

SUMMARY
Fanconi anemia (FA) is a human genetic disease characterized by chromosome instability, cancer predisposition, and cellular hypersensitivity to DNA crosslinking agents. The FA pathway regulates the repair of DNA crosslinks. A critical step in this pathway is the monoubiquitination and deubiquitination of FANCD2. Deubiquitination of FANCD2 is mediated by the ubiquitin protease, USP1. Here, we demonstrate that targeted deletion of mouse Usp1 results in elevated perinatal lethality, male infertility, crosslinker hypersensitivity, and a FA phenotype. Usp1−/− mouse embryonic fibroblasts had heightened levels of monoubiquitinated Fancd2 in chromatin. Usp1−/− cells exhibited impaired Fancd2 foci assembly and a defect in homologous recombination repair. Double knockout of Usp1 and Fancd2 resulted in a more severe phenotype than either single knockout. Our results indicate that mouse Usp1 functions downstream in the FA pathway. Deubiquitination is a critical event required for Fancd2 nuclear foci assembly, release from chromatin, and function in DNA repair.
Keywords: Fanconi Anemia, Usp1, Deubiquitinating Enzymes, Mouse Knockout Models, Mitomycin C
INTRODUCTION
The FA pathway mediates DNA repair and promotes normal cellular resistance to DNA crosslinking agents (Wang, 2007). The FA pathway is regulated by thirteen FA proteins (FANC-A, B, C, D1, D2, E, F, G, I, J, L, M and N) (Grompe and van de Vrugt, 2007). Eight of the FA proteins are assembled in a nuclear ubiquitin E3 ligase complex (FANCA/B/C/E/F/G/L/M), known as the FA core complex, which monoubiquitinates FANCD2 and FANCI (Dorsman et al., 2007; Sims et al., 2007; Smogorzewska et al., 2007). The monoubiquitinated FANCD2/FANCI complex is targeted to chromatin (Montes De Oca et al., 2004) where it interacts, either directly or indirectly, with additional downstream FA proteins (FANCD1, FANCN and FANCJ). The downstream FA genes are also breast cancer susceptibility genes (Grompe and van de Vrugt, 2007).
Recent studies have indicated that the FANCD2/FANCI complex is deubiquitinated by the ubiquitin-specific protease, USP1 (Cohn et al., 2007; Huang et al., 2006; Nijman et al., 2005a). USP1 is not a FA gene per se, since no human FA patients harboring mutations in the USP1 gene have been identified. Disruption of the Usp1 gene in chicken cells (DT40) results in crosslinker hypersensitivity, and the chicken Usp1 and Fancl genes are epistatic for crosslink repair (Oestergaard et al., 2007).
The human genome encodes over ninety deubiquitinating enzymes (DUBs) (Nijman et al., 2005b). In rare cases, disruption of a Ubiquitin E3 ligase results in a similar outcome to disruption of a corresponding DUB (Nijman et al., 2005b), suggesting that coupled ubiquitination and deubiquitination may be essential for the function of some pathways.
In the current study, we disrupted the murine Usp1 gene. Interestingly, Usp1−/−mice had a strong resemblance to FA mice (small size, infertility, Mitomycin C (MMC) hypersensitivity, and chromosome instability). In addition, Usp1−/− mice exhibited a higher rate of perinatal lethality and absence of male germ cells. Our results indicate that Usp1 is required for Fancd2 foci assembly and contributes to homologous recombination (HR) repair, suggesting a regulatory role of Usp1 in the FA pathway.
RESULTS
Perinatal lethality and growth retardation in Usp1 null mutation
To address the physiological role of Usp1, we performed a targeted gene deletion of Usp1 in the mouse (Figure S1). Usp1+/− mice were grossly normal and fertile. Murine Usp1 was detected as two distinct protein products of different size in wild-type mouse embryonic fibroblasts (MEFs), and Usp1+/− MEFs displayed approximately 2-fold reduction in Usp1 protein level (Figure S1C). From Usp1+/− intercrosses, Usp1−/− mice were obtained at a low frequency (6%, instead of the expected 25%; Figure 1A). When analyzed during gestation (E13.5-E18.5) or after cesarean delivery at E19.5-E20.5, Usp1−/− embryos, although significantly smaller than their wild-type littermates (Figure 1C and data not shown), were present at the expected Mendelian ratio. Thus, deletion of the Usp1 gene is not embryonic lethal; rather, it led to perinatal lethality, since within 1–2 days after birth, up to 80% of the Usp1−/− mice were dead. Usp1−/− pups found dead displayed several abnormalities with incomplete penetrance; cyanosis, bilateral hydronephrosis, and hemorrhagic edema. When closely monitored at the moment of delivery, most of the Usp1−/− pups (75%) became progressively cyanotic and died within 2 hr of birth. Thus, cyanosis may be one of the causes of perinatal lethality in newborn Usp1−/− mice. The surviving Usp1-deficient mice were consistently smaller than their wild-type littermates, at weaning and as adults, suggesting that growth retardation occurred prenatally and persisted into adulthood (Figures 1B and 1C).
