

Abstract
Medullary thyroid carcinoma occurs in both sporadic (75%) and hereditary (25%) forms. The missense mutations of RET proto-oncogene in MTC development have been well demonstrated. To investigate the spectrum of predominant RET germline mutations in exons 10, 11, and 16 in hereditary MTC in Iranian population, 217 participants were included. Genomic DNAs were extracted from the leukocytes using the standard Salting Out/Proteinase K method. Mutation detection was performed through PCR-RFLP and DNA sequencing. In 217 participants, 43 missense mutations were identified in exons 10 (6%), 11 (13%), and 16 (0.9%). Moreover, a novel germline mutation was detected in exon 11 (S686N). Also four different polymorphisms were found in intron 16 in eight patients. The obtained data showed the frequency profile of RET mutations in Iranian individuals with MTC (19.8%). The most frequent mutation in our population was C634G whereas in most population it was C634R. Altogether, these results underline the importance of the genetic background of family members of any patient with MTC.
1. Introduction
Thyroid carcinoma is the most frequent malignant tumor of the endocrine system and accounts for nearly 1% of total human cancers [1]. Medullary thyroid carcinoma (MTC) is a malignancy of the parafollicular C cells derived from neural crest. MTC represents 5–10% of all types of thyroid cancers [2–4] and occurs in both sporadic (75%) and hereditary (25%) forms. The latter has an autosomal dominant mode of inheritance with variable expressivity and an age-related penetrance [5, 6]. This form of MTC is divided into 3 subtypes: isolated Familial MTC (FMTC) and multiple endocrine neoplasia type 2A and 2B (MEN2A, 2B). Affected individuals in FMTC develop MTC without any other abnormalities. MEN2A is characterized by MTC, pheochromocytoma, and parathyroid hyperplasia (75% of hereditary MTC) and MEN2B is characterized by MTC, pheochromocytoma, mucosal neuromas, ganglioneuromatosis of the gut, and a Marfanoid habitus (MEN2B) [5–8]. The gene(s) responsible for FMTC, MEN2A, and MEN2B were mapped on chromosome 10q11.2 by genetic linkage analysis [9]. Rearranged during transfection (RET) proto-oncogene is located on 10q11.21 chromosome [10] within the candidate region and has 21 exons and its point mutations have been identified in FMTC, MEN2A, and MEN2B. This proto-oncogene encodes a single-pass transmembrane receptor with a tyrosine kinase activity that is crucial in signal transduction during cell growth and differentiation [11]. The RET receptor in cell membrane is composed of one cystein rich residue, four cadherin-like repeats, and one calcium binding site in extracellular portion and contains tyrosine kinase domain in intracellular portion [12–14]. Upon ligand binding, RET dimerization is induced and mutual transphosphorylation of tyrosine residues occurs [15].
RET proto-oncogene loss of function results in Hirchsprung disease and its gain of function is implicated in a number of cancer syndromes such as MTC [13, 15]. As RET is a proto-oncogene, a single activating mutation in its one allele is sufficient to cause neoplastic changes [16]. Hereditary MTC is caused by germline mutations in the RET proto-oncogene and its somatic mutations is implicated in the sporadic MTC [2, 17]. The most frequent mutations in the RET proto-oncogene have been found in five cysteine codons 609, 611, 618, and 620 of exon 10 and codon 634 of exon 11. In addition, some other mutations have also been identified in noncystein codons such as 804 in exon 14, 883 in exon 15, and 918 in exon 16 [18, 19]. A germline mutation in this proto-oncogene has been observed in more than 95% of MEN2 patients [3] and several studies have found that point mutations are the extracellular domain in more than 96% MEN2A and 86% FMTC patients [13, 20]. These mutations induce RET proto-oncogene catalytic activity through disulfide homodimerization even in ligand absence [21–24]. Germline mutations also occur in the RET intracellular domain in codons 768, 790, and 791 (exon 13), codons 804, 844 (exon 14), and codon 891 (exon 15) in the FMTC [25] and codon 918 (exon 16) in MEN2B patients [16].
