

Abstract
Amphetamine (AMPH) is thought to disrupt normal patterns of action potential-dependent dopaminergic signaling by depleting dopamine (DA) vesicular stores and promoting non-exocytotic DA efflux. Voltammetry in brain slices concurrently demonstrates these key drug effects, along with competitive inhibition of neuronal DA uptake. Here we perform comparable kinetic and voltammetric analyses in vivo to determine whether AMPH acts qualitatively and quantitatively similar in the intact brain. Fast-scan cyclic voltammetry measured extracellular DA in dorsal and ventral striata of urethane-anesthetized rats. Electrically evoked recordings were analyzed to determine Km and Vmax for DA uptake and vesicular DA release, while background voltammetric current indexed basal DA concentration. AMPH (0.5, 3, and 10 mg/kg i.p.) robustly increased evoked DA responses in both striatal subregions. The predominant contributor to these elevated levels was competitive uptake inhibition, as exocytotic release was unchanged in the ventral striatum and only modestly decreased in the dorsal striatum. Increases in basal DA levels were not detected. These results are consistent with AMPH augmenting action potential-dependent dopaminergic signaling in vivo across a wide, behaviorally relevant dose range. Future work should be directed at possible causes for the distinct in vitro and in vivo pharmacology of AMPH.
INTRODUCTION
Because of prominent clinical and recreational uses, the effects of amphetamine (AMPH) on behavior and brain function have been extensively studied. In the rat, AMPH increases locomotion at low doses and stereotypic movements at high doses (Gaytan et al. 1998). These drug-elicited behaviors are due, at least in part, to AMPH acting on midbrain dopaminergic neurons, with the projections to the nucleus accumbens (ventral striatum) involved in locomotor activation and the caudate-putamen (dorsal striatum) in stereotypy (Kelly et al. 1975). Not unexpectedly, AMPH robustly increases basal extracellular dopamine (DA) levels in both striatal subregions as measured by microdialysis (Kuczenski et al. 1991;Kuczenski et al. 1997). Comparatively greater increases in dialysate DA suggest that AMPH acts distinctly from other psychostimulants, such as cocaine, whose primary mechanism is inhibition of the dopamine transporter (DAT) (Kuczenski et al. 1991).
A large body of work indicates that AMPH elevates striatal extracellular levels of DA through a concerted action on multiple sites of dopaminergic neurotransmission (Sulzer et al. 2005;Fleckenstein et al. 2007). Early studies demonstrated DA synthesis activation (Kuczenski 1975), monoamine oxidase inhibition (Miller et al. 1980), competitive blockade of DA uptake (Krueger 1990), depletion of vesicular DA stores (Floor and Meng 1996), and increased non-exocytotic DA release or DA efflux via reversal of DAT (Sulzer et al. 1995). More recent investigations have extended these initial findings by resolving the effects of AMPH on DA uptake blockade from DAT reversal and from exocytotic DA release (Jones et al. 1998;Schmitz et al. 2001), by examining the effects of AMPH on cytosolic DA pools directly (Mosharov et al. 2003), and by demonstrating new actions of AMPH on DA uptake including DAT insertion (Johnson et al. 2005) and internalization (Saunders et al. 2000), and activation of DAT channel-like activity (Kahlig et al. 2005).
By compromising vesicular DA release and promoting DA efflux, AMPH should disrupt normal patterns of action potential-dependent dopaminergic neurotransmission. Indeed, it is these effects that may distinguish AMPH and its analogs from other addictive substances, which are thought to augment phasic DA signals elicited by burst firing of dopaminergic neurons (Stuber et al. 2005;Cheer et al. 2007;Robinson et al. 2009) and to cause an overlearning of cues predicting drug availability (Hyman 2005;Wanat et al. 2009). However, because most mechanistic investigations have utilized in vitro preparations, there is a great need to establish whether AMPH acts qualitatively and quantitatively similar in vivo. Several lines of evidence suggest important differences between the two preparations. For example, deafferentation of inputs to midbrain dopaminergic neurons potentiates AMPH-induced increases in extracellular levels of striatal DA (Miller et al. 2002), indicating that intact circuitry plays an important role in drug action. While brain slice measurements demonstrate robust decreases in electrically evoked DA levels with AMPH (Jones et al. 1998;Schmitz et al. 2001), some, but not all, studies in the anesthetized rat actually show an augmentation (Kuhr et al. 1986;May et al. 1988;Dugast et al. 1994). Finally, when monitored by fast-scan cyclic voltammetry (FSCV) at a carbon-fiber microelectrode (CFM), AMPH elicits a large, micromolar increase in basal DA levels in the brain slice attributed to DA efflux (Jones et al. 1998;Schmitz et al. 2001), but no detectable signal in vivo (Wiedemann et al. 1990).
The present study assessed the mechanism of AMPH action on dopaminergic signaling in vivo. FSCV at a CFM was used, in the urethane-anesthetized rat, to monitor extracellular DA levels in the dorsal and ventral striatum elicited by electrical stimulation of the medial forebrain bundle (MFB). Parameters for exocytotic DA release and neuronal DA uptake were determined by kinetic evaluation of these evoked responses. Background voltammetric current was used to index changes in basal DA concentration. This experimental design, which is comparable to previous work in brain slices (Jones et al. 1998;Schmitz et al. 2001), fosters comparisons between AMPH actions in vitro and in vivo on the basis of similar voltammetric and kinetic analyses. We found that AMPH robustly increased electrically evoked DA levels in both dorsal and ventral striata across a wide dose range (0.5, 3, and 10 mg/kg i.p.). Consistent with the brain-slice work, AMPH competitively inhibited DA uptake in a dose-dependent manner. Unlike in vitro studies, however, vesicular DA release was either unchanged in the ventral striatum or only modestly decreased in the dorsal striatum, while increases in basal DA levels were not detected in either striatal subregion. Taken together with previous work, these results suggest that AMPH acts substantively different in urethane-anesthetized rats compared to brain slices, with competitive uptake inhibition predominating over vesicular depletion and DA efflux. These results additionally demonstrate that AMPH robustly augments action potential-dependent dopaminergic signaling in vivo.
METHODS
Animals
Male Sprague-Dawely rats (300–500 g) were purchased from Harlan Industries (Indianapolis, IN, U.S.A.). Food and water were provided ad libitum. Animals were housed in temperature-controlled facility with a 12 hour light/dark cycle, and care was in accordance with NIH guidelines (Institute of Laboratory Animal Resources 1996) and approved by the Institutional Animal Care and Use Committee of Illinois State University.
Surgical Procedure
Rats were anesthetized using urethane (1.5 g/kg, i.p.) and immobilized in a stereotaxic frame (Kopf Instrumentation, Tujunga, CA, USA). Skin and connective tissue were removed, exposing the skull. Holes were drilled through the skull, and electrodes were positioned according to flat-skull stereotaxic coordinates (Paxinos G and Watson C 1986) using bregma and top of brain as landmarks. Core body temperature was maintained at approximately 37° C with a Deltaphase Isothermal Pad (Braintree Scientific, Braintree, MA, USA). The stimulating electrode was placed just dorsal to the MFB (−4.6 AP, +1.4 ML, −6.5 DV). Recording electrodes were positioned in the dorsal (+1.2 AP, +2.0 or 3.0 ML, −4.5 DV) or ventral striatum (+1.2 AP, +2.0 or 3.0 ML, −6.5 DV). Electrodes positioned at +3.0 ML were angled at 6°. A Ag/AgCl reference electrode (i.e., chloridized silver wire) was placed contralaterally in superficial cortex. The stimulating electrode was incrementally lowered until robust DA release was achieved in both striatal regions. Recording electrodes were then lowered until a fast, high rate of evoked DA release was observed. After optimization, the positions of the stimulating and recording electrodes were not changed for the duration of the experiment.