Figure 1. Perinatal lethality, growth retardation, and impaired germ cell development in Usp1-deficient mice.
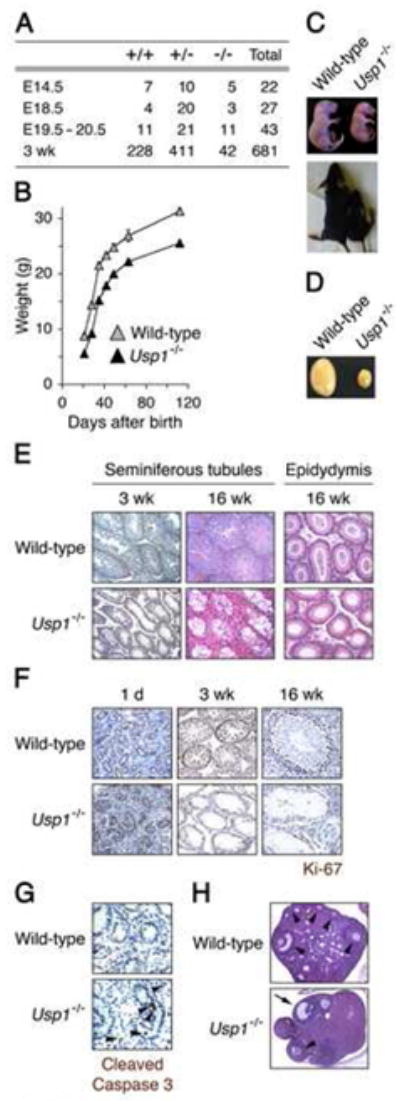
(A) Viability of Usp1-deficient mice at different stages of development. (B) Growth curve of wild-type (gray squares) and Usp1−/− (filled squares) mice. (C) Gross appearances of newborns and 2-week-old wild-type and Usp1−/− mice. (D) Comparison of testis size from 16-week-old wild-type and Usp1−/− mice. (E) Sections of wild-type and Usp1−/− seminiferous tubules and epididymis stained with hematoxylin-eosin. Magnification, 10 x. (F) Detection of Ki-67 by immunohistochemistry in seminiferous tubules from wild-type and Usp1−/− mice. Magnification, 40 x. (G) Detection of apoptosis by cleaved Caspase-3 staining in seminiferous tubules from 1-day-old testes. Magnification, 63 x. (H) Histological analysis of ovaries of wild-type (upper panel) and Usp1−/− (lower panel) females. Reduced number of oocytes (indicated by arrowhead) was observed in Usp1−/− ovaries, but note the presence of follicle (indicated by arrow) from which the oocyte was released during ovulation. Magnification, 5 x. For the quantitative analysis described in the text, we counted oocytes from 5 females from each genotype. We counted 4 sections per ovary (8 sections per each female; total 5 females (8X5=40 sections). All visible oocytes (including small oocytes and immediately visible oocytes) were counted.
Gonadal dysfunction in Usp1-deficient male mice
Usp1−/− male mice were sterile. The Usp1−/− testis was strikingly smaller than that of wild-type or Usp1+/− littermates (Figure 1D and data not shown). Histology of adult testes from Usp1−/− mice revealed that their seminiferous tubules were markedly atrophic and mostly devoid of spermatogenic cells (e.g., spermatogonia, spermatocytes, spermatids and spermatozoa; Figure 1E). Consistently, the epidydymis of Usp1−/− mice was devoid of spermatozoa (Figure 1E). We examined seminiferous tubules for cellular proliferation by Ki-67 immunostaining (Figure 1F). At 1 day after birth, Ki-67-positive cells were detected in wild-type and Usp1−/− tubules with similar frequency, and the intensity of Ki-67 staining in Usp1−/− tubules was even stronger than that of the wild-type tubules. At 3 and 16 weeks, however, cellularity of Usp1−/− tubules was greatly decreased and remaining cells were mostly negative for Ki-67 staining (Figure 1F). Immunostaining for cleaved Caspase 3 revealed that Usp1−/− tubules had a slightly increased frequency of apoptosis 1 day after birth (Figure 1G). However, the majority of Usp1−/− cells did not express cleaved Caspase 3, suggesting that apoptosis is not the major cause of testicular atrophy in Usp1−/− mice.