The early diagnosis of carrier RET mutation individuals, which are susceptible to develop MTC later in life, is possible. Genetic screening especially is useful for first-degree kindred of MTC patients. The aim of this study was to determine the allele frequency of predominant RET germline mutations in exons 10, 11, and 16 among Iranian hereditary MTC patients.
2. Materials and Methods
2.1. Patients
The study population consisted of 217 individuals, including 151 patients and 66 their first-degree relatives diagnosed for MTC between 2002 and 2010. They were referred to Research Institute for Endocrine Sciences, Shahid Beheshti University of Medical Science and the volunteer individuals were included in the survey after obtaining an informed consent. The diagnosis of MTC was confirmed by histopathologic documents. After germline RET mutation analysis, the first-degree relatives of MTC patients with positive mutations were also examined for RET mutations. This study has been approved by the Institutional Review Board and Ethics Committee of Obesity Research Center, Research Institute for Endocrine Sciences, Shahid Beheshti University of Medical Sciences.
2.2. RET Genetic Analysis
Blood samples were collected in EDTA from all 217 subjects. Genomic DNA was extracted from peripheral leucocytes samples according to a Standard Salting-out/Proteinase K method. An aliquot of DNA for each individual was stored at −20°C.
The RET gene exons 10, 11, and 16 were analyzed in all subjects using PCR-RFLP methods [22, 23]. For positive patients, sequencing was carried out in both sense and antisense direction.
For amplification of the DNA segment containing RET exon 10, the following primers were used: (10F 5′GCGCCCCAGGAGGCTGATGC3′) and (10R 5′CGTGGTGGTCCCGGCCGCC3′). The RET exon 11 was amplified using following primers: (11AF 5′CCTCTGCGGTGCCAAGCCTC3′) and (11AR 5′CACCGGAAGAGGAGTAGCTG3′) [23, 24, 26]. Amplification was carried out in a volume of 50 μL containing 1.5 μL of 10×buffer, 50 ng DNA, 0.3 μL of each dNTPs (10 mM) (Boehringer Mannheim Co.), 1 μL of each exons 10 and 11 primers (10 μM) (TIB MOLBIOL Synthesalabor Co.), 0.25 μL MgCl2 (50 mM), and one U Taq polymerase (Boehringer Mannheim Co.). PCR reaction for both exons 10 and 11 was 30 cycles and performed in an automatic thermocycler (Omnigene & Hybaid Co.) under the following conditions: denaturation at 93°C for 45 seconds, annealing at 67°C for 30 seconds and extension at 72°C for another 45 seconds, and final extension at 72°C for 10 minutes [18, 27].
For amplification of the DNA segment containing RET exon 16, the following primers were used: (16F 5′GTGCCCAGGAGTGTCTACCA3′) and (16R 5′CAGGACCACAGGAGGGTAAC3′). A PCR reaction of exon 16 was performed in a 15 μL mixture containing 50 ng DNA, 0.35 μL of MgCl2 (50 mM), 0.5 μL of each dNTPs (10 mM) (Boehringer Mannheim Co.), 0.6 μL of each exon 16 primers (10 μM) (TIB MOLBIOL Synthesalabor Co.), 1.5 μL of 10 × buffer, and one U Taq polymerase (Boehringer Mannheim Co.). PCR reaction for exon 16 was 30 cycles and performed in an automatic thermocycler (Omnigene & Hybaid Co.) under the following conditions: denaturation at 92°C for 10 minutes and 93°C for 45 seconds, annealing at 59.5°C for 30 seconds, extension at 72°C for 55 seconds, and final extension at 72°C for 10 minutes [27–29].
The amplified PCR products were digested by each of Taq I, BstU I, Mbo II, Rsa I, Nla IV (England Biolabs), and Cfo I (Roehe Molecular Biochemicals) restriction enzymes for exon 10. The products were digested with the following restriction enzymes Cfo I, Rsa I, Hae III, and Dde I for exon 11, and FokI for exon 16 (England Biolabs) in the restriction buffer according to the manufacturer's instructions [10, 27, 28]. The RFLP-produced patterns by these restriction enzymes in the presence and absence of each RET exon 10, 11, and 16 mutations have been shown in Table 1. The digested samples were separated by electrophoresis through a 10% nondenaturing polyacrylamide gel electrophoresis and then detected by silver staining method. The positive samples for RET mutation then were sequences.