Fast-Scan Cyclic Voltammetry
FSCV was recorded at a glass-sealed CFM. Individual carbon fibers (3.5 µm-radius, Cytec Engineering Materials, West Patterson, NJ, USA) were aspirated into borosilicate capillary tubes (1.0 or 1.2 mm o.d., Sutter Instrument, Novato, CA, USA) and pulled in a micropipette puller (Narishige, Tokyo, Japan). The carbon fiber was cut so that approximately 100 µm extended beyond the glass/carbon-fiber seal. Electrical connection between the carbon fiber and 26-gauge lead wire (Newark, Palatine, IL, USA) was provided by a low-melting point bismuth alloy (Small Parts, Inc., Miramar, FL, USA), which was melted with a heat gun (Master Appliance Corp., Racine, WI, USA) and allowed to cool. A triangle waveform (−0.4 V to 1.0 V, 300 V/s) was applied at 10 Hz, with a bias potential of −0.4 V. Electrochemistry was performed by a EI400 (Ensman Instrumentation, Bloomington, IN, USA) or UEI (University of North Carolina at Chapel Hill, Department of Chemistry Electronics Facility, Chapel Hill, NC, USA) bipotentiostat and controlled by TH-1 software (ESA, Chelmsford, MA, USA). Background subtracted cyclic voltammograms were used for electrochemical identification. Evoked DA dynamics were obtained by monitoring current at the DA oxidation peak (~+0.6 V) and converting to concentration based on postcalibration, as previously described (Wu et al. 2001b).
Electrical Stimulation
Stimulus pulses were applied to a twisted bipolar electrode (Plastics One, Roanoke, VA, USA), with tips separated approximately 1 mm. Stimulus parameters were ±300 – 350 µA, 4 ms biphasic pulses (2 ms each phase), either 20 Hz or 60 Hz, and 2 s trains. Electrical stimulation was computer generated and passed through an optical isolator and constant-current generator (Neurolog NL800, Digitimer Limited, Letchworth Garden City, UK).
Experimental Design
After evoked DA responses stabilized, a time course using 20-Hz trains was collected. For the predrug responses, 4 trains were applied every 5 minutes. For the first 20 min after the injection of AMPH (0, 0.5, 3, or 10 mg/kg i.p.; n = 5 to 9 and 4 to 6 for dorsal and ventral striatum, respectively), trains were applied every 5 min. Thereafter, trains were applied every 15 min. Two 60-Hz trains were also applied. The first occurred immediately after response stabilization and 10 min before collecting the 20 Hz predrug trains. This predrug 60 Hz response and the average of the four predrug 20-Hz responses were used to obtain predrug kinetic parameters for DA release and uptake. The second 60-Hz train was applied 25 min postdrug. This train and the 20-Hz train applied at 35 min were used to obtain postdrug kinetic parameters for DA release and uptake. The different dynamics elicited by these two stimulation frequencies improve the accuracy of curve fitting (Wu et al. 2001b). Background current at the DA-oxidation potential, an index of basal DA levels (Jones et al. 1998;Schmitz et al. 2001), was also determined from the pre-stimulation portion of each evoked recording during the time course. With the exception of Figure 5, all averaged postdrug effects were expressed as a percent of predrug responses.
Figure 5. Time course of the effects of AMPH on background voltammetric current.
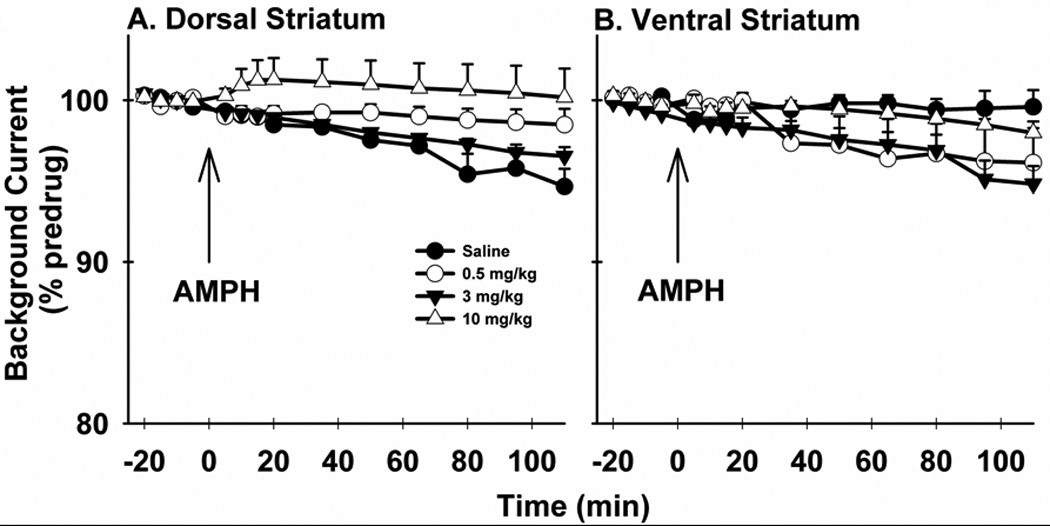
Background currents were measured during the pre-stimulation (i.e., baseline) portion of evoked recordings collected in the dorsal (A) and ventral (B) striatum and used to generate the time courses in Figure 2. Background Current is expressed as a percent of the predrug value (% predrug).
Data Analysis
Evoked DA responses were mathematically described as a balance between DA release and uptake (Wightman et al. 1988):
| (1) |
where [DA]p is the concentration of DA released per stimulus pulse, f is the frequency of stimulation, Vmax is the Michaelis-Menten uptake term related to DAT number and turnover rate, and Km is the Michaelis-Menten uptake term inversely related to the affinity of DA for DAT. Curve fitting, to determine [DA]p, Vmax, and Km, employed a simplex-minimization algorithm (Wu et al. 2001b). Two recordings, evoked by a 20- and 60-Hz train, were analyzed simultaneously for each animal during both predrug and postdrug conditions. One set of DA release and uptake parameters were obtained for each pair of recordings.
Statistical Analysis
When appropriate, data are presented as the mean±SEM. Statistical analysis was performed using SAS version 9.1.3 (SAS Institute Inc. 2004). Specifically, a repeated-measures MANOVA with pairwise multivariate contrasts was used to analyze time courses in Figure 2 and Figure 6. Grouped data in Figure 3 and Figure 4 and in Sup. Table S1 were compared using a one-way ANOVA followed by Ryan-Einot-Gabriel-Welsch Multiple Range Test (REGWQ) as a post-hoc test. Student’s t-test was used to compare linear-regression slopes in Figure 5 and the combined groups in Sup. Table S1. Significance was set at p < 0.05.
Figure 2. Time course of the effects of AMPH on evoked DA levels.
