Atherosclerotic abdominal aortic aneurysm and the interaction between autologous human plaque-derived vascular smooth muscle cells, type 1 NKT, and helper T cells. Circ Res. 2005;96:675–683. doi: 10.1161/01.RES.0000160543.84254.f1. [DOI] [PubMed] [Google Scholar]
- Chapman HA, Riese RJ, Shi GP. Emerging roles for cysteine proteases in human biology. Annu Rev Physiol. 1997;59:63–88. doi: 10.1146/annurev.physiol.59.1.63. [DOI] [PubMed] [Google Scholar]
- Chatzizisis YS, Baker AB, Sukhova GK, Koskinas KC, Papafaklis MI, Beigel R, Jonas M, Coskun AU, Stone BV, Maynard C, Shi G-P, Libby P, Feldman CL, Edelman ER, Stone PH. Augmented expression and activity of extracellular matrix-degrading enzymes in regions of low endothelial shear stress colocalize with coronary atheromata with thin fibrous caps in pigs. Circulation. 2011;123:621–630. doi: 10.1161/CIRCULATIONAHA.110.970038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Tung CH, Mahmood U, Ntziachristos V, Gyurko R, Fishman MC, Huang PL, Weissleder R. In vivo imaging of proteolytic activity in atherosclerosis. Circulation. 2002;105:2766–2771. doi: 10.1161/01.cir.0000017860.20619.23. [DOI] [PubMed] [Google Scholar]
- Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- Cheng XW, Kuzuya M, Nakamura K, Di Q, Liu Z, Sasaki T, Kanda S, Jin H, Shi GP, Murohara T, Yokota M, Iguchi A. Localization of cysteine protease, cathepsin S, to the surface of vascular smooth muscle cells by association with integrin alphanubeta3. Am J Pathol. 2006;168:685–694. doi: 10.2353/ajpath.2006.050295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury SF, Sivaraman J, Wang J, Devanathan G, Lachance P, Qi H, Menard R, Lefebvre J, Konishi Y, Cygler M, Sulea T, Purisima EO. Design of noncovalent inhibitors of human cathepsin L. From the 96-residue proregion to optimized tripeptides. J Med Chem. 2002;45:5321–5329. doi: 10.1021/jm020238t. [DOI] [PubMed] [Google Scholar]
- Chwieralski CE, Welte T, Buhling F. Cathepsin-regulated apoptosis. Apoptosis. 2006;11:143–149. doi: 10.1007/s10495-006-3486-y. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinides P. Mast cells and susceptibility to experimental atherosclerosis. Science. 1953;117:505–506. doi: 10.1126/science.117.3045.505. [DOI] [PubMed] [Google Scholar]
- Deliargyris EN, Upadhya B, Sane DC, Dehmer GJ, Pye J, Smith SC, Jr, Boucher WS, Theoharides TC. Mast cell tryptase: a new biomarker in patients with stable coronary artery disease. Atherosclerosis. 2005;178:381–386. doi: 10.1016/j.atherosclerosis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Dell’Italia LJ, Husain A. Dissecting the role of chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol. 2002;17:374–379. doi: 10.1097/00001573-200207000-00009. [DOI] [PubMed] [Google Scholar]
- de Nooijer R, Bot I, von der Thusen JH, Leeuwenburgh MA, Overkleeft HS, Kraaijeveld AO, Dorland R, van Santbrink PJ, van Heiningen SH, Westra MM, Kovanen PT, Jukema JW, van der Wall EE, van Berkel TJ, Shi GP, Biessen EA. Leukocyte cathepsin S is a potent regulator of both cell and matrix turnover in advanced atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:188–194. doi: 10.1161/ATVBAHA.108.181578. [DOI] [PubMed] [Google Scholar]
- Di Girolamo N, Indoh I, Jackson N, Wakefield D, McNeil HP, Yan W, Geczy C, Arm JP, Tedla N. Human mast cell-derived gelatinase B (matrix metalloproteinase-9) is regulated by inflammatory cytokines: role in cell migration. J Immunol. 2006;177:2638–2650. doi: 10.4049/jimmunol.177.4.2638. [DOI] [PubMed] [Google Scholar]
- Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–819. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin G. Proteinaceous cysteine protease inhibitors. Cell Mol Life Sci. 2005;62:653–669. doi: 10.1007/s00018-004-4445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duftner C, Seiler R, Klein-Weigel P, Gobel H, Goldberger C, Ihling C, Fraedrich G, Schirmer M. High prevalence of circulating CD4+CD28- T-cells in patients with small abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2005;25:1347–1352. doi: 10.1161/01.ATV.0000167520.41436.c0. [DOI] [PubMed] [Google Scholar]
- Faleiro LC, Machado CR, Gripp A, Jr, Resende RA, Rodrigues PA. Cerebral vasospasm: presence of mast cells in human cerebral arteries after aneurysm rupture. J Neurosurg. 1981;54:733–735. doi: 10.3171/jns.1981.54.6.0733. [DOI] [PubMed] [Google Scholar]
- Fang KC, Raymond WW, Lazarus SC, Caughey GH. Dog mastocytoma cells secrete a 92-kD gelatinase activated extracellularly by mast cell chymase. J Clin Invest. 1996;97:1589–1596. doi: 10.1172/JCI118583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipiak KJ, Tarchalska-Krynska B, Opolski G, Rdzanek A, Kochman J, Kosior DA, Czlonkowski A. Tryptase levels in patients after acute coronary syndromes: the potential new marker of an unstable plaque? Clin Cardiol. 2003;26:366–372. doi: 10.1002/clc.4950260804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furubayashi K, Takai S, Jin D, Muramatsu M, Ibaraki T, Nishimoto M, Fukumoto H, Katsumata T, Miyazaki M. The significance of chymase in the progression of abdominal aortic aneurysms in dogs. Hypertens Res. 2007;30:349–357. doi: 10.1291/hypres.30.349. [DOI] [PubMed] [Google Scholar]