Table 1.
Characterization and distribution of RET proto-oncogene germ-line mutations in exons 10, 11, 16, and intron 16 among patients with MTC and their families.
| RET mutation |
Exon | Changed bp | ||
|---|---|---|---|---|
| Normal (Mutant) | Frequency | Families | ||
| C611W | 10 | TGC (TGG) | 0 | 0 |
| C618Y | 10 | TGC (TAC) | 5 | 2 |
| C618R | 10 | TGC (CGC) | 1 | 1 |
| C618F | 10 | TGC (TTC) | 4 | 4 |
| C618S | 10 | TGC (AGC) | 1 | 1 |
| C620R | 10 | TGC (CGC) | 1 | 1 |
| C620F | 10 | TGC (TTC) | 1 | 1 |
| C634R | 11 | TGC (CGC) | 1 | 1 |
| C634Y | 11 | TGC (TAC) | 5 | 3 |
| C634G | 11 | TGC (GGC) | 11 | 9 |
| C634W | 11 | TGC (TGG) | 1 | 1 |
| C634S | 11 | TGC (AGC) | 9 | 2 |
| S686N | 11 | AGC>AAC | 1 | 1 |
| M918T | 16 | ATG (ACG) | 2 | 2 |
| Intron 16 | A>T (rs3026772) | 2 | 2 | |
| 45044G>A | 1 | 1 | ||
| 45095C>A | 4 | 1 | ||
| 45190C>A | 1 | 1 | ||
3. Results
Altogether, 217 individuals, including 126 females and 91 males, participated in this study and the overall female-to-male ratio was 1.4 : 1. Among these, 151 individuals were diagnosed with MTC (88 females and 63 males) and 66 individuals were their first-degree relatives. The mean age of individuals was 33.4 ± 15.8 years. Genetic analyses revealed a germline RET missense mutation in 43 out of 217 (19.8%) individuals that 24 mutations occurred in female and 19 mutations were in male. From RET positive individuals, 36 mutations were in patients and seven mutations were in their first-degree relatives. Moreover, the mutations found in this study are belonging to the independent families.
In this study the majority of RET mutations (28 of 43, 65.1%) were located in exon 11 (11 C634G, nine C634S, five C634Y, one C634R, one C634W, and one S686N) (Figure 1). Interestingly, one of the positive RET patients had a new restriction site in exon 11 for CfoI restriction enzyme, but its cut fragments on a poly acryl amid gel were differed from another positive patients who had this restriction site (C634R). With direct DNA sequencing of exon 11 of this patient, a new missense mutation was detected at codon 686 (Ser686Asn, AGC>AAC) that has been not reported yet. This patient was an 18-year-old girl who had underwent thyroidectomy for thyroid nodules about 2 years ago, and her father suffered from very aggressive MTC.
Figure 1.
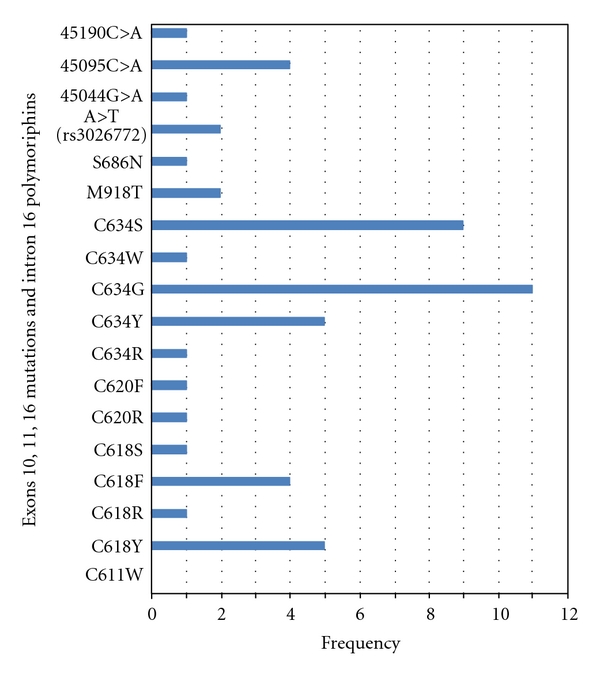
The allele frequency of the RET proto-oncogene mutations in Iranian patients with MTC.