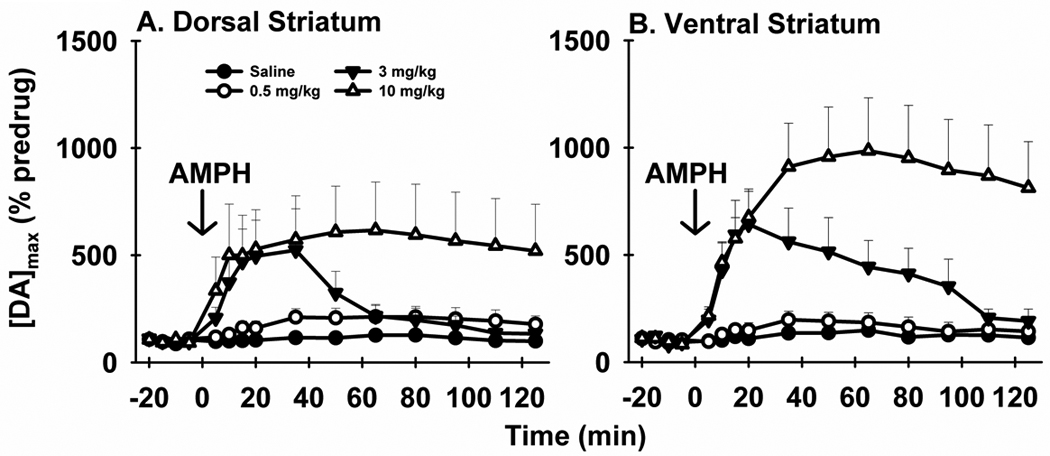
AMPH elicits time- and dose-dependent effects on [DA]max in the dorsal (A) and ventral (B) striatum. [DA]max is the maximal concentration of DA evoked by MFB stimulation with a 20-Hz train. Data are expressed as a percent of the predrug value (% predrug).
Figure 6. FSCV analysis of the change in background current elicited by AMPH.
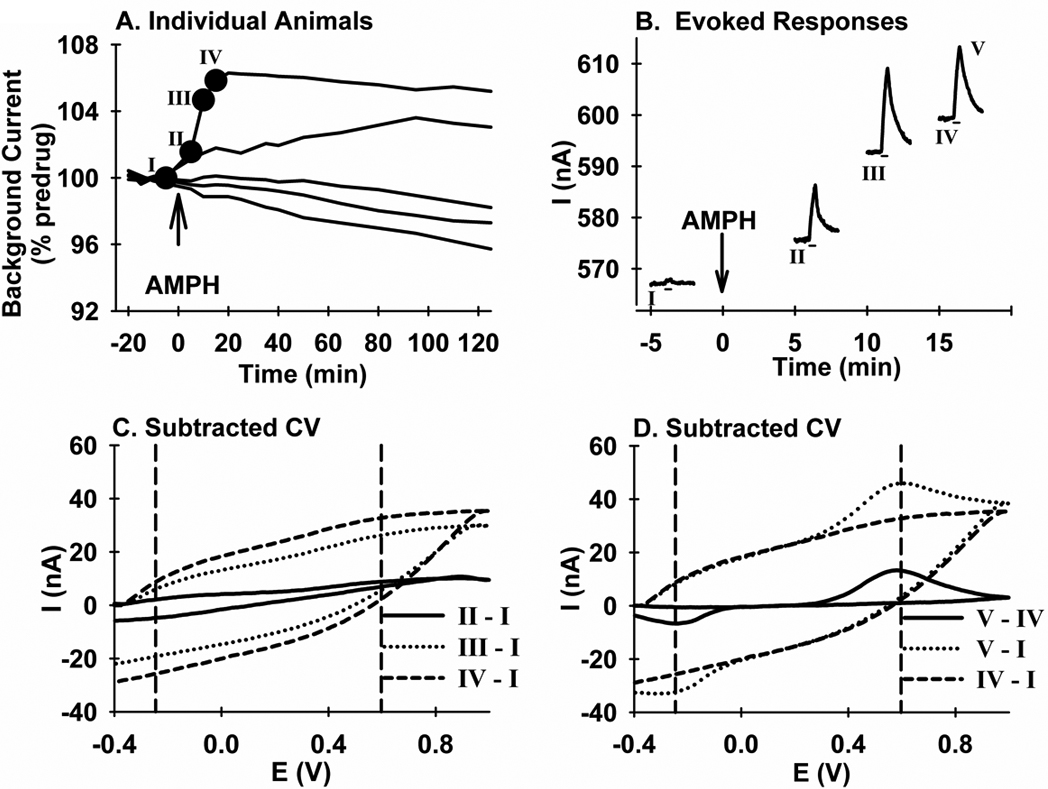
A. Data are all responses from the 10 mg/kg-AMPH group collected in the dorsal striatum and averaged in Figure 6. B. Data are the evoked responses designated with the closed circles and Roman Numerals in A. Absolute current (i.e., not background subtracted) is plotted (I). C. Data are background-cyclic voltammograms (“Subtracted CV”) with current (I) as the ordinate, and voltage (E) as the abscissa. The pre-stimulation background current from time point I was subtracted from the corresponding region in time points II, III, and IV, and designated “II - I”, “III- I”, and “IV - I” , respectively. D. Data are background-cyclic voltammograms (“Subtracted CV”). The “IV - I” voltammogram from B is replicated, along with two additional voltammograms. The first, “V - IV”, was calculated by subtracting the pre-stimulation background current in IV from the peak of the evoked response also in IV, labeled in B as V. The second, “V - I”, was calculated by subtracting the pre-stimulation background current in I from the peak of the evoked response, V, in recording IV.
Figure 3. Effects of AMPH on best-fit parameters for DA release and uptake.
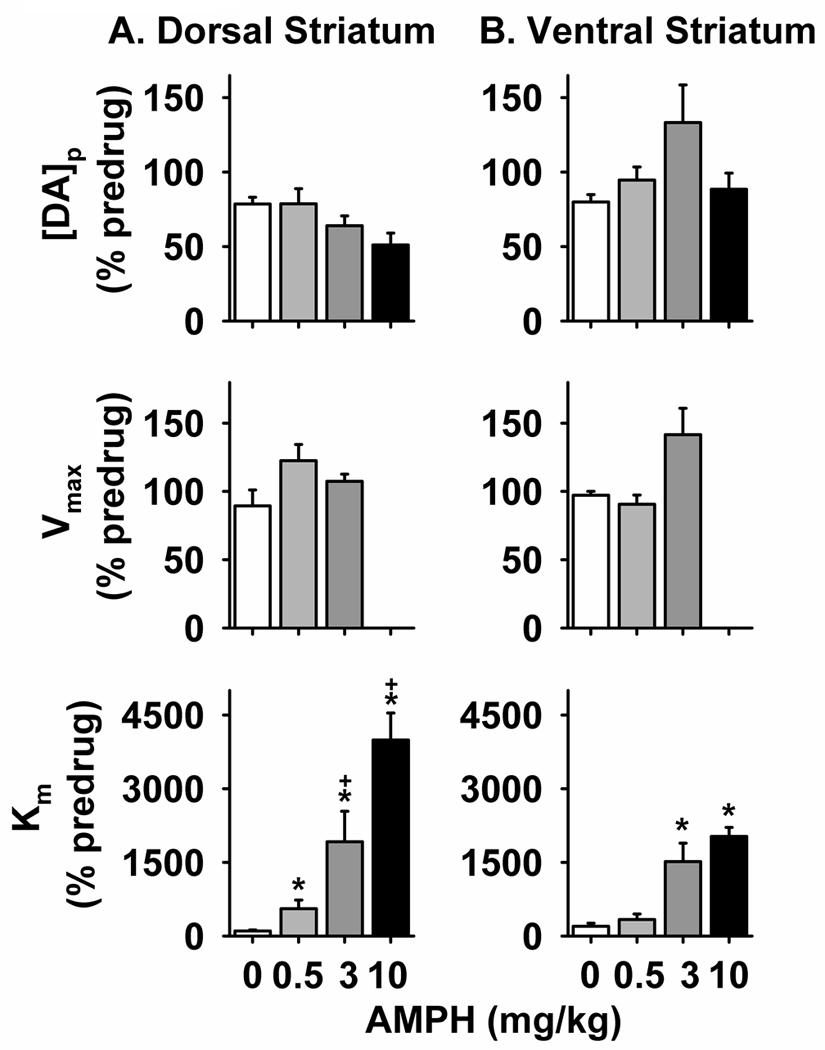
The 20- and 60-Hz responses represented in Figure 1 and Figure S1 were kinetically analyzed to determine parameters for exocytotic DA release ([DA]p, top) and neuronal DA uptake (Vmax, middle; Km, bottom). These values, determined for both the dorsal (A) and ventral (B) striatum, were expressed as a percent of the predrug value (% predrug). *significantly different than saline values (p < 0.05). +significantly different than 0.5 mg/kg (p < 0.05).