Usp1−/− female mice showed a reduced fertility as compared to wild-type females. Usp1−/− ovaries had less oocytes than wild-type ovaries (8.5 ± 0.6 and 2.9 ± 0.3 per section for wild-type and Usp1−/− 12-week-old ovaries, respectively; 5 females each, Figure 1H).
MMC hypersensitivity and enhanced Fancd2 and PCNA monoubiquitination in Usp1-deficient cells
There were no significant differences in cell cycle distribution between wild-type and Usp1−/− primary MEFs (either asynchronous or after release from synchronization in G0 by serum starvation) (Figures S2A and S2B). As predicted from studies with transformed human cell lines (Huang et al., 2006; Nijman et al., 2005a), Usp1−/− MEFs had a compensatory increase in total Fancd2 levels and in its monoubiquitination as well as PCNA monoubiquitination (Figure 2A). Usp1+/− MEFs exhibited an intermediate increase in Fancd2- and PCNA-monoubiquitination levels, suggesting a haploinsufficiency phenotype (Figure S3).
Figure 2. Hypersensitivity to DNA crosslinking agents in Usp1-deficient cells.
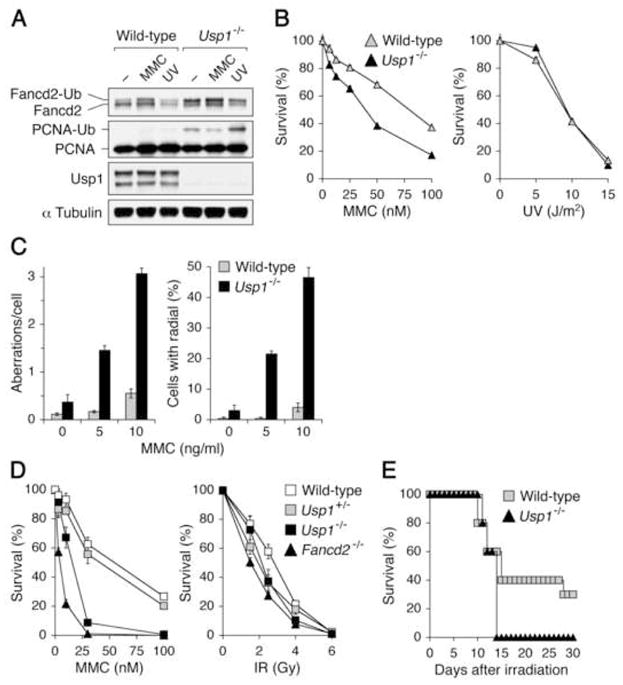
(A) Increased Fancd2-Ub and PCNA-Ub in Usp1-deficient cells. Wild-type and Usp1−/−MEFs were either left untreated, treated with MMC (500 ng/ml for 20 hr) or UV (30 J/m2, harvested at 3 hr after irradiation). Cell lysates were immunoblotted with indicated antibodies. (B) Survival rates of wild-type and Usp1−/− MEFs to MMC and UV treatment are plotted as the percentage of viable cells relative to that for respective untreated cells. (C) MMC-induced chromosomal aberrations in wild-type (open bars), Usp1+/− (gray bars) and Usp1−/− (filled bars) MEFs following the treatment with MMC for 48 hr. The numbers of chromosomal aberrations (left panel) and radial forms (right panel) per metaphase spread were scored. (D) Clonogenic survival assay of BM cells from wild-type (open squares), Usp1+/− (gray squares), Usp1−/− (filled squares) and Fancd2−/− (filled triangles) mice. BM cells were treated with increasing doses of MMC (left) or IR (right). After 7–10 days in culture, the numbers of hematopoietic colonies were compared. (E) Survival of wild-type (n = 10; gray squares) and Usp1−/− (n = 5; filled triangles) mice following 8.25 Gy of whole-body irradiation.
We next analyzed the response of Usp1−/− MEFs to DNA crosslinking agents. Usp1−/− MEFs showed elevated sensitivity to MMC treatment, whereas the sensitivity of Usp1−/− MEFs to ultraviolet (UV) irradiation was similar to that of wild-type cells (Figure 2B). Furthermore, following MMC treatment, Usp1−/− MEFs exhibited increased chromosomal breaks and radial forms that distinguish FA from other chromosomal breakage syndromes (Figure 2C). Retroviral transduction of wild-type Usp1 partially corrected the MMC-induced chromosome instability of Usp1−/− MEFs (Figure S2D). Since FA cells show an increased accumulation in G2/M following exposure to DNA interstrand crosslinkers, we examined whether Usp1-deficient cells show G2/M accumulation in response to MMC. After a short exposure of MMC, followed by recovery for 24 hr, Usp1−/− MEFs had an increase in G2/M content (57%) compared to that of wild-type MEFs (40%) (Figure S2C). Thus, Usp1−/− MEFs have a specific defect in DNA crosslink repair, similar to FA cells.