- Gacko M, Glowinski S. Cathepsin D and cathepsin L activities in aortic aneurysm wall and parietal thrombus. Clin Chem Lab Med. 1998;36:449–452. doi: 10.1515/CCLM.1998.075. [DOI] [PubMed] [Google Scholar]
- Gacko M, Chyczewski L, Chrostek L. Distribution, activity and concentration of cathepsin B and cystatin C in the wall of aortic aneurysm. Pol J Pathol. 1999;50:83–86. [PubMed] [Google Scholar]
- Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273:1236–1238. doi: 10.1126/science.273.5279.1236. [DOI] [PubMed] [Google Scholar]
- Gelb BD, Shi GP, Heller M, Weremowicz S, Morton C, Desnick RJ, Chapman HA. Structure and chromosomal assignment of the human cathepsin K gene. Genomics. 1997;41:258–262. doi: 10.1006/geno.1997.4631. [DOI] [PubMed] [Google Scholar]
- Grubb A, Abrahamson M, Olafsson I, Trojnar J, Kasprzykowska R, Kasprzykowski F, Grzonka Z. Synthesis of cysteine proteinase inhibitors structurally based on the proteinase interacting N-terminal region of human cystatin C. Biol Chem Hoppe Seyler. 1990;371 Suppl:137–144. [PubMed] [Google Scholar]
- Guinec N, Dalet-Fumeron V, Pagano M. "In vitro" study of basement membrane degradation by the cysteine proteinases, cathepsins B, B-like and L. Digestion of collagen IV, laminin, fibronectin, and release of gelatinase activities from basement membrane fibronectin. Biol Chem Hoppe Seyler. 1993;374:1135–1146. doi: 10.1515/bchm3.1993.374.7-12.1135. [DOI] [PubMed] [Google Scholar]
- Hall A, Ekiel I, Mason RW, Kasprzykowski F, Grubb A, Abrahamson M. Structural basis for different inhibitory specificities of human cystatins C and D. Biochemistry. 1998;37:4071–4079. doi: 10.1021/bi971197j. [DOI] [PubMed] [Google Scholar]
- Hamada H, Terai M, Kimura H, Hirano K, Oana S, Niimi H. Increased expression of mast cell chymase in the lungs of patients with congenital heart disease associated with early pulmonary vascular disease. Am J Respir Crit Care Med. 1999;160:1303–1308. doi: 10.1164/ajrccm.160.4.9810058. [DOI] [PubMed] [Google Scholar]
- Heikkila HM, Latti S, Leskinen MJ, Hakala JK, Kovanen PT, Lindstedt KA. Activated mast cells induce endothelial cell apoptosis by a combined action of chymase and tumor necrosis factor-alpha. Arterioscler Thromb Vasc Biol. 2008;28:309–314. doi: 10.1161/ATVBAHA.107.151340. [DOI] [PubMed] [Google Scholar]
- Higgins WJ, Fox DM, Kowalski PS, Nielsen JE, Worrall DM. Heparin enhances serpin inhibition of the cysteine protease cathepsin L. J Biol Chem. 2010;285:3722–3729. doi: 10.1074/jbc.M109.037358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino F, Urata H, Inoue Y, Saito Y, Yahiro E, Ideishi M, Arakawa K, Saku K. Chymase inhibitor improves survival in hamsters with myocardial infarction. J Cardiovasc Pharmacol. 2003;41 Suppl 1:S11–S18. [PubMed] [Google Scholar]
- Ichinose S, Usuda J, Hirata T, Inoue T, Ohtani K, Maehara S, Kubota M, Imai K, Tsunoda Y, Kuroiwa Y, Yamada K, Tsutsui H, Furukawa K, Okunaka T, Oleinick NL, Kato H. Lysosomal cathepsin initiates apoptosis, which is regulated by photodamage to Bcl-2 at mitochondria in photodynamic therapy using a novel photosensitizer, ATX-s10 (Na) Int J Oncol. 2006;29:349–355. [PubMed] [Google Scholar]
- Ihara M, Urata H, Kinoshita A, Suzumiya J, Sasaguri M, Kikuchi M, Ideishi M, Arakawa K. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension. 1999;33:1399–1405. doi: 10.1161/01.hyp.33.6.1399. [DOI] [PubMed] [Google Scholar]
- Inoue N, Muramatsu M, Jin D, Takai S, Hayashi T, Katayama H, Kitaura Y, Tamai H, Miyazaki M. Effects of chymase inhibitor on angiotensin II-induced abdominal aortic aneurysm development in apolipoprotein E-deficient mice. Atherosclerosis. 2009;204:359–364. doi: 10.1016/j.atherosclerosis.2008.09.032. [DOI] [PubMed] [Google Scholar]
- Irie O, Kosaka T, Ehara T, Yokokawa F, Kanazawa T, Hirao H, Iwasaki A, Sakaki J, Teno N, Hitomi Y, Iwasaki G, Fukaya H, Nonomura K, Tanabe K, Koizumi S, Uchiyama N, Bevan SJ, Malcangio M, Gentry C, Fox AJ, Yaqoob M, Culshaw AJ, Allan H. Discovery of orally bioavailable cathepsin S inhibitors for the reversal of neuropathic pain. J Med Chem. 2008;51:5502–5505. doi: 10.1021/jm800839j. [DOI] [PubMed] [Google Scholar]
- Jaffer FA, Kim DE, Quinti L, Tung CH, Aikawa E, Pande AN, Kohler RH, Shi GP, Libby P, Weissleder R. Optical visualization of cathepsin K activity in atherosclerosis with a novel, protease-activatable fluorescence sensor. Circulation. 2007;115:2292–2298. doi: 10.1161/CIRCULATIONAHA.106.660340. [DOI] [PubMed] [Google Scholar]
- James IE, Marquis RW, Blake SM, Hwang SM, Gress CJ, Ru Y, Zembryki D, Yamashita DS, McQueney MS, Tomaszek TA, Oh HJ, Gowen M, Veber DF, Lark MW. Potent and selective cathepsin L inhibitors do not inhibit human osteoclast resorption in vitro. J Biol Chem. 2001;276:11507–11511. doi: 10.1074/jbc.M010684200. [DOI] [PubMed] [Google Scholar]