Figure 4. In vivo determination of a Ki for competitive inhibition of DA uptake by AMPH.
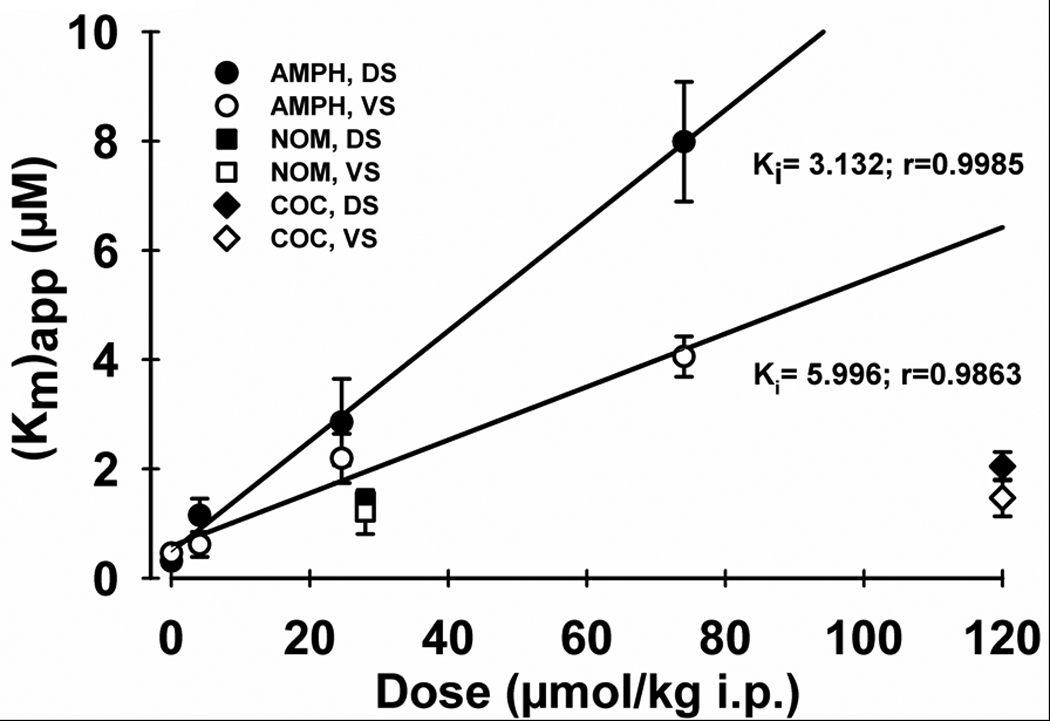
The x axis describes AMPH dose converted from mg/kg to µmol/kg. The y axis is apparent Km ((Km)app or postdrug Km) determined from the kinetic analysis shown in Figure 4. r is the coefficient for the linear regression. (Km)app values for cocaine and nomifensine were taken from Wu et al. (2001a) after converting to µmol/kg (original drug dose: 40 and 10 mg/kg i.p., respectively). Abbreviations: AMPH, amphetamine; NOM, nomifensine; COC, cocaine; DS, dorsal striatum; VS, ventral striatum.
Drugs
Urethane and d-amphetamine sulfate were purchased from Sigma (St. Louis, MO, USA). Both drugs were dissolved in 150 mM saline prepared with nanopure water (Barnstead/Thermolyne Coporation, Dubuque, IA, USA) prior to injection. d-AMPH dose was determined as base weight.
RESULTS
Effects of AMPH on evoked DA levels
Figure 1 shows representative effects of AMPH on extracellular DA dynamics electrically evoked in the dorsal (Fig. 1A) and ventral (Fig. 1B) striatum by MFB stimulation. These responses were collected 35 minutes after drug administration. In each recording, the stimulus pulse train (20 Hz, 2 s) was delivered at 5 s, as demarcated by the solid line underneath. In both striatal subregions, AMPH elicited robust increases in evoked DA levels at all doses examined. The slowed extracellular clearance of released DA indicated that uptake inhibition contributed to observed increases. Changes in exocytotic DA release cannot be ruled out, however, because the amplitude of the evoked response is determined by the balance of release and uptake processes (Wightman et al. 1988;Wu et al. 2001b). Interestingly, the effects of AMPH on signal amplitude were greatest in the ventral striatum at the highest dose (10 mg/kg), but plateaued in the dorsal striatum going at 3 mg/kg. In the dorsal striatum, the further slowing of extracellular DA clearance without a change in signal amplitude as AMPH dose is increased from 3 to 10 mg/kg is consistent with a decrease in exocytotic DA release. Voltammograms (Insets) indicate that DA was the origin of these evoked responses.
Figure 1. Effects of AMPH on evoked DA signals.
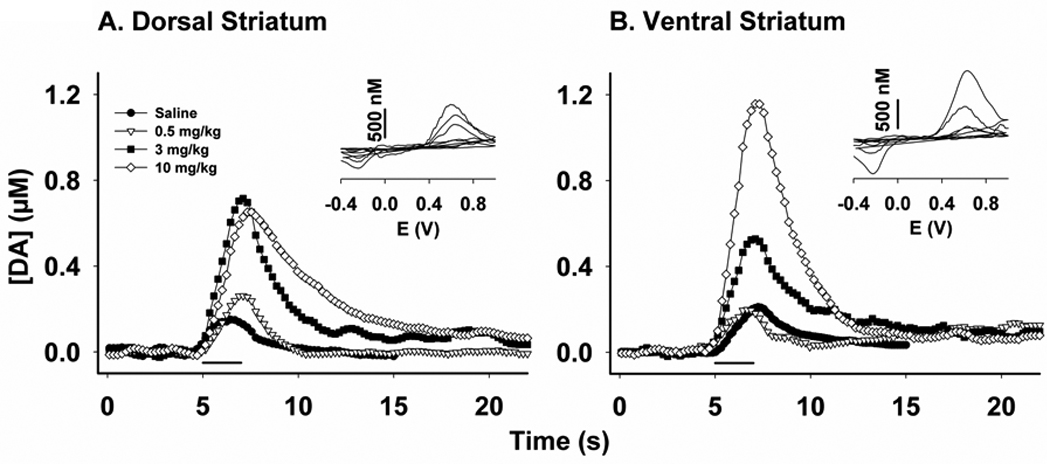
AMPH alters the amplitude and dynamics of extracellular DA levels in the dorsal (A) and ventral (B) striatum measured by FSCV at a CFM and elicited by electrical stimulation of the MFB. The stimulus train (20 Hz, 2 s) was applied at 5 seconds, as indicated by the solid line underneath these representative responses. Recordings were collected 35 minutes after drug injection (i.p.). Insets: Background-subtracted cyclic voltammograms with voltage (E) as the abscissa.