FA phenotype in Usp1-deficient lymphocytes
Usp1−/− bone marrow (BM) cells had increased monoubiquitination of Fancd2 and PCNA, as observed in Usp1−/− MEFs (data not shown). The comparison of the number of BM mononuclear cells per hind limb indicated a significant reduction in Usp1−/− mice, whereas peripheral blood from Usp1−/− mice showed normal values for hemoglobin and white blood cell count (data not shown). Following methylcellulose culture in the presence of increasing concentrations of MMC, Usp1−/− BM cells, like Fancd2−/− BM cells, exhibited hypersensitivity to MMC compared to wild-type cells (Figure 2D). Both Usp1−/− and Fancd2−/− BM cells showed only mild sensitivity to ionizing radiation (IR) compared to wild-type controls. However, Usp1-deficient mice displayed hypersensitivity to total body irradiation (Figure 2E), as has been described for Fancd2-deficient mice (Houghtaling et al., 2003).
Increased chromatin accumulation of monoubiquitinated Fancd2 but decreased nuclear foci in Usp1-deficient MEFs
Monoubiquitinated FANCD2 and FANCI are targeted to chromatin and form DNA repair foci in response to DNA damage (Garcia-Higuera et al., 2001; Smogorzewska et al., 2007). Since Usp1 depletion resulted in increased monoubiquitination of Fancd2, we examined whether this monoubiquitinated Fancd2 is localized to chromatin by using biochemical cell fractionation (Figure 3A). The “S1” fraction contains cytoplasmic and nucleoplasmic proteins, the “S2” fraction contains proteins bound to chromatin, and the “P2” fraction contains proteins tightly associated with nuclear structures (chromatin, nuclear matrix, etc.). In Usp1−/− MEFs, monoubiquitinated Fancd2 efficiently localized in chromatin even in the absence of DNA damage (Figure 3A), suggesting that monoubiquitination is sufficient for chromatin targeting of Fancd2. A similar increase in chromatin-associated Fancd2 has been demonstrated in Usp1-depleted DT40 cells (Oestergaard et al., 2007).
Figure 3. Increased chromatin accumulation of monoubiquitinated Fancd2 and impaired Fancd2 foci assembly in Usp1-deficient MEFs.
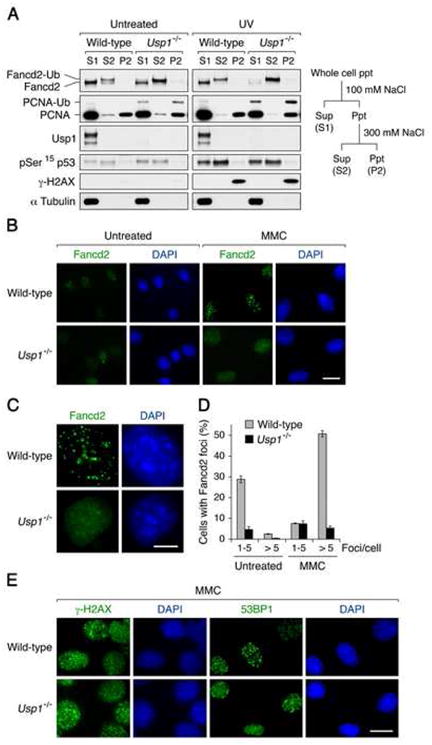
(A) Wild-type and Usp1−/− MEFs were either left untreated or treated with UV (30 J/m2). Cells were collected 3 hr after UV irradiation and fractionated. Each fraction was immunoblotted with the indicated antibodies. (B and C) Wild-type and Usp1−/− MEFs were stained for Fancd2 following either mock treatment (untreated) or 500 ng/ml of MMC for 20 hr. Bar, 10 μm (B) and 5 μm (C). (D) Quantification of cells with Fancd2 foci. Values represent the mean ± SEM, examined at least 700 nuclei each in three independent experiments. (E) Wild-type (upper panels) and Usp1−/− (lower panels) MEFs were stained for γ-H2AX and 53BP1 foci following MMC treatment (500 ng/ml) for 20 hr. Bar, 10 μm.