- Jin D, Takai S, Yamada M, Sakaguchi M, Miyazaki M. Beneficial effects of cardiac chymase inhibition during the acute phase of myocardial infarction. Life Sci. 2002;71:437–446. doi: 10.1016/s0024-3205(02)01689-2. [DOI] [PubMed] [Google Scholar]
- Jin D, Takai S, Yamada M, Sakaguchi M, Kamoshita K, Ishida K, Sukenaga Y, Miyazaki M. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc Res. 2003;60:413–420. doi: 10.1016/s0008-6363(03)00535-2. [DOI] [PubMed] [Google Scholar]
- Jin D, Takai S, Sakaguchi M, Okamoto Y, Muramatsu M, Miyazaki M. An antiarrhythmic effect of a chymase inhibitor after myocardial infarction. J Pharmacol Exp Ther. 2004;309:490–497. doi: 10.1124/jpet.103.061465. [DOI] [PubMed] [Google Scholar]
- Jones SE, Gilbert RE, Kelly DJ. Tranilast reduces mesenteric vascular collagen deposition and chymase-positive mast cells in experimental diabetes. J Diabetes Complications. 2004;18:309–315. doi: 10.1016/j.jdiacomp.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Jormsjo S, Wuttge DM, Sirsjo A, Whatling C, Hamsten A, Stemme S, Eriksson P. Differential expression of cysteine and aspartic proteases during progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2002;161:939–945. doi: 10.1016/S0002-9440(10)64254-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M, Hanahan D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- Kaminska R, Helisalmi P, Harvima RJ, Naukkarinen A, Horsmanheimo M, Harvima IT. Focal dermal-epidermal separation and fibronectin cleavage in basement membrane by human mast cell tryptase. J Invest Dermatol. 1999;113:567–573. doi: 10.1046/j.1523-1747.1999.00738.x. [DOI] [PubMed] [Google Scholar]
- Kanemitsu H, Takai S, Tsuneyoshi H, Nishina T, Yoshikawa K, Miyazaki M, Ikeda T, Komeda M. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens Res. 2006;29:57–64. doi: 10.1291/hypres.29.57. [DOI] [PubMed] [Google Scholar]
- Kanemitsu H, Takai S, Tsuneyoshi H, Yoshikawa E, Nishina T, Miyazaki M, Ikeda T, Komeda M. Chronic chymase inhibition preserves cardiac function after left ventricular repair in rats. Eur J Cardiothorac Surg. 2008;33:25–31. doi: 10.1016/j.ejcts.2007.09.040. [DOI] [PubMed] [Google Scholar]
- Katsuda S, Kaji T. Atherosclerosis and extracellular matrix. J Atheroscler Thromb. 2003;10:267–274. doi: 10.5551/jat.10.267. [DOI] [PubMed] [Google Scholar]
- Katunuma N, Murata E, Kakegawa H, Matsui A, Tsuzuki H, Tsuge H, Turk D, Turk V, Fukushima M, Tada Y, Asao T. Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett. 1999;458:6–10. doi: 10.1016/s0014-5793(99)01107-2. [DOI] [PubMed] [Google Scholar]
- Kido H, Nakano A, Okishima N, Wakabayashi H, Kishi F, Nakaya Y, Yoshizumi M, Tamaki T. Human chymase, an enzyme forming novel bioactive 31-amino acid length endothelins. Biol Chem. 1998;379:885–891. doi: 10.1515/bchm.1998.379.7.885. [DOI] [PubMed] [Google Scholar]
- Kim MK, Kim HD, Park JH, Lim JI, Yang JS, Kwak WY, Sung SY, Kim HJ, Kim SH, Lee CH, Shim JY, Bae MH, Shin YA, Huh Y, Han TD, Chong W, Choi H, Ahn BN, Yang SO, Son MH. An orally active cathepsin K inhibitor, furan-2-carboxylic acid, 1-{1-[4-fluoro-2-(2-oxo-pyrrolidin-1-yl)-phenyl]-3-oxo-piperidin-4-ylcarba moyl}-cyclohexyl)-amide (OST-4077), inhibits osteoclast activity in vitro and bone loss in ovariectomized rats. J Pharmacol Exp Ther. 2006;318:555–562. doi: 10.1124/jpet.106.102798. [DOI] [PubMed] [Google Scholar]
- Kitamoto S, Sukhova GK, Sun J, Yang M, Libby P, Love V, Duramad P, Sun C, Zhang Y, Yang X, Peters C, Shi GP. Cathepsin L deficiency reduces diet-induced atherosclerosis in low-density lipoprotein receptor-knockout mice. Circulation. 2007;115:2065–2075. doi: 10.1161/CIRCULATIONAHA.107.688523. [DOI] [PubMed] [Google Scholar]
- Koch AE, Haines GK, Rizzo RJ, Radosevich JA, Pope RM, Robinson PG, Pearce WH. Human abdominal aortic aneurysms. Immunophenotypic analysis suggesting an immune-mediated response. Am J Pathol. 1990;137:1199–1213. [PMC free article] [PubMed] [Google Scholar]
- Kofford MW, Schwartz LB, Schechter NM, Yager DR, Diegelmann RF, Graham MF. Cleavage of type I procollagen by human mast cell chymase initiates collagen fibril formation and generates a unique carboxyl-terminal propeptide. J Biol Chem. 1997;272:7127–7131. doi: 10.1074/jbc.272.11.7127. [DOI] [PubMed] [Google Scholar]