The other 15 mutations were found in exons 10 and 16. In particular, 13 of 43 mutations (30.2%) were in exon 10 (five C618Y, four C618F, one C618S, one C618R, one C620F, and one C620R). In addition, two of 43 mutations (4.6%) were identified at codon 918 of exon 16 (M918T). One of these patients was a 14-year-old girl with early-diagnosed MTC. This mutation was found only in this patient although her parents were normal suggesting that it may be a de novo mutation. The other patient with M918T mutation was a boy that had been thyroidectomized when he was 10-year-old and he was diagnosed for early MTC. Unfortunately, he died when he was 19-year-old, because of distant metastasis to lung and brain.
Additionally, in the present study four sequence variations were detected in intron 16 of the RET proto-oncogene in seven patients and one their relatives, which three of those were new variations. These polymorphisms include A>T (rs3026772), 45044G>A, 45095C>A, and 45190C>A. A first variant, A>T (rs3026772), was identified in intron 16 in 2 patients affected to MTC. A second variant, 45044G>A, was detected in a patient that had a mutation in codon 634 (C634S). A third variant, 45095C>A, was identified in a family (4 individuals) with MEN2B where all of them carried a mutation in codon 611 (C611Y). The last variant, 45190C>A, was in a patient diagnosed for MTC with additional mutation in codon 620 (C620R).
4. Discussion
In present study, by mutational screening of the RET proto-oncogene we found 43 germline mutations in the predominant codons of exons 10, 11, and 16 among 217 individuals with MTCs. All of these encode cysteine codon 618 or 620 in exon 10 (30.2%) and cysteine codon 634 in exon 11 (65.1%), except of two (4.6%) mutations that occurred in codon 918 in exon 16 (M918T) and one new mutation that found in codon 686 in exon 11 (S686N). In this investigation, overall frequency profile of RET proto-oncogene germline mutation in Iranian MTC patients was estimated 19.8%.
Mutations of the extracellular RET cysteine-rich domain at codons 634, 609, 611, 618, and 620 resulted in ligand-independent dimerization of receptor molecules, enhanced phosphorylation of intracellular substrates, and cell transformation. Germline mutations in codons 609, 611, 618, 620, 634, and 768 have been discovered predominantly in MEN 2A and FMTC [27–29]. Mutation of the intracellular tyrosine kinase (codon 918) has no effect on receptor dimerization but causes constitutive activation of intracellular signaling pathways and also results in cellular transformation. It is demonstrated that patients with codon 918 mutations and MEN2B have a high risk of aggressive MTC occurring at a young age [29].
The mutations at codon 634, known as a common mutation in Caucasians [26], accounted for 65.1% of all mutations found in Iranian patients with MTC, in our study. Among five different types of nucleotide substitution found in this codon, changes from Cys to Gly (11 of 43) were the most common, followed by Cys to Ser (9 of 43), Cys to Tyr (5 of 43), Cys to Arg (one of 43), and Cys to Trp (one of 43). A comparison of our data with those available in the literature on other Caucasians indicates that the common alteration from Cys634Gly in this study may represent a founder effect. Indeed it has been reported that Cys634Arg mutation-that is the most common mutation in MTC patients in many population—is related to parathyroid diseases [30]. However, this mutation is rare in our population (identified in one patient, only). However, in the very recent study that carried out by Alvandi et al. in Iran, the most frequent mutation was Cys634Arg (five mutations in 55 patients). This different result in comparison with our study may be related to different genetic background of those studied population [31].