We next examined whether Fancd2 forms subnuclear foci in Usp1-deficient MEFs. Notably, Usp1−/− MEFs were defective in both spontaneous (S phase-specific) and DNA damage-inducible Fancd2 foci assembly (Figures 3B and 3C). Fancd2 foci formation was severely impaired in Usp1-deficient MEFs (Figure 3D), despite high levels of monoubiquitinated Fancd2 detected in the chromatin. The absence of Fancd2 foci therefore correlated with the MMC hypersensitivity of Usp1−/− cells. In contrast, Usp1-deficient MEFs exhibited normal levels of γ-H2AX foci and 53BP1 foci in response to MMC treatment (Figure 3E).
HR defect in Usp1-deficient cells
HR constitutes a central pathway to mediate repair of MMC-induced DNA damage (Niedzwiedz et al., 2004). Cells deficient for upstream or downstream FA proteins, including Fancd2, are deficient in HR (Litman et al., 2005; Nakanishi et al., 2005; Niedzwiedz et al., 2004; Sims et al., 2007; Smogorzewska et al., 2007; Xia et al., 2006; Yamamoto et al., 2005). The severe defect in Fancd2 foci formation in Usp1−/− MEFs raises the possibility of an HR defect. To determine whether Usp1 is required for HR, we generated Usp1fl/fl MEFs in which each allele of Usp1 is flanked by loxP sites (Figure 4A). We established four independent clones of Usp1fl/fl MEFs containing a single copy of the HR reporter DR-GFP (Pierce et al., 2001). These Usp1fl/fl DR-GFP clones were infected with adenovirus expressing Cre recombinase to generate Usp1−/− DR-GFP MEFs. Cre-mediated excision of the loxP-flanked Usp1 gene was confirmed by PCR and immunoblotting (Figure 4B). Following the induction of HR by transfection with an I-SceI expression plasmid, Usp1−/− DR-GFP MEFs showed a 50% reduction in the frequency of GFP+ cells compared to that of Usp1fl/fl DR-GFP MEFs (Figure 4C). Thus, the deficiency of Usp1 results in a defect in HR repair.
Figure 4. HR defect in Usp1-deficient MEFs.
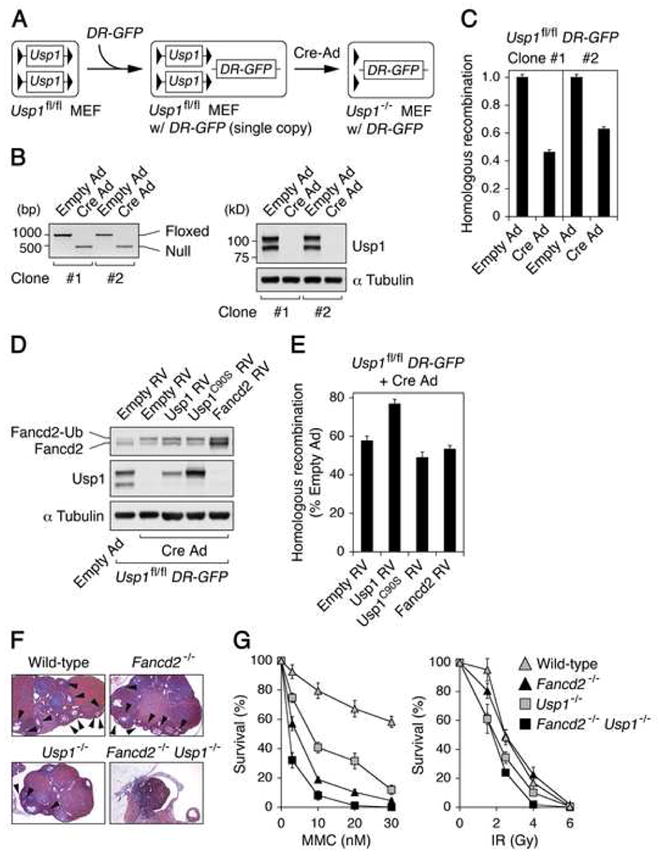
(A) The scheme for the generation of Usp1−/− MEFs with DR-GFP reporter. (B) Confirmation of Cre-mediated excision of the loxP-flanked Usp1 gene by genomic PCR (left panel) and Western blot (right panel). (C) Two independent Usp1fl/fl DR-GFP MEF clones were analyzed for HR frequencies, each with triplicates. Values were normalized for the transfection efficiency and were displayed as mean ± SEM GFP+ frequencies relative to that of each Usp1fl/fl DR-GFP clone. (D) Immunoblotting of Usp1fl/fl DR-GFP MEFs expressing either wild-type Usp1, Usp1C90S or Fancd2 following Cre-expressing adenovirus infection. (E) I-SceI-induced HR frequencies in Usp1−/− DR-GFP MEFs transduced with empty retroviral vector or the retroviral vector encoding wild-type Usp1, Usp1C90S or Fancd2. Values were normalized for the transfection efficiency and were displayed as mean ± SEM GFP+ frequencies relative to that of each Usp1fl/fl DR-GFP MEFs. (F) Histopathology of ovaries from 12-week-old wild-type, Fancd2−/−, Usp1−/− and Usp1−/−Fancd2−/− siblings generated by Usp1+/−Fancd2+/− crosses. Usp1−/−Fancd2−/−double knockout females had a more severe ovarian atrophy than either single knockout mice. Magnification, 5 x. Arrowheads, oocytes. (G) Survival rates of BM cells from wild-type (gray triangles), Usp1−/− (gray squares), Fancd2−/− (filled triangles) and Usp1−/− Fancd2−/− (filled squares) mice following the treatment with increasing doses of MMC (left panel) or IR (right panel). After 7–10 days in culture, the numbers of hematopoietic colonies were compared.