- Koskinas KC, Feldman CL, Chatzizisis YS, Coskun AU, Jonas M, Maynard C, Baker AB, Papafaklis MI, Edelman ER, Stone PH. Natural history of experimental coronary atherosclerosis and vascular remodeling in relation to endothelial shear stress: a serial, in vivo intravascular ultrasound study. Circulation. 2010;121:2092–2101. doi: 10.1161/CIRCULATIONAHA.109.901678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Dare L, Vasko-Moser JA, James IE, Blake SM, Rickard DJ, Hwang SM, Tomaszek T, Yamashita DS, Marquis RW, Oh H, Jeong JU, Veber DF, Gowen M, Lark MW, Stroup G. A highly potent inhibitor of cathepsin K (relacatib) reduces biomarkers of bone resorption both in vitro and in an acute model of elevated bone turnover in vivo in monkeys. Bone. 2007;40:122–131. doi: 10.1016/j.bone.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Lazaar AL, Plotnick MI, Kucich U, Crichton I, Lotfi S, Das SK, Kane S, Rosenbloom J, Panettieri RA, Jr, Schechter NM, Pure E. Mast cell chymase modifies cell-matrix interactions and inhibits mitogen-induced proliferation of human airway smooth muscle cells. J Immunol. 2002;169:1014–1020. doi: 10.4049/jimmunol.169.2.1014. [DOI] [PubMed] [Google Scholar]
- Lee M, Sommerhoff CP, von Eckardstein A, Zettl F, Fritz H, Kovanen PT. Mast cell tryptase degrades HDL and blocks its function as an acceptor of cellular cholesterol. Arterioscler Thromb Vasc Biol. 2002;22:2086–2091. doi: 10.1161/01.atv.0000041405.07367.b5. [DOI] [PubMed] [Google Scholar]
- Lee-Rueckert M, Kovanen PT. Mast cell proteases: physiological tools to study functional significance of high density lipoproteins in the initiation of reverse cholesterol transport. Atherosclerosis. 2006;189:8–18. doi: 10.1016/j.atherosclerosis.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Lewiecki EM. Odanacatib, a cathepsin K inhibitor for the treatment of osteoporosis and other skeletal disorders associated with excessive bone remodeling. IDrugs. 2009;12:799–809. [PubMed] [Google Scholar]
- Li W, Kornmark L, Jonasson L, Forssell C, Yuan XM. Cathepsin L is significantly associated with apoptosis and plaque destabilization in human atherosclerosis. Atherosclerosis. 2009;202:92–102. doi: 10.1016/j.atherosclerosis.2008.03.027. [DOI] [PubMed] [Google Scholar]
- Li W, Yuan XM. Increased expression and translocation of lysosomal cathepsins contribute to macrophage apoptosis in atherogenesis. Ann N Y Acad Sci. 2004;1030:427–433. doi: 10.1196/annals.1329.053. [DOI] [PubMed] [Google Scholar]
- Lijnen HR. Extracellular proteolysis in the development and progression of atherosclerosis. Biochem Soc Trans. 2002;30:163–167. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Lindholt JS, Erlandsen EJ, Henneberg EW. Cystatin C deficiency is associated with the progression of small abdominal aortic aneurysms. Br J Surg. 2001;88:1472–1475. doi: 10.1046/j.0007-1323.2001.01911.x. [DOI] [PubMed] [Google Scholar]
- Lindstedt KA, Kovanen PT. Mast cells in vulnerable coronary plaques: potential mechanisms linking mast cell activation to plaque erosion and rupture. Curr Opin Lipidol. 2004;15:567–573. doi: 10.1097/00041433-200410000-00011. [DOI] [PubMed] [Google Scholar]
- Lindstedt L, Lee M, Oorni K, Bromme D, Kovanen PT. Cathepsins F and S block HDL3-induced cholesterol efflux from macrophage foam cells. Biochem Biophys Res Commun. 2003;312:1019–1024. doi: 10.1016/j.bbrc.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Linke M, Gordon RE, Brillard M, Lecaille F, Lalmanach G, Bromme D. Degradation of apolipoprotein B-100 by lysosomal cysteine cathepsins. Biol Chem. 2006;387:1295–1303. doi: 10.1515/BC.2006.160. [DOI] [PubMed] [Google Scholar]
- Liu J, Sukhova GK, Sun JS, Xu WH, Libby P, Shi GP. Lysosomal cysteine proteases in atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1359–1366. doi: 10.1161/01.ATV.0000134530.27208.41. [DOI] [PubMed] [Google Scholar]
- Liu J, Ma L, Yang J, Ren A, Sun Z, Yan G, Sun J, Fu H, Xu W, Hu C, Shi GP. Increased serum cathepsin S in patients with atherosclerosis and diabetes. Atherosclerosis. 2006a;186:411–419. doi: 10.1016/j.atherosclerosis.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Liu J, Sukhova GK, Yang JT, Sun J, Ma L, Ren A, Xu WH, Fu H, Dolganov GM, Hu C, Libby P, Shi GP. Cathepsin L expression and regulation in human abdominal aortic aneurysm, atherosclerosis, and vascular cells. Atherosclerosis. 2006b;184:302–311. doi: 10.1016/j.atherosclerosis.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Lutgens E, Lutgens SP, Faber BC, Heeneman S, Gijbels MM, de Winther MP, Frederik P, van der Made I, Daugherty A, Sijbers AM, Fisher A, Long CJ, Saftig P, Black D, Daemen MJ, Cleutjens KB. Disruption of the cathepsin K gene reduces atherosclerosis progression and induces plaque fibrosis but accelerates macrophage foam cell formation. Circulation. 2006;113:98–107. doi: 10.1161/CIRCULATIONAHA.105.561449. [DOI] [PubMed] [Google Scholar]
- Maehr R, Mintern JD, Herman AE, Lennon-Dumenil AM, Mathis D, Benoist C, Ploegh HL. Cathepsin L is essential for onset of autoimmune diabetes in NOD mice. J Clin Invest. 2005;115:2934–2943. doi: 10.1172/JCI25485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallen-St Clair J, Shi GP, Sutherland RE, Chapman HA, Caughey GH, Wolters PJ. Cathepsins L and S are not required for activation of dipeptidyl peptidase I (cathepsin C) in mice. Biol Chem. 2006;387:1143–1146. doi: 10.1515/BC.2006.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis RW, James I, Zeng J, Trout RE, Thompson S, Rahman A, Yamashita DS, Xie R, Ru Y, Gress CJ, Blake S, Lark MA, Hwang SM, Tomaszek T, Offen P, Head MS, Cummings MD, Veber DF. Azepanone-based inhibitors of human cathepsin L. J Med Chem. 2005;48:6870–6878. doi: 10.1021/jm0502079. [DOI] [PubMed] [Google Scholar]