Fernández et al. in a study showed that the most frequent RET mutation in MEN 2A Spanish families is C634Y [32]. The RET proto-oncogene mutation analyses in French hereditary MEN2A and their first-degree relatives revealed that the most frequent mutation in this population was C634R and C634Y [29, 33]. In contrast, more prevalent mutation in FMTC in Sardinia was observed in codon 804 (V804M) and the less frequent mutant allele was present in codon 634 [34]. Also, high prevalence of RET mutations in the hereditary type of MTC has been found in codons 634 (C634R), 918 (M918T), 768, and 804 in American population [19]. Another study in China showed that the highest frequency of the RET mutation in patients with hereditary MTC was in codon 634 (C634Y) and 918 (M918T) in MEN2A and MEN2B, respectively. However, the most frequent RET proto-oncogene mutations in Saudi's families with MEN2A and FMTC [35] and in the Netherlands population with FMTC were at codon 618 [19]. A mutation rate in codon 918 (M918T) was high in sporadic type of MTC in Portugal, Czech Republic, and Italy population [2–4, 33, 36]. However, we identified only two M918T germline mutations in studied population, which in comparison with other population is low.
In general, it is apparent that the prevalence of RET proto-oncogene mutations in most Caucasian population may be related to codon 634 and codon 918. The present study also is in agreement with these reports, except for codon 918.
We showed the frequency profile of RET proto-oncogene mutations in a sample of 151 Iranian MTCs and 66 their relatives. These results underline the importance of the genetic background in the distribution of RET mutations and should be taken into account in genetic evaluation of MTC patients.
Finally, it is suggested that other RET exons especially those with high frequency of mutations such as exons 13, 14, and 15 should be examined. Direct sequencing analysis is also an accurate method to detect unknown RET mutations. Furthermore, a transforming activity and functional effect(s) of a new RET mutants such as S686N and intronic polymorphisms remain to be elucidated.
Acknowledgments
This study was supported by a research grant from the Endocrine Research Center, Shahid Beheshti University of Medical Sciences. The authors are indebted to kind collaboration of several endocrinology specialists. They express their gratitude to the staff of molecular genetic laboratory at the Endocrine Research Center, for their skillful technical assistance.
References
- 1.Nikiforova MN, Nikiforov YE. Molecular genetics of thyroid cancer: implications for diagnosis, treatment and prognosis. Expert Review of Molecular Diagnostics. 2008;8(1):83–95. doi: 10.1586/14737159.8.1.83. [DOI] [PubMed] [Google Scholar]
- 2.Dvoráká S, Václavíková E, Sýkorová V, et al. New multiple somatic mutations in the RET proto-oncogene associated with a sporadic medullary thyroid carcinoma. Thyroid. 2006;16(3):311–316. doi: 10.1089/thy.2006.16.311. [DOI] [PubMed] [Google Scholar]
- 3.Moura MM, Cavaco BM, Pinto AE, et al. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. British Journal of Cancer. 2009;100(11):1777–1783. doi: 10.1038/sj.bjc.6605056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marsh DJ, Learoyd DL, Robinson BG. Medullary thyroid carcinoma: recent advances and management update. Thyroid. 1995;5(5):407–424. doi: 10.1089/thy.1995.5.407. [DOI] [PubMed] [Google Scholar]
- 5.Elisei R, Cosci B, Romei C, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. Journal of Clinical Endocrinology & Metabolism. 2008;93(3):682–687. doi: 10.1210/jc.2007-1714. [DOI] [PubMed] [Google Scholar]
- 6.Sippel RS, Kunnimalaiyaan M, Chen H. Current management of medullary thyroid cancer. Oncologist. 2008;13(5):539–547. doi: 10.1634/theoncologist.2007-0239. [DOI] [PubMed] [Google Scholar]