Figure 2 shows the time course of AMPH effects on the maximal DA concentration ([DA]max) elicited by 20-Hz stimulation. Both the amplitude and duration of the effects of AMPH on [DA]max in the dorsal (Fig. 2A) and ventral (Fig. 2B) striatum were dose related. Similar to representative recordings, averaged [DA]max responses did not further increase in the dorsal striatum from 3 to 10 mg/kg AMPH. Also similar to the recordings shown in Figure 1, [DA]max appear to increase in a dose-dependent fashion in the ventral striatum. Dissimilar from other doses, which elicited more transient elevations, 10 mg/kg AMPH caused [DA]max to plateau for the duration of the time course. A repeated-measures MANOVA revealed a significant effect of time in the dorsal (F14,6 = 5.95, p = 0.0187) but not ventral (F14,2 = 9.37, p = 0.1005) striatum, and no significant interaction (F42,24 = 1.75, p = 0.0727 and F42,12 = 1.45, p = 0.2473, respectively). There was a main effect of drug treatment in both the dorsal and ventral striatum (F3,19 = 3.84, p = 0.0264 and F3,15 = 8.30, p = 0.0017, respectively), and post-hoc tests determined significant differences between 10 mg/kg and all other doses in both subregions (p < 0.05).
Effects of AMPH on DA release and uptake
Real-time recordings evoked by 20 and 60 Hz stimulation (Sup. Fig. S1 and Sup. Fig. S2) were kinetically analyzed to resolve the relative contributions of DA release and uptake mechanisms to the AMPH-induced alterations in DA dynamics and [DA]max. Pre-drug values for all measures were similar across group and consistent with previous descriptions (Sup. Table S1). The effects of AMPH on best-fit parameters for DA release and uptake, expressed as a percent of the predrug response, are found in Figure 3. As shown in the top panel, ANOVA revealed no significant main effect of AMPH on [DA]p in either the dorsal (Fig. 4A) or ventral (Fig. 4B) striatum (F3, 21 = 2.76, p = 0.0678 and F3, 19 = 1.41, p = 0.2711, respectively). Because the p value (0.0678) approached significance (0.05), the modest decrease in exocytotic DA release in the dorsal subregion at 3 and 10 mg/kg AMPH, with the highest dose eliciting the greatest reduction, is considered in this study a statistical trend. In contrast, the p value (0.2711) in the ventral striatum is considerably higher, and trend is not used to describe these data.
Figure 3, middle and bottom panels, shows the effects of AMPH on the Michaelis-Menten parameters for neuronal DA uptake. Because curve fitting of 10 mg/kg responses was not stable when all three parameters ([DA]p, Vmax and Km) were permitted to float, Vmax was fixed at the predrug level for analysis of postdrug responses measured with the highest AMPH dose. This procedure assumes that AMPH competitively inhibits DA uptake at the highest dose, which is consistent with drug action observed at the two lower doses (see below). As a result, statistical analysis of Vmax only used the AMPH doses of 0, 0.5 and 3 mg/kg. As shown in the middle panel, there were no significant main effects of AMPH on Vmax in the dorsal (Fig. 4A; F2, 17 = 1.30, p = 0.2983) or ventral (Fig. 4B; F2, 14 = 3.19, p = 0.0724) striatum. The effects of AMPH on Km are shown in the bottom panel. Overall, AMPH appeared to inhibit DA uptake to a greater extent in the dorsal (Fig. 3A) compared to the ventral (Fig. 3B) striatum. Moreover, in contrast to Vmax, AMPH elicited highly significant main effects on Km in both the dorsal (F3, 21 = 26.39, p < 0.0001) and ventral (F3, 19 = 17.31, p < 0.0001) striatum. In the dorsal striatum, post-hoc tests revealed a significant difference between all AMPH doses and saline and a significant difference between 0.5 mg/kg and the higher doses (p < 0.05). In the ventral striatum, 3 and 10 mg/kg AMPH were significantly different from saline and 0.5 mg/kg (p < 0.05). Overall, these results suggest that AMPH competitively inhibits DA uptake in both striatal subregions.
Determination of a Ki for the Effects of AMPH on DA uptake
To further assess the regional potency of AMPH on DA uptake, a dissociation constant (Ki) for the binding of the competitive inhibitor to DAT was determined by plotting apparent Km ((Km)app or postdrug Km) versus dose. In this plot, the slope of the linear regression line is Km/Ki (Jones et al. 1995). Instead of mg/kg, drug dose is expressed as micromoles (µmol)/kg (i.e., 0.5, 3.0, and 10 mg/kg equals 4.1, 24.6, and 74.0 µmol/kg, respectively). This conversion permits comparisons between AMPH and two other psychostimulants, cocaine and nomifensine, on a mole-drug dose basis (see below). Figure 4 shows a significant linear relationship between (Km)app and AMPH dose for both the dorsal (solid circles, DS; r = 0.9985, p = 0.0015) and ventral (open circles, VS; r = 0.9863, p = 0.0137) striatum. The slope of the linear regression is steeper for the dorsal (0.101±0.0039 µM/µmol/kg) compared to the ventral (0.0487±0.0058 µM/µmol/kg) striatum, and statistical analysis demonstrated a significance difference (t = 7.483, p < 0.001). Based on these slopes, Ki for AMPH inhibiting DA uptake was calculated to be 3.123 and 5.996 µmol/kg in the dorsal and ventral striatum, respectively. Thus, AMPH is ~2 times more potent for competitively inhibiting DA uptake in the dorsal striatal subregion.
Also shown in Figure 4 is the apparent Km for two other psychostimulants that inhibit DA uptake, nomifensine (NOM, squares) and cocaine (COC, diamonds). These data were previously, but comparably determined in vivo (Wu et al. 2001a). AMPH appears substantively more efficacious than cocaine for inhibiting DA uptake in both the dorsal (solid circles, DS) and ventral (open circles, VS) striatum, but more similar to nomifensine (solid squares, DB; open squares, VS). Taken together, these results suggest that AMPH is inhibiting neuronal DA uptake quantitatively and qualitatively similar in vivo and in vitro. AMPH also appears more efficacious in inhibiting DA uptake in vivo than cocaine.