- Martinet W, Kockx MM. Apoptosis in atherosclerosis: focus on oxidized lipids and inflammation. Curr Opin Lipidol. 2001;12:535–541. doi: 10.1097/00041433-200110000-00009. [DOI] [PubMed] [Google Scholar]
- Mayranpaa MI, Heikkila HM, Lindstedt KA, Walls AF, Kovanen PT. Desquamation of human coronary artery endothelium by human mast cell proteases: implications for plaque erosion. Coron Artery Dis. 2006;17:611–621. doi: 10.1097/01.mca.0000224420.67304.4d. [DOI] [PubMed] [Google Scholar]
- Mayranpaa MI, Trosien JA, Fontaine V, Folkesson M, Kazi M, Eriksson P, Swedenborg J, Hedin U. Mast cells associate with neovessels in the media and adventitia of abdominal aortic aneurysms. J Vasc Surg. 2009;50:388–395. doi: 10.1016/j.jvs.2009.03.055. discussion 395–386. [DOI] [PubMed] [Google Scholar]
- McGrath ME. The lysosomal cysteine proteases. Annu Rev Biophys Biomol Struct. 1999;28:181–204. doi: 10.1146/annurev.biophys.28.1.181. [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Takai S. Local angiotensin II-generating system in vascular tissues: the roles of chymase. Hypertens Res. 2001;24:189–193. doi: 10.1291/hypres.24.189. [DOI] [PubMed] [Google Scholar]
- Mizutani H, Schechter N, Lazarus G, Black RA, Kupper TS. Rapid and specific conversion of precursor interleukin 1 beta (IL-1 beta) to an active IL-1 species by human mast cell chymase. J Exp Med. 1991;174:821–825. doi: 10.1084/jem.174.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Omoto Y, Tokime K, Yamanaka K, Habe K, Morioka T, Kurokawa I, Tsutsui H, Yamanishi K, Nakanishi K, Mizutani H. Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J Immunol. 2006;177:8315–8319. doi: 10.4049/jimmunol.177.12.8315. [DOI] [PubMed] [Google Scholar]
- Oorni K, Sneck M, Bromme D, Pentikainen MO, Lindstedt KA, Mayranpaa M, Aitio H, Kovanen PT. Cysteine protease cathepsin F is expressed in human atherosclerotic lesions, is secreted by cultured macrophages, and modifies low density lipoprotein particles in vitro. J Biol Chem. 2004;279:34776–34784. doi: 10.1074/jbc.M310814200. [DOI] [PubMed] [Google Scholar]
- Ortlepp JR, Janssens U, Bleckmann F, Lauscher J, Merkelbach-Bruse S, Hanrath P, Hoffmann R. A chymase gene variant is associated with atherosclerosis in venous coronary artery bypass grafts. Coron Artery Dis. 2001;12:493–497. doi: 10.1097/00019501-200109000-00008. [DOI] [PubMed] [Google Scholar]
- Otto HH, Schirmeister T. Cysteine Proteases and Their Inhibitors. Chem Rev. 1997;97:133–172. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- Pagano MB, Bartoli MA, Ennis TL, Mao D, Simmons PM, Thompson RW, Pham CT. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci U S A. 2007;104:2855–2860. doi: 10.1073/pnas.0606091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol Sci. 2008;29:22–28. doi: 10.1016/j.tips.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Pang L, Nie M, Corbett L, Sutcliffe A, Knox AJ. Mast cell beta-tryptase selectively cleaves eotaxin and RANTES and abrogates their eosinophil chemotactic activities. J Immunol. 2006;176:3788–3795. doi: 10.4049/jimmunol.176.6.3788. [DOI] [PubMed] [Google Scholar]
- Pejler G, Abrink M, Ringvall M, Wernersson S. Mast cell proteases. Adv Immunol. 2007;95:167–255. doi: 10.1016/S0065-2776(07)95006-3. [DOI] [PubMed] [Google Scholar]
- Platt MO, Ankeny RF, Jo H. Laminar shear stress inhibits cathepsin L activity in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1784–1790. doi: 10.1161/01.ATV.0000227470.72109.2b. [DOI] [PubMed] [Google Scholar]
- Potts W, Bowyer J, Jones H, Tucker D, Freemont AJ, Millest A, Martin C, Vernon W, Neerunjun D, Slynn G, Harper F, Maciewicz R. Cathepsin L-deficient mice exhibit abnormal skin and bone development and show increased resistance to osteoporosis following ovariectomy. Int J Exp Pathol. 2004;85:85–96. doi: 10.1111/j.0959-9673.2004.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punturieri A, Filippov S, Allen E, Caras I, Murray R, Reddy V, Weiss SJ. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin K-deficient human macrophages. J Exp Med. 2000;192:789–799. doi: 10.1084/jem.192.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy VY, Zhang QY, Weiss SJ. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc Natl Acad Sci U S A. 1995;92:3849–3853. doi: 10.1073/pnas.92.9.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinheckel T, Deussing J, Roth W, Peters C. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol Chem. 2001;382:735–741. doi: 10.1515/BC.2001.089. [DOI] [PubMed] [Google Scholar]