- 7.Thakker RV. Multiple endocrine neoplasia—syndromes of the twentieth century. Journal of Clinical Endocrinology & Metabolism. 1998;83(8):2617–2620. doi: 10.1210/jcem.83.8.5045. [DOI] [PubMed] [Google Scholar]
- 8.Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Human Molecular Genetics. 1993;2(7):851–856. doi: 10.1093/hmg/2.7.851. [DOI] [PubMed] [Google Scholar]
- 9.Mathew CGP, Chin KS, Easton DF, et al. A linked genetic marker for multiple endocrine neoplasia type 2A on chromosome 10. Nature. 1987;328(6130):527–528. doi: 10.1038/328527a0. [DOI] [PubMed] [Google Scholar]
- 10.Shirahama S, Ogura K, Takami H, et al. Mutational analysis of the RET proto-oncogene in 71 Japanese patients with medullary thyroid carcinoma. Journal of Human Genetics. 1998;43(2):101–106. doi: 10.1007/s100380050048. [DOI] [PubMed] [Google Scholar]
- 11.Manié S, Santoro M, Fusco A, Billaud M. The RET receptor: function in development and dysfunction in congenital malformation. Trends in Genetics. 2001;17(10):580–589. doi: 10.1016/s0168-9525(01)02420-9. [DOI] [PubMed] [Google Scholar]
- 12.Cosma MP, Cardone M, Carlomagno F, Colantuoni V. Mutations in the extracellular domain cause RET loss of function by a dominant negative mechanism. Molecular and Cellular Biology. 1998;18(6):3321–3329. doi: 10.1128/mcb.18.6.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bethanis S, Koutsodontis G, Palouka T, et al. A newly detected mutation of the RET protooncogene in exon 8 as a cause of multiple endocrine neoplasia type 2A. Hormones. 2007;6(2):152–156. doi: 10.14310/horm.2002.1111011. [DOI] [PubMed] [Google Scholar]
- 14.Grimm J, Sachs M, Britsch S, et al. Novel p62dok family members, dok-4 and dok-5, are substrates of the c-Ret receptor tyrosine kinase and mediate neuronal differentiation. Journal of Cell Biology. 2001;154(2):345–354. doi: 10.1083/jcb.200102032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lips CJM, Höppener JWM, Thijssen JHH. Medullary thyroid carcinoma: role of genetic testing and calcitonin measurement. Annals of Clinical Biochemistry. 2001;38, part 3(3):168–179. doi: 10.1258/0004563011900614. [DOI] [PubMed] [Google Scholar]
- 16.Elisei R, Romei C, Cosci B, Agate L, Bottici V, Molinaro E, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. Journal of Clinical Endocrinology & Metabolism. 2007;92(12):4725–4729. doi: 10.1210/jc.2007-1005. [DOI] [PubMed] [Google Scholar]
- 17.Wohllk N, Cote GJ, Bugalho MMJ, et al. Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. Journal of Clinical Endocrinology & Metabolism. 1996;81(10):3740–3745. doi: 10.1210/jcem.81.10.8855832. [DOI] [PubMed] [Google Scholar]
- 18.Lallier M, St-Vil D, Giroux M, et al. Prophylactic thyroidectomy for medullary thyroid carcinoma in gene carriers of MEN2 syndrome. Journal of Pediatric Surgery. 1998;33(6):846–848. doi: 10.1016/s0022-3468(98)90656-x. [DOI] [PubMed] [Google Scholar]
- 19.Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. Journal of the American Medical Association. 1996;276(19):1575–1579. [PubMed] [Google Scholar]
- 20.Chappuis-Flament S, Pasini A, de Vita G, et al. Dual effect on the RET receptor of MEN 2 mutations affecting specific extracytoplasmic cysteines. Oncogene. 1998;17(22):2851–2861. doi: 10.1038/sj.onc.1202202. Laboratoire de Génétique, CNRS UMR 5641, Domaine Rockefeller, Lyon, France. [DOI] [PubMed] [Google Scholar]
- 21.Santoro M, Carlomagno F, Romano A, et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science. 1995;267(5196):381–383. doi: 10.1126/science.7824936. [DOI] [PubMed] [Google Scholar]