Effects of AMPH on background voltammetric current
Similar to previous brain slice studies using FSCV at a CFM (Jones et al., 1998; Schmitz et al., 2001), AMPH-induced changes in basal DA levels were indexed by background voltammetric current measured at the peak oxidation potential for DA. As shown in Figure 5, only the highest dose of AMPH elicited an increase in background current in the dorsal striatum, and this increase (~5 %) was relatively small (Fig. 5A). ANOVA demonstrated significant main effects of drug treatment (F3, 19 = 4.36, p = 0.0169) and time (F14,6 = 5.97, p = 0.0186), but no significant interaction between time and treatment (F42,24 = 1.69, p = 0.0862). Post-hoc tests indicated a significant difference between 10 mg/kg and both saline and 3 mg/kg (p < 0.5). No effects of AMPH on background current were apparent in the ventral striatum (Fig. 5B), and no significant main effects of drug (F3,15 = 3.02, p = 0.0629) or time (F14,2 = 11.81, p = 0.0807), and no significant interaction (F42,12 = 1.52, p = 0.22) were determined.
Figure 6 describes an analysis of voltammograms to establish the electrochemical origin of the increase in background current elicited in the dorsal striatum by 10 mg/kg AMPH. Figure 6A shows individual time-course responses for the entire high-dose group. Three out five responses appeared to show no increase with AMPH. The individual time-course response that demonstrated the greatest increase was further analyzed. Shown in Figure 6B are the four evoked responses at the time points demarcated by the closed circles and Roman Numerals in 6A. Notice the increase in the amplitude of the evoked response, consistent with Figure 1 and Figure 2. Stimulus delivery is demarcated by the line underneath each recording. Also notice the increase in background current (i.e., pre-stimulus signal) with time, consistent with 6A. In contrast to recordings shown in Figure 1, background current is not subtracted and is rather reported as an absolute value in the y-axis. The effects of stimulation are still observed on top of this rising background, which changes more slowly than the evoked DA signal. Figure 6C shows the individual cyclic voltammogram (“Subtracted CV”) determined by subtracting the background response (i.e., pre-stimulation) in recording I from the corresponding background responses in recordings II through IV. All voltammograms showed non-specific changes in the electrochemical signal, perhaps due to electrode drift, but no evidence for DA.
Figure 6D shows additional cyclic voltammograms calculated to determine whether a change in DA, if present, could contribute to these non-specific changes. The solid line describes the characteristic voltammogram for DA, with an upward peak at ~+0.6 V, due to the oxidation of DA to a quinone, and a downward peak at ~−0.2 V, due to the reduction of the electroformed quinone. This voltammogram, calculated by subtracting the background response in IV from the peak of the evoked response in this same recording, identified as V, demonstrates that DA is the source of the evoked signal, consistent with Figure 1 (Insets). The long-dashed line is the same voltammogram shown for “IV - I” in 6C, which describes the change in background electrochemistry between these two time points and whose origin appears to be non-specific drift of the CFM.
The dotted line in Figure 6D is the voltammogram calculated by subtracting the background response in I from the peak of the evoked response V. This voltammogram clearly shows the non-specific change in background signal, as well as the characteristic redox peaks for DA. In terms of absolute current, the non-specific background change (~30 nA) is about two-times the amplitude of the evoked signal (~15 nA). Thus, if present at this concentration, approximately 1 µM after postcalibration conversion, DA should also appear on top of the non-specific voltammogram. Based on an estimated detection limit for the present in vivo measurements of ~150 nM (i.e., three times the standard deviation of baseline noise or a signal-to-noise ratio of three), even lower DA concentrations would appear in the voltammograms as well. Taken together, these results suggest that AMPH-induced increases in basal levels of extracellular DA were not detected in either the dorsal or ventral striatum.
DISCUSSION
We examined the mechanism of AMPH action in vivo. Voltammetric and kinetic analyses were comparable to those used previously in brain slices demonstrating key drug effects acting concurrently on presynaptic dopaminergic signaling: competitive inhibition of DA uptake, depletion of vesicular DA stores, as indexed by exocytotic DA release, and increased DA efflux, as indexed by increases in basal DA levels. We were able to replicate robust competitive inhibition of DA uptake. However, no effect of AMPH on vesicular DA release was observed in the ventral striatum and only modest decreases were found in the dorsal striatum. Moreover, increases in basal DA levels were not detected in either striatal subregion. Taken together, these results suggest distinct in vivo and in vitro pharmacology of AMPH.
Effects of AMPH on electrically evoked DA levels are complex
While voltammetry in brain slices consistently demonstrates a decrease in electrically evoked DA levels with AMPH (Jones et al. 1998;Schmitz et al. 2001;Patel et al. 2003), results with in vivo voltammetry are mixed. Robust (~70 %) decreases have been observed in the dorsal striatum with very long (10 s), high-frequency (60 Hz) trains (Ewing et al. 1983;Kuhr et al. 1985;Kuhr et al. 1986), but with shorter trains (0.25 to 2 s) an increase has been reported (May et al. 1988). In the ventral striatum, the same long, high-frequency trains elicited no change (Kuhr et al. 1986), and increases have been demonstrated with short, low-frequency trains (20 Hz, 0.3 s; Dugast et al. 1994). We report here that AMPH increases DA levels evoked by 20-Hz stimulation in both the dorsal and ventral striatum across a wide range of doses (Fig. 2). It thus appears that in vivo, the use of long-duration, high-frequency trains is associated with AMPH-induced decreases in evoked DA levels in the dorsal striatum, while shorter-duration, low-frequency stimulation supports augmented responses in both striatal subregions. The ventral striatum also appears more resistant to AMPH-induced decreases in evoked DA levels.
Dose-dependent, AMPH-induced increases in evoked DA signals were observed in both striatal regions, but to different degrees. In contrast to the ventral striatum, no further increase in signal was observed going from 3 to 10 mg/kg (Fig. 2). A similar response plateau has been reported in the olfactory tubercle, also with low-frequency stimulation (Suaud-Chagny et al. 1989). We additionally show in both dorsal and ventral striata that 10 mg/kg elevated evoked DA levels for the entire 2-h time course, in contrast to lower doses that increased signals more transiently. This effect, which has not been reported previously to our knowledge, could be due to the combination of high AMPH dose (10 mg/kg) and low-frequency (20 Hz) stimulation. It thus appears that multiple aspects of AMPH-induced changes in evoked DA levels are dose dependent. Interestingly, this extended time course closely parallels the stereotypy observed in awake rats, which remains high for >2 h and is elicited by high doses of AMPH (Kelley et al., 1975). In contrast, low doses of AMPH have more transient effects on both locomotor activity (Clausing et a., 1995) and electrically evoked DA levels. Thus, these voltammetric measurements may have implications for AMPH-induced changes in motor behavior.
Effects of AMPH on exocytotic DA release
To our knowledge, this is the first study to isolate AMPH effects on dopaminergic signaling in vivo to a parameter for vesicular DA release. We show that AMPH does not alter exocytotic DA release in the ventral striatum, and only a trend for a modest decrease was observed in the dorsal striatum (Fig. 3). The further reduction in exocytotic DA release from 3 to 10 mg/kg AMPH could mediate the similar [DA]max observed for the two does (Fig. 2), however, because this small decrease in release is additive with each stimulus pulse across the entire train. Our in vivo results are in sharp contrast to those obtained with voltammetry in brain slices, which demonstrate that AMPH robustly (~75 to 100 %) decreases exocytotic DA release in the dorsal striatum (Jones et al. 1998;Schmitz et al. 2001;Patel et al. 2003). The origin of this decrease in the brain slice is debated. Schmitz et al. (2001) argue that 47 % is due to vesicular DA depletion, with the remaining 35 % stemming from autoreceptor activation, while Jones et al. (1998) and Patel et al. (2003) found no evidence for presynaptic DA inhibition.