- Riese RJ, Wolf PR, Bromme D, Natkin LR, Villadangos JA, Ploegh HL, Chapman HA. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity. 1996;4:357–366. doi: 10.1016/s1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- Riese RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER, De Sanctis GT, Ploegh HL, Chapman HA. Cathepsin S activity regulates antigen presentation and immunity. J Clin Invest. 1998;101:2351–2363. doi: 10.1172/JCI1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizas KD, Ippagunta N, Tilson MD., 3rd Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol Rev. 2009;17:201–210. doi: 10.1097/CRD.0b013e3181b04698. [DOI] [PubMed] [Google Scholar]
- Roberts R. Lysosomal cysteine proteases: structure, function and inhibition of cathepsins. Drug News Perspect. 2005;18:605–614. doi: 10.1358/dnp.2005.18.10.949485. [DOI] [PubMed] [Google Scholar]
- Rodgers KJ, Watkins DJ, Miller AL, Chan PY, Karanam S, Brissette WH, Long CJ, Jackson CL. Destabilizing role of cathepsin S in murine atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2006;26:851–856. doi: 10.1161/01.ATV.0000203526.75772.4b. [DOI] [PubMed] [Google Scholar]
- Russo A, Russo G, Peticca M, Pietropaolo C, Di Rosa M, Iuvone T. Inhibition of granuloma-associated angiogenesis by controlling mast cell mediator release: role of mast cell protease-5. Br J Pharmacol. 2005;145:24–33. doi: 10.1038/sj.bjp.0706112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, Moritz JD, Schu P, von Figura K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A. 1998;95:13453–13458. doi: 10.1073/pnas.95.23.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen-Mankonen HJ, Morko J, Vuorio E. Role of cathepsin K in normal joints and in the development of arthritis. Curr Drug Targets. 2007;8:315–323. doi: 10.2174/138945007779940188. [DOI] [PubMed] [Google Scholar]
- Samokhin AO, Wong A, Saftig P, Bromme D. Role of cathepsin K in structural changes in brachiocephalic artery during progression of atherosclerosis in apoE-deficient mice. Atherosclerosis. 2008;200:58–68. doi: 10.1016/j.atherosclerosis.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Sanker S, Chandrasekharan UM, Wilk D, Glynias MJ, Karnik SS, Husain A. Distinct multisite synergistic interactions determine substrate specificities of human chymase and rat chymase-1 for angiotensin II formation and degradation. J Biol Chem. 1997;272:2963–2968. doi: 10.1074/jbc.272.5.2963. [DOI] [PubMed] [Google Scholar]
- Satomura K, Yin M, Shimizu S, Kato Y, Nagano T, Komeichi H, Ohsuga M, Katsuta Y, Aramaki T, Omoto Y. Increased chymase in livers with autoimmune disease: colocalization with fibrosis. J Nippon Med Sch. 2003;70:490–495. doi: 10.1272/jnms.70.490. [DOI] [PubMed] [Google Scholar]
- Schulte S, Sun J, Libby P, Macfarlane L, Sun C, Lopez-Ilasaca M, Shi GP, Sukhova GK. Cystatin C deficiency promotes inflammation in angiotensin II-induced abdominal aortic aneurisms in atherosclerotic mice. Am J Pathol. 2010;177:456–463. doi: 10.2353/ajpath.2010.090381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurigt U, Sevenich L, Vannier C, Gajda M, Schwinde A, Werner F, Stahl A, von Elverfeldt D, Becker AK, Bogyo M, Peters C, Reinheckel T. Trial of the cysteine cathepsin inhibitor JPM-OEt on early and advanced mammary cancer stages in the MMTV-PyMT-transgenic mouse model. Biol Chem. 2008;389:1067–1074. doi: 10.1515/BC.2008.115. [DOI] [PubMed] [Google Scholar]
- Schwartz LB, Bradford TR, Littman BH, Wintroub BU. The fibrinogenolytic activity of purified tryptase from human lung mast cells. J Immunol. 1985;135:2762–2767. [PubMed] [Google Scholar]
- Shah PP, Myers MC, Beavers MP, Purvis JE, Jing H, Grieser HJ, Sharlow ER, Napper AD, Huryn DM, Cooperman BS, Smith AB, 3rd, Diamond SL. Kinetic characterization and molecular docking of a novel, potent, and selective slow-binding inhibitor of human cathepsin L. Mol Pharmacol. 2008;74:34–41. doi: 10.1124/mol.108.046219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11:3061–3072. doi: 10.2174/1381612054865064. [DOI] [PubMed] [Google Scholar]
- Shenoy RT, Chowdhury SF, Kumar S, Joseph L, Purisima EO, Sivaraman J. A combined crystallographic and molecular dynamics study of cathepsin L retrobinding inhibitors. J Med Chem. 2009;52:6335–6346. doi: 10.1021/jm900596y. [DOI] [PubMed] [Google Scholar]
- Shi GP, Munger JS, Meara JP, Rich DH, Chapman HA. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J Biol Chem. 1992;267:7258–7262. [PubMed] [Google Scholar]
- Shi GP, Webb AC, Foster KE, Knoll JH, Lemere CA, Munger JS, Chapman HA. Human cathepsin S: chromosomal localization, gene structure, and tissue distribution. J Biol Chem. 1994;269:11530–11536. [PubMed] [Google Scholar]
- Shi GP, Chapman HA, Bhairi SM, DeLeeuw C, Reddy VY, Weiss SJ. Molecular cloning of human cathepsin O, a novel endoproteinase and homologue of rabbit OC2. FEBS Lett. 1995;357:129–134. doi: 10.1016/0014-5793(94)01349-6. [DOI] [PubMed] [Google Scholar]
- Shi GP, Villadangos JA, Dranoff G, Small C, Gu L, Haley KJ, Riese R, Ploegh HL, Chapman HA. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity. 1999a;10:197–206. doi: 10.1016/s1074-7613(00)80020-5. [DOI] [PubMed] [Google Scholar]