- 22.Eng C, Crossey PA, Mulligan LM, et al. Mutations in the RET proto-oncogene and the von Hippel-Lindau disease tumour suppressor gene in sporadic and syndromic phaeochromocytomas. Journal of Medical Genetics. 1995;32(12):934–937. doi: 10.1136/jmg.32.12.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hedayati M, Nabipour I, Rezaei-Ghaleh N, Azizi F. Germline RET mutations in exons 10 and 11: an Iranian survey of 57 medullary thyroid carcinoma cases. Medical Journal of Malaysia. 2006;61(5):564–569. [PubMed] [Google Scholar]
- 24.Hedayati M, Zarif Yeganeh M, Daneshpour M, Ahmadi M, Azizi F. Frequent germline mutation in RET proto-oncogene exons 10 and 11 in hereditary MTC of Iranian patients. Kowsar Medical Journal. 2010;15(1):17–21. [Google Scholar]
- 25.Zhou Y, Zhao Y, Cui B, et al. RET proto-oncogene mutations are restricted to codons 634 and 918 in mainland chinese families with MEN2A and MEN2B. Clinical Endocrinology. 2007;67(4):570–576. doi: 10.1111/j.1365-2265.2007.02927.x. [DOI] [PubMed] [Google Scholar]
- 26.Mulligan LM, Eng C, Healey CS, et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nature Genetics. 1994;6(1):70–74. doi: 10.1038/ng0194-70. [DOI] [PubMed] [Google Scholar]
- 27.Machens A, Niccoli-Sire P, Hoegel J, et al. Early malignant progression of hereditary medullary thyroid cancer. The New England Journal of Medicine. 2003;349(16):1517–1525. doi: 10.1056/NEJMoa012915. [DOI] [PubMed] [Google Scholar]
- 28.Frank-Raue K, Machens A, Scheuba C, Niederle B, Dralle H, Raue F. Difference in development of medullary thyroid carcinoma among carriers of RET mutations in codons 790 and 791. Clinical Endocrinology. 2008;69(2):259–263. doi: 10.1111/j.1365-2265.2008.03215.x. [DOI] [PubMed] [Google Scholar]
- 29.Schuffenecker I, Virally-Monod M, Brohet R, et al. Risk and penetrance of primary hyperparathyroidism in multiple endocrine neoplasia type 2A families with mutations at codon 634 of the RET proto-oncogene. Groupe d'etude des tumeurs à calcitonine. Journal of Clinical Endocrinology & Metabolism. 1998;83(2):487–491. doi: 10.1210/jcem.83.2.4529. [DOI] [PubMed] [Google Scholar]
- 30.Saggiorato E, Rapa I, Garino F, et al. Absence of RET gene point mutations in sporadic thyroid C-cell hyperplasia. Journal of Molecular Diagnostics. 2007;9(2):214–219. doi: 10.2353/jmoldx.2007.060166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvandi E, Akrami SM, Chiani M, et al. Molecular analysis of the RET proto-oncogene key exons in patients with medullary thyroid carcinoma: a comprehensive study of the Iranian population. Thyroid. 2011;2(4):373–382. doi: 10.1089/thy.2010.0267. [DOI] [PubMed] [Google Scholar]
- 32.Fernández RM, Navarro E, Antiñolo G, Ruiz-Ferrer M, Borrego S. Evaluation of the role of RET polymorphisms/haplotypes as modifier loci for MEN 2, and analysis of the correlation with the type of RET mutation in a series of Spanish patients. International Journal of Molecular Medicine. 2006;17(4):575–581. [PubMed] [Google Scholar]
- 33.Berard I, Kraimps JL, Savagner F, et al. Germline-sequence variants S836S and L769L in the RE arranged during transfection (RET) proto-oncogene are not associated with predisposition to sporadic medullary carcinoma in the french population. Clinical Genetics. 2004;65(2):150–152. doi: 10.1111/j.0009-9163.2004.00172.x. [DOI] [PubMed] [Google Scholar]
- 34.Pinna G, Orgiana G, Riola A, et al. RET proto-oncogene in Sardinia: V804M is the most frequent mutation and may be associated with FMTC/MEN-2A phenotype. Thyroid. 2007;17(2):101–104. doi: 10.1089/thy.2006.0198. [DOI] [PubMed] [Google Scholar]
- 35.Nasser T, Qari F, Karawgh A, Al Aama J. RET codon 618 mutations is the most frequent phenotype in Saudi families with multiple endocrine neoplasia type 2A. In: Proceedings of the European Congress of Endocrinology, vol. 22; April 2010; Prague, Czech Republic. European Society of Endocrinology; p. 380. [Google Scholar]
- 36.Robledo M, Gil L, Pollán M, et al. Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Research. 2003;63(8):1814–1817. [PubMed] [Google Scholar]