To confirm this effect in human cells, we used DR-U2OS cells, a human osteosarcoma cell line containing an integrated DR-GFP (Xia et al., 2006). siRNA against USP1 reduced the HR frequency in these cells to 45% of control, similar to the siRNA against FANCI (Figure S4).
The defect in HR observed in Usp1-deficient cells could occur because the cellular pool of Fancd2 is primarily ubiquitinated and bound to chromatin, leaving inadequate cellular levels of free Fancd2. If so, forced expression of Fancd2 could reverse the defect. To clarify this, Usp1fl/fl DR-GFP MEFs were stably transfected with empty retroviral vector or the retroviral vector encoding either wild-type Usp1, Usp1C90S (i.e., catalytically inactive form of the Usp1), or Fancd2, and these cells were subsequently infected with Cre-expressing adenovirus (Figures 4D and 4E). Expression of the heterologous proteins in the absence of endogenous Usp1 was confirmed by immunoblotting (Figure 4D). Immunoblotting for Fancd2 revealed a partially reverted L/S ratio (i.e., FANCD2-L (monoubiquitinated FANCD2) to FANCD2-S (unubiquitinated Fancd2) ratio) in Usp1−/− MEFs expressing wild-type Usp1 but not Usp1C90S. Consistent with this pattern, wild-type Usp1, but not Usp1C90S, corrected the HR defect of Usp1-deficient MEFs (Figure 4E). Interestingly, expression of excess Fancd2 did not restore HR activity, indicating that the HR defect in Usp1-deficient cells is not due to low cellular levels of free Fancd2.
Double knockout of Usp1 and Fancd2 results in a more severe phenotype
We next generated Usp1−/−Fancd2−/− double knockout mice by crossing Usp1+/−Fancd2+/− mice. Interestingly, Usp1−/−Fancd2−/− females showed more severe ovarian atrophy than either single knockout mice (Figure 4F). Western blots showed that DNA damage signaling in response to MMC or UV irradiation is not significantly different from that of either of the single mutants or wild-type controls (Figure S5). However, an increase in baseline phosphorylation of Chk2 was observed in Usp1−/−Fancd2−/−fibroblasts, suggesting that spontaneous DNA damage may accumulate in Usp1−/−Fancd2−/− mice (Figure S5). Furthermore, BM cells from Usp1−/−Fancd2−/− mice were more sensitive to MMC than either single knockout mice (Figure 4G). Although it has been reported that mice deficient in Fanc proteins tend to display more severe phenotype in C57B6 background (Agoulnik et al., 2002; Chen et al., 1996; Houghtaling et al., 2003; Wong et al., 2003), our Usp1−/−Fancd2−/− mice in C57BL/6 x 129Sv mixed genetic background consistently showed enhanced defects compared to their either single knockout littermates or Usp1−/− mice in C57B6 background (Figure 4G and data not shown). Therefore, it is unlikely that the severe phenotype observed for Usp1−/−Fancd2−/−mice is solely due to the difference in genetic background. Taken together, these data suggest that Usp1 may regulate additional DNA repair pathways in addition to the FA pathway, perhaps by controlling the ubiquitination state of substrates other than Fancd2 or Fanci.