- Shi GP, Sukhova GK, Grubb A, Ducharme A, Rhode LH, Lee RT, Ridker PM, Libby P, Chapman HA. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J Clin Invest. 1999b;104:1191–1197. doi: 10.1172/JCI7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–583. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhova GK, Zhang Y, Pan JH, Wada Y, Yamamoto T, Naito M, Kodama T, Tsimikas S, Witztum JL, Lu ML, Sakara Y, Chin MT, Libby P, Shi GP. Deficiency of cathepsin S reduces atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;111:897–906. doi: 10.1172/JCI14915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhova GK, Wang B, Libby P, Pan JH, Zhang Y, Grubb A, Fang K, Chapman HA, Shi GP. Cystatin C deficiency increases elastic lamina degradation and aortic dilatation in apolipoprotein E-null mice. Circ Res. 2005;96:368–375. doi: 10.1161/01.RES.0000155964.34150.F7. [DOI] [PubMed] [Google Scholar]
- Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, MacFarlane LA, Mallen-St Clair J, Shi GP. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007a;13:719–724. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, Sun C, Zhang Y, Liu J, Ennis TL, Knispel R, Xiong W, Thompson RW, Baxter BT, Shi GP. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest. 2007b;117:3359–3368. doi: 10.1172/JCI31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Zhang J, Lindholt JS, Sukhova GK, Liu J, He A, Abrink M, Pejler G, Stevens RL, Thompson RW, Ennis TL, Gurish MF, Libby P, Shi GP. Critical role of mast cell chymase in mouse abdominal aortic aneurysm formation. Circulation. 2009;120:973–982. doi: 10.1161/CIRCULATIONAHA.109.849679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270:4689–4696. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- Takai S, Jin D, Nishimoto M, Yuda A, Sakaguchi M, Kamoshita K, Ishida K, Sukenaga Y, Sasaki S, Miyazaki M. Oral administration of a specific chymase inhibitor, NK3201, inhibits vascular proliferation in grafted vein. Life Sci. 2001;69:1725–1732. doi: 10.1016/s0024-3205(01)01255-3. [DOI] [PubMed] [Google Scholar]
- Takai S, Sakonjo H, Fukuda K, Jin D, Sakaguchi M, Kamoshita K, Ishida K, Sukenaga Y, Miyazaki M. A novel chymase inhibitor, 2-(5-formylamino-6-oxo-2-phenyl-1,6-dihydropyrimidine-1-yl)-N-[[,4-dioxo-1 -phenyl-7-(2-pyridyloxy)]2-heptyl]acetamide (NK3201), suppressed intimal hyperplasia after balloon injury. J Pharmacol Exp Ther. 2003;304:841–844. doi: 10.1124/jpet.102.042580. [DOI] [PubMed] [Google Scholar]
- Taleb S, Lacasa D, Bastard JP, Poitou C, Cancello R, Pelloux V, Viguerie N, Benis A, Zucker JD, Bouillot JL, Coussieu C, Basdevant A, Langin D, Clement K. Cathepsin S, a novel biomarker of adiposity: relevance to atherogenesis. Faseb J. 2005;19:1540–1542. doi: 10.1096/fj.05-3673fje. [DOI] [PubMed] [Google Scholar]
- Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. 2005;280:9291–9296. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- Tertov VV, Orekhov AN. Metabolism of native and naturally occurring multiple modified low density lipoprotein in smooth muscle cells of human aortic intima. Exp Mol Pathol. 1997;64:127–145. doi: 10.1006/exmp.1997.2216. [DOI] [PubMed] [Google Scholar]
- Toomes C, James J, Wood AJ, Wu CL, McCormick D, Lench N, Hewitt C, Moynihan L, Roberts E, Woods CG, Markham A, Wong M, Widmer R, Ghaffar KA, Pemberton M, Hussein IR, Temtamy SA, Davies R, Read AP, Sloan P, Dixon MJ, Thakker NS. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet. 1999;23:421–424. doi: 10.1038/70525. [DOI] [PubMed] [Google Scholar]
- Tsunemi K, Takai S, Nishimoto M, Yuda A, Hasegawa S, Sawada Y, Fukumoto H, Sasaki S, Miyazaki M. Possible roles of angiotensin II-forming enzymes, angiotensin converting enzyme and chymase-like enzyme, in the human aneurysmal aorta. Hypertens Res. 2002;25:817–822. doi: 10.1291/hypres.25.817. [DOI] [PubMed] [Google Scholar]
- Tsunemi K, Takai S, Nishimoto M, Jin D, Sakaguchi M, Muramatsu M, Yuda A, Sasaki S, Miyazaki M. A specific chymase inhibitor, 2-(5-formylamino-6-oxo-2-phenyl-1,6-dihydropyrimidine-1-yl)-N-[[3,4-dioxo- 1-phenyl-7-(2-pyridyloxy)]-2-heptyl]acetamide (NK3201), suppresses development of abdominal aortic aneurysm in hamsters. J Pharmacol Exp Ther. 2004;309:879–883. doi: 10.1124/jpet.103.063974. [DOI] [PubMed] [Google Scholar]
- Tu C, Ortega-Cava CF, Chen G, Fernandes ND, Cavallo-Medved D, Sloane BF, Band V, Band H. Lysosomal cathepsin B participates in the podosome-mediated extracellular matrix degradation and invasion via secreted lysosomes in v-Src fibroblasts. Cancer Res. 2008;68:9147–9156. doi: 10.1158/0008-5472.CAN-07-5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk D, Guncar G. Lysosomal cysteine proteases (cathepsins): promising drug targets. Acta Crystallogr D Biol Crystallogr. 2003;59((Pt 2)):203–213. doi: 10.1107/s0907444902021479. [DOI] [PubMed] [Google Scholar]
- Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 2001;20:4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara Y, Urata H, Ideishi M, Arakawa K, Saku K. Chymase inhibition suppresses high-cholesterol diet-induced lipid accumulation in the hamster aorta. Cardiovasc Res. 2002;55:870–876. doi: 10.1016/s0008-6363(02)00458-3. [DOI] [PubMed] [Google Scholar]
- Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr Pharm Des. 2007;13:387–403. doi: 10.2174/138161207780162962. [DOI] [PubMed] [Google Scholar]
- Villadangos JA, Bryant RA, Deussing J, Driessen C, Lennon-Dumenil AM, Riese RJ, Roth W, Saftig P, Shi GP, Chapman HA, Peters C, Ploegh HL. Proteases involved in MHC class II antigen presentation. Immunol Rev. 1999;172:109–120. doi: 10.1111/j.1600-065x.1999.tb01360.x. [DOI] [PubMed] [Google Scholar]
- Wang B, Sun J, Kitamoto S, Yang M, Grubb A, Chapman HA, Kalluri R, Shi GP. Cathepsin S controls angiogenesis and tumor growth via matrix-derived angiogenic factors. J Biol Chem. 2006;281:6020–6029. doi: 10.1074/jbc.M509134200. [DOI] [PubMed] [Google Scholar]
- Welss T, Sun J, Irving JA, Blum R, Smith AI, Whisstock JC, Pike RN, von Mikecz A, Ruzicka T, Bird PI, Abts HF. Hurpin is a selective inhibitor of lysosomal cathepsin L and protects keratinocytes from ultraviolet-induced apoptosis. Biochemistry. 2003;42:7381–7389. doi: 10.1021/bi027307q. [DOI] [PubMed] [Google Scholar]
- Wiener JJ, Sun S, Thurmond RL. Recent advances in the design of cathepsin S inhibitors. Curr Top Med Chem. 2010;10:717–732. doi: 10.2174/156802610791113432. [DOI] [PubMed] [Google Scholar]
- Wolters PJ, Pham CT, Muilenburg DJ, Ley TJ, Caughey GH. Dipeptidyl peptidase I is essential for activation of mast cell chymases, but not tryptases, in mice. J Biol Chem. 2001;276:18551–18556. doi: 10.1074/jbc.M100223200. [DOI] [PubMed] [Google Scholar]
- Xiang M, Sun J, Lin Y, Zhang J, Chen H, Yang D, Wang J, Shi GP. Usefulness of serum tryptase level as an independent biomarker for coronary plaque instability in a Chinese population. Atherosclerosis. 2011 doi: 10.1016/j.atherosclerosis.2011.01.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Ishimaru N, Arakaki R, Katunuma N, Hayashi Y. Cathepsin L inhibition prevents murine autoimmune diabetes via suppression of CD8(+) T cell activity. PLoS One. 2010;5:e12894. doi: 10.1371/journal.pone.0012894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita DS, Marquis RW, Xie R, Nidamarthy SD, Oh HJ, Jeong JU, Erhard KF, Ward KW, Roethke TJ, Smith BR, Cheng HY, Geng X, Lin F, Offen PH, Wang B, Nevins N, Head MS, Haltiwanger RC, Narducci Sarjeant AA, Liable-Sands LM, Zhao B, Smith WW, Janson CA, Gao E, Tomaszek T, McQueney M, James IE, Gress CJ, Zembryki DL, Lark MW, Veber DF. Structure activity relationships of 5-, 6-, and 7-methyl-substituted azepan-3-one cathepsin K inhibitors. J Med Chem. 2006;49:1597–1612. doi: 10.1021/jm050915u. [DOI] [PubMed] [Google Scholar]
- Yang H, Kala M, Scott BG, Goluszko E, Chapman HA, Christadoss P. Cathepsin S is required for murine autoimmune myasthenia gravis pathogenesis. J Immunol. 2005;174:1729–1737. doi: 10.4049/jimmunol.174.3.1729. [DOI] [PubMed] [Google Scholar]
- Yang M, Zhang Y, Pan J, Sun J, Liu J, Libby P, Sukhova GK, Doria A, Katunuma N, Peroni OD, Guerre-Millo M, Kahn BB, Clement K, Shi GP. Cathepsin L activity controls adipogenesis and glucose tolerance. Nat Cell Biol. 2007;9:970–977. doi: 10.1038/ncb1623. [DOI] [PubMed] [Google Scholar]
- Yang M, Sun J, Zhang T, Liu J, Zhang J, Shi MA, Darakhshan F, Guerre-Millo M, Clement K, Gelb BD, Dolgnov G, Shi GP. Deficiency and inhibition of cathepsin K reduce body weight gain and increase glucose metabolism in mice. Arterioscler Thromb Vasc Biol. 2008;28:2202–2208. doi: 10.1161/ATVBAHA.108.172320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Li Z, Greenbaum D, Bogyo M, Weber E, Bromme D. Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J Biol Chem. 2004;279:36761–36770. doi: 10.1074/jbc.M403986200. [DOI] [PubMed] [Google Scholar]
- Yasuda Y, Kaleta J, Bromme D. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005;57:973–993. doi: 10.1016/j.addr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Yeong P, Ning Y, Xu Y, Li X, Yin L. Tryptase promotes human monocyte-derived macrophage foam cell formation by suppressing LXRalpha activation. Biochim Biophys Acta. 2010;1801:567–576. doi: 10.1016/j.bbalip.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Imada T, Nakakubo H, Nakamura N, Naito K. Rat mast cell protease-I enhances immunoglobulin E production by mouse B cells stimulated with interleukin-4. Immunology. 2001;104:333–340. doi: 10.1046/j.1365-2567.2001.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi M, Troen B, Moonga BS, Abe E. Cathepsin K, osteoclastic resorption, and osteoporosis therapy. J Bone Miner Res. 2001;16:1747–1749. doi: 10.1359/jbmr.2001.16.10.1747. [DOI] [PubMed] [Google Scholar]
- Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res. 1983;53:502–514. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]