While there is a precedent for discrepant effects of drugs on DA release between brain-slice and whole-animal preparations (e.g., Wu et al 2000; Kulagina et al 2001), it is difficult to fully reconcile the marked decrease in exocytotic DA release observed in vitro with the minimal to modest effects of AMPH in vivo. AMPH dose does not appear to be the origin of this discrepancy. A similar inhibition of DA uptake was achieved in the two preparations, and brain AMPH concentrations sufficient to decrease exocytotic DA release in the slice, are detected after systemic administration of similar doses used in the present study (see Results; Clausing et al. 1995;Kuczenski et al. 1997). This discrepancy could be due to differences in the responsiveness of isolated terminals versus intact neurons to AMPH. Circuit-level modulation may play a role as well (Miller et al. 2002). Another potential origin for this discrepancy is electrical stimulation. A single pulse was applied directly to slice dopaminergic terminals, as opposed to stimulus trains activating action potentials in ascending dopaminergic axons in vivo. Perhaps as a result, per-stimulus-pulse DA release differs as well, reaching ~2.5 µM (Schmitz et al. 2001) in the slice, but only ~0.05 µM in vivo (Sup. Table S1). How this ~50-fold difference in eliciting exocytotic DA release might alter AMPH action, however, is not established. It is known that intense stimulation recruits vesicles from the reserve pool (Rizzoli and Betz 2005) and that AMPH differentially depletes different classes of DA vesicles (Anderson et al. 1998). Thus, the slice stimulation may be preferentially interrogating AMPH-sensitive DA pools.
AMPH competitively inhibits DA uptake
Consistent with classical uptake studies (Harris and Baldessarini 1973;Richelson and Pfenning 1984;Krueger 1990) and voltammetry in brain slices (Jones et al. 1998;Schmitz et al. 2001;John and Jones 2007), AMPH competitively inhibits DA uptake in vivo (Fig. 3C). Taken together with the analysis of exocytotic DA release (Fig. 3A), these results indicate that inhibition of neuronal DA uptake is the predominant mechanism underlying AMPH-induced increases in electrically evoked DA levels in the ventral striatum in vivo. The situation is more complex in the dorsal striatum, where both decreased exocytotic DA release and DA uptake inhibition contribute. Although reduced exocytotic DA release was modest, inhibited DA uptake was robust and significant at all AMPH doses, suggesting that the latter mechanism also predominates in the dorsal striatum.
One important caveat is that other AMPH effects besides competitive inhibition may cause an apparent change in Km. Indeed, AMPH has been shown to reverse DAT action (Sulzer et al., 1995) and induce channel-like activity in DAT (Kahlig et a., 2005). Both of these phenomena could conceivably cause an apparent slowing of DA uptake. Because neither is accounted for by the kinetic model used in the present study to analyze evoked DA signals, we cannot rule out a contribution of non-exocyctotic DA release to our measurements of DA uptake. However, it would also seem that all studies using simple Michaelis-Menten kinetics, as we do here, would suffer from the same criticism. Thus, we argue that there still is value comparing AMPH-induced changes Km across studies, even though the exact mechanism by which this drug alters DA uptake is not fully established.
A related issue is that for responses recorded after administration of 10 mg/kg AMPH, curve fitting by permitting all three parameters, [DA]p, Vmax, and Km to float independently was not possible. Instead, competitive inhibition was assumed by fixing Vmax to predrug levels. We justify this manipulation on the following grounds. First, previous studies using voltammetry in brain slices did not demonstrate a change in Vmax with AMPH (Jones et al. 1999;Schmitz et al. 2001). Second, competitive inhibition has similarly been assumed for analyzing AMPH and related drugs that elicit non-exocytotic DA release also using slice voltammetry (John and Jones 2007). And third, all values for Km, whether determined independently or by fixing Vmax, are linearly related to AMPH dose in both the dorsal and ventral striatum. This linear relationship is a hallmark of competitive inhibition. Although AMPH has been shown to alter Vmax in vitro (Johnson et al. 2005) and ex vivo (Fleckenstein et al. 1999), further study is required to determine whether AMPH acts as a uncompetitive inhibitor in vivo.
Interestingly, we demonstrate that AMPH inhibition of DA uptake is ~2-fold more potent in the dorsal compared to ventral striatum (Fig. 4). The regional potency of AMPH parallels another DA uptake inhibitor, nomifensine, which has been shown using voltammetry in vivo (Wu et al., 2001) and in brain slices (Jones et al., 1995) to increase Km to a greater extent in the dorsal than ventral striatum, but not cocaine, which has similar effects. Different effects of cocaine and nomifensine on DA uptake have been suggested to be mediated by different binding sites on DAT (Meiergerd and Schenk, 1994). One likely explanation for the regional effects of DA uptake inhibitors is that DAT may differ in the dorsal and ventral striatum. For example, DAT in the ventral striatum exhibits a higher molecular weight than in the dorsal striatum due to greater glycosylation (Lew et al., 1992). Another possibility is that with a higher Vmax (Sup. Table S1; Wu et al., 2001), the reverse transport of DA, if present, would have a more prominent effect on slowing DA uptake in the dorsal compared to the ventral striatum.
By itself, more potent inhibition by AMPH leads to greater increases in evoked DA levels. However, because of decreased exocytotic DA release in the dorsal subregion, greater increases were observed in the ventral subregion (Fig. 1 and Fig. 2). This result is consistent with AMPH-induced increases in dialysate DA (Kuczenski and Segal, 1992), suggesting convergent measurements by these two different analytical techniques. AMPH also appears more efficacious than cocaine in inhibiting DA uptake (Fig. 4). While AMPH is a poorer binder to DAT relative to cocaine (Ritz et al. 1987), in vitro studies are equivocal with regards to which is a more potent inhibitor of DA uptake (Harris and Baldessarini 1973;Richelson and Pfenning 1984;Krueger 1990;John and Jones 2007). It is important to consider that, with systemic injections, pharmacokinetics and brain access also factor into potency.
It may be possible to quantitatively relate the Ki for AMPH inhibiting DA uptake determined in the present study in vivo to previously reported values obtained in brain slices by using comparable voltammetric measurements in the two preparations to equate doses. For example, Jones et al. (1999) demonstrated that 10 µM AMPH increased Km from 0.2 µM to 8 µM, which is nearly identical to the 40-fold increase at 10 mg/kg from 0.2 µM to 7.98 µM that we report here. Also, there is a similar ~25-fold increase in Km for DA uptake measured by Schmitz et al. (2001) from 0.89 to 24 µM. Assuming that a similar effect on Km reflects similar drug concentrations acting on DAT, then the in vivo and in vitro AMPH doses are equivalent. This equivalence seems reasonable, because systemic AMPH doses in the range administered in the present study result in low micromolar drug levels measured in striatal extracellular fluid (Clausing et al. 1995;Kuczenski et al. 1997). Accordingly, the Ki of 3.123 µmol/kg measured in the dorsal striatum in vivo thus converts to 0.425 µM, which is quite similar to values determined in vitro with voltammetry (0.42 µM, John and Jones 2007) and classical uptake measurements using 3H-dopamine (0.22 µM, Krueger 1990); 0.28 µM, Lin and Uhl 2002).