DISCUSSION
We have demonstrated that Usp1-deficient mice have a similar phenotype to other FA mouse models. FA-deficient mice are generally small, exhibit reduced fertility, and have heightened crosslinker sensitivity of their primary cells (Agoulnik et al., 2002; Chen et al., 1996; Cheung et al., 2004; Houghtaling et al., 2003; Koomen et al., 2002; McAllister et al., 2002; Pellas et al., 1991; Whitney et al., 1996; Wong et al., 2003; Yang et al., 2001). Usp1-deficient mice are small and exhibit male infertility, chromosomal instability, and cellular hypersensitivity to crosslinking agents. Moreover, the Usp1-deficient mice exhibit a more severe phenotype than most FA mouse models, with approximate 80% perinatal lethality, testicular atrophy, and depletion of male germ cells. Such a similarity in phenotypes of mice deficient in Usp1 and Fanc genes further argue for their genetic (epistatic) relationships. At the same time, the elevated perinatal lethality and heightened cellular levels of PCNA-Ub in Usp1−/− cells, which is not observed in FA cells, suggest that Usp1 may have other functions (and perhaps other substrates) in somatic cells, which may account, at least in part, for the absence of humans with USP1 deficiency.
The mechanism by which USP1 depletion leads to MMC hypersensitivity is unknown, and several models are possible. First, loss of USP1 results in elevated FANCD2-Ub and depletion of the unubiquitinated form of FANCD2 (FANCD2-S). Loss of FANCD2-S may impair the ability of a cell to respond to additional crosslink damage. Accordingly, the normal function of USP1 may be to recycle free FANCD2-S (and free Ubiquitin) in the cell. However, forced expression of Fancd2 in Usp1−/− MEFs did not correct the HR defect (Figures 4A-4C). Alternatively, excessive chromatin accumulation of Fancd2-Ub in Usp1−/− cells may recruit an additional repair factor to chromatin, reducing its availability to participate at sites of damage. Second, elevated Fancd2-Ub levels in the Usp1-deficient MEFs may be toxic to cells and promote MMC sensitivity. However, double knockout of Usp1 and Fancd2 in mice did not improve the MMC sensitivity, suggesting that high levels of FANCD2 ubiquitination do not account for the cellular phenotype of Usp1−/− mice (Figures 4F and G). Third, coupled ubiquitination and deubiquitination may be necessary for proper FA pathway function, suggesting that USP1 acts at a critical later step in the FA pathway itself (Figure S6). For instance, FANCD2 monoubiquitination by the FA core complex promotes chromatin loading and assembly of DNA repair complexes. USP1-mediated deubiquitination of FANCD2-Ub may catalyze additional later events in the DNA repair process. Consistent with this model, Usp1-deficient cells have elevated Fancd2-Ub in chromatin but impaired foci formation of Fancd2.
Usp1 depletion also results in prolonged, elevated levels of PCNA-Ub. Depletion of USP1 in human cell lines, by siRNA, results in elevated PCNA-Ub levels and elevated mutagenesis (Huang et al., 2006), secondary to increased recruitment of error prone polymerases such as DNA Polη. One might expect, therefore, that Usp1-deficient mice will exhibit an increased point mutation frequency and perhaps an increased cancer incidence.
EXPERIMENTAL PROCEDURES
Generation of Usp1 knockout mice
Usp1 conditional knockout mice (in C57BL/6J genetic background) were generated by Ingenko under a consortium agreement. A targeting vector was engineered to disrupt the Usp1 gene in C57BL/6J-derived ES cells. The exon 3 was targeted for replacement by the exon 3 flanked by loxP sites and neor cassette flanked by FRT sites. EIIa-Cre transgenic female mice (JAX stock no. 003724) were used to generate heterozygous Usp1+/−Cre mutant mice. The Cre transgene was removed by back-crossing to wild-type C57BL/6J mice.
Fancd2 knockout mice (C57BL/6 x 129/Sv mixed genetic background) were generated by a gene-trap method (K. Parmar and A. D’Andrea, manuscript in preparation), and this mouse had a phenotype similar to that observed in previous report (Houghtaling et al., 2003). Mice deficient for both Usp1 and Fancd2 were generated by intercrossing of Usp1+/−Fancd2+/− mice.
HR assay
For HR assay with MEFs, the DR-GFP reporter, hprtDRGFPhygro (Nakanishi et al., 2005), was integrated into the genome of Usp1fl/fl MEFs. Clones with a single copy of DR-GFP were identified by Southern blotting. These clones were then used for infection with either an “empty” pBABE-puro retrovirus or the pBABE-puro retrovirus into which wild-type Usp1, Usp1C90S or Fancd2 cDNA was inserted. Retrovirus-infected cells were selected in puromycin before infection with adenovirus expressing Cre recombinase. Usp1fl/fl DR-GFP MEFs were transfected with I-SceI expression plasmid (pCBASce) in triplicate wells of 24-well plates using Lipofectamine 2000. The parallel transfection with pEGFP-C1 was used to normalize transfection efficiency. 48 hr after transfection, cells were trypsinized and single cell suspensions were analyzed by flow cytometry.
Supplementary Material
Acknowledgments
We thank Drs. Maria Jasin and Hisao Masai for DR-U2OS and for Cre recombinase-expressing adenovirus, respectively. We thank Lisa Moreau for chromosomal breakage analysis, Kenneth Law for immunohistochemistry, Dr. Eunmi Park for help in characterization of newborn mice and Dr. Tony Huang for helpful discussions. We thank Patricia Stuckert, Mary Kathryn DeLoach and Kaya Zhu for technical assistance. This study was supported by NIH grants RO1DK43889, R01HL52725, PO1DK50654 and U19A1067751.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agoulnik AI, Lu B, Zhu Q, Truong C, Ty MT, Arango N, Chada KK, Bishop CE. A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum Mol Genet. 2002;11:3047–3053. doi: 10.1093/hmg/11.24.3047. [DOI] [PubMed] [Google Scholar]
- Chen M, Tomkins DJ, Auerbach W, McKerlie C, Youssoufian H, Liu L, Gan O, Carreau M, Auerbach A, Groves T, et al. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nature Genetics. 1996;12:448–451. doi: 10.1038/ng0496-448. [DOI] [PubMed] [Google Scholar]
- Cheung AM, Elia A, Tsao MS, Done S, Wagner KU, Hennighausen L, Hakem R, Mak TW. Brca2 deficiency does not impair mammary epithelium development but promotes mammary adenocarcinoma formation in p53+/− mutant mice. Cancer Res. 2004;64:1959–1965. doi: 10.1158/0008-5472.can-03-2270. [DOI] [PubMed] [Google Scholar]
- Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D'Andrea AD. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell. 2007;28:786–797. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, et al. Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncol. 2007;29:211–218. doi: 10.1155/2007/151968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Grompe M, van de Vrugt H. The Fanconi family adds a fraternal twin. Dev Cell. 2007;12:661–662. doi: 10.1016/j.devcel.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17:2021–2035. doi: 10.1101/gad.1103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8:339–347. doi: 10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- Kim JM, Kee Y, Gurtan A, D'Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koomen M, Cheng NC, van De Vrugt HJ, Godthelp BC, van Der Valk MA, Oostra AB, Zdzienicka MZ, Joenje H, Arwert F. Reduced fertility and hypersensitivity to mitomycin C characterize Fancg/Xrcc9 null mice. Hum Mol Genet. 2002;11:273–281. doi: 10.1093/hmg/11.3.273. [DOI] [PubMed] [Google Scholar]
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Bennett LM, Houle CD, Ward T, Malphurs J, Collins NK, Cachafeiro C, Haseman J, Goulding EH, Bunch D, et al. Cancer susceptibility of mice with a homozygous deletion in the COOH-terminal domain of the Brca2 gene. Cancer Res. 2002;62:990–994. [PubMed] [Google Scholar]
- Montes De Oca R, Andreassen PR, Margossian SP, Gregory RG, Taniguchi T, Wang X, Houghtaling S, Grompe M, D'Andrea AD. Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood. 2004;105:1003–1009. doi: 10.1182/blood-2003-11-3997. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D'Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15:607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005a;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005b;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell. 2007;28:798–809. doi: 10.1016/j.molcel.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellas TC, Ramachandran B, Duncan M, Pan SS, Marone M, Chada K. Germ-cell deficient (gcd), an insertional mutation manifested as infertility in transgenic mice. Proc Natl Acad Sci U S A. 1991;88:8787–8791. doi: 10.1073/pnas.88.19.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims AE, Spiteri E, Sims RJ, 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ. Identification of the FANCI Protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Whitney MA, Royle G, Low MJ, Kelly MA, Axthelm MK, Reifsteck C, Olson S, Braun RE, Heinrich MC, Rathbun RK, et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88:49–58. [PubMed] [Google Scholar]
- Wong JC, Alon N, McKerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 2003;12:2063–2076. doi: 10.1093/hmg/ddg219. [DOI] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Hirano S, Ishiai M, Morishima K, Kitao H, Namikoshi K, Kimura M, Matsushita N, Arakawa H, Buerstedde JM, et al. Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Mol Cell Biol. 2005;25:34–43. doi: 10.1128/MCB.25.1.34-43.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kuang Y, De Oca RM, Hays T, Moreau L, Lu N, Seed B, D'Andrea AD. Targeted disruption of the murine Fanconi anemia gene, Fancg/Xrcc9. Blood. 2001;98:3435–3440. doi: 10.1182/blood.v98.12.3435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.