AMPH-induced increases in basal DA levels
Consistent with previous work using FSCV at a CFM in the urethane-anesthetized rat (Wiedemann et al. 1990), AMPH-induced increases in basal DA levels were not detected in the striatum (Fig. 5 and Fig. 6). These results are clearly at variance with studies using slice voltammetry and microdialysis in awake animals. Slice studies report a ~4 µM increase in basal DA levels after 10 µM AMPH (Jones et al. 1998;Schmitz et al. 2001). Because identical FSCV parameters were used, discrepancies are most likely not due to differences in DA sensitivity. Indeed, the estimated detection limit for the present in vivo measurements (~150 nM) is more than sufficient to measure the large DA efflux recorded in the slice. Basal DA levels measured in the brain slice preparation appear to be a good index of AMPH-induced DA efflux, because, without input from cell bodies, action potential-dependent DA release would not occur at the excised dopaminergic terminals. Interestingly, with ongoing dopaminergic activity in the urethane-anesthetized rat (Tepper et al. 1995), one might expect a greater increase in basal DA levels in vivo, due to the combined actions of AMPH eliciting DA efflux and inhibiting DA uptake, which indirectly augments exocytotic DA release.
Our result of no increase in basal DA levels is also surprising given that the robust effects of AMPH on dialysate DA levels are well documented (Kuczenski et al. 1991;Kuczenski et al. 1997). However, quantitative comparisons between these results with microdialysis and the present in vivo voltammetry study are more challenging. The critical issue is that determination of an absolute basal concentration for DA is made difficult by interactions between the implanted probe and surrounding brain tissue (Bungay et al. 2003). If, according to the no-net-flux method, basal DA levels are ~5 nM (Justice, Jr. 1993), however, then the ~30-fold increase in dialysate DA recorded in awake animals with moderate to high AMPH doses (Kuczenski et al. 1991;Kuczenski et al. 1997) might fall below the present in vivo detection limit. This postulate can be tested with more sensitive FSCV (Heien et al. 2003). Anesthesia may also play a role, but AMPH robustly increases dialysate DA in the striatum under urethane as well (Yamamoto and Pehek 1990).
Conclusion
We show using voltammetry in the urethane-anesthetized rat that AMPH augments action potential-dependent presynaptic dopaminergic signaling. This activation occurred across a wide dose range that elicits behaviors spanning the psychostimulant spectrum from locomotion at the low end to stereotypy at the high end (Kelly et al. 1975;Gaytan et al. 1998). This result also starkly contrasts the near-complete disruption of normal patterns of action potential-dependent signaling observed with slice voltammetry and mediated by depletion of vesicular DA stores and robust DA efflux (Jones et al. 1998;Schmitz et al. 2001). Future work should be directed at determining the origin of the discrepant results obtained in the two preparations. Possible contributing factors are anesthesia, different responsiveness of intact versus severed neurons, and dissimilar stimulation characteristics.
Due to the combined actions of robust inhibition of neuronal DA uptake and minimal to modest reductions of exocytotic DA release, AMPH may thus function like other drugs of abuse and potentiate phasic DA signaling (Stuber et al. 2005;Cheer et al. 2007;Robinson et al. 2009). These transient elevations of brain extracellular DA are evoked by burst firing of dopaminergic neurons in response to primary rewards and cues predictive of these rewards (Carelli and Wightman 2004;Schultz 2007). A DA-mediated overlearning of cues predicting drug availability could thus be a general mechanism of abused drugs in the addiction process (Hyman 2005;Wanat et al. 2009).
Supplementary Material
Acknowledgement
This work was supported by the National Institute on Drug Abuse [DA 021770 and DA 024036] and ISU POENB.
Abbreviations
- DA
dopamine
- DAT
dopamine transporter
- AMPH
amphetamine
- FSCV
fast-scan cyclic voltammetry
- CFM
carbon-fiber microelectrode
- [DA]p
concentration of dopamine released per stimulus pulse
- [DA]max
maximal evoked concentration of dopamine
Footnotes
The authors declare no conflict of interest.
References
- Anderson BB, Chen G, Gutman DA, Ewing AG. Dopamine levels of two classes of vesicles are differentially depleted by amphetamine. Brain Res. 1998;788:294–301. doi: 10.1016/s0006-8993(98)00040-7. [DOI] [PubMed] [Google Scholar]
- Bungay PM, Newton-Vinson P, Isele W, Garris PA, Justice JB. Microdialysis of dopamine interpreted with quantitative model incorporating probe implantation trauma. J Neurochem. 2003;86:932–946. doi: 10.1046/j.1471-4159.2003.01904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carelli RM, Wightman RM. Functional microcircuitry in the accumbens underlying drug addiction: insights from real-time signaling during behavior. Curr Opin Neurobiol. 2004;14:763–768. doi: 10.1016/j.conb.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Cheer JF, Wassum KM, Sombers LA, Heien ML, Ariansen JL, Aragona BJ, Phillips PE, Wightman RM. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci. 2007;27:791–795. doi: 10.1523/JNEUROSCI.4152-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausing P, Gough B, Holson RR, Slikker W, Jr, Bowyer JF. Amphetamine levels in brain microdialysate, caudate/putamen, substantia nigra and plasma after dosage that produces either behavioral or neurotoxic effects. J Pharmacol Exp Ther. 1995;274:614–621. [PubMed] [Google Scholar]
- Dugast C, Suaud-Chagny MF, Gonon F. Continuous in vivo monitoring of evoked dopamine release in the rat nucleus accumbens by amperometry. Neuroscience. 1994;62:647–654. doi: 10.1016/0306-4522(94)90466-9. [DOI] [PubMed] [Google Scholar]
- Ewing AG, Bigelow JC, Wightman RM. Direct in vivo monitoring of dopamine released from two striatal compartments in the rat. Science. 1983;221:169–171. doi: 10.1126/science.6857277. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Floor E, Meng L. Amphetamine releases dopamine from synaptic vesicles by dual mechanisms. Neurosci Lett. 1996;215:53–56. doi: 10.1016/s0304-3940(96)12963-3. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Haughey HM, Metzger RR, Kokoshka JM, Riddle EL, Hanson JE, Gibb JW, Hanson GR. Differential effects of psychostimulants and related agents on dopaminergic and serotonergic transporter function. Eur J Pharmacol. 1999;382:45–49. doi: 10.1016/s0014-2999(99)00588-9. [DOI] [PubMed] [Google Scholar]
- Gaytan O, Swann A, Dafny N. Diurnal differences in rat's motor response to amphetamine. Eur J Pharmacol. 1998;345:119–128. doi: 10.1016/s0014-2999(97)01558-6. [DOI] [PubMed] [Google Scholar]
- Harris JE, Baldessarini RJ. Uptake of (3H)-catecholamines by homogenates of rat corpus striatum and cerebral cortex: effects of amphetamine analogues. Neuropharmacology. 1973;12:669–679. doi: 10.1016/0028-3908(73)90120-2. [DOI] [PubMed] [Google Scholar]
- Heien ML, Phillips PE, Stuber GD, Seipel AT, Wightman RM. Overoxidation of carbon-fiber microelectrodes enhances dopamine adsorption and increases sensitivity. Analyst. 2003;128:1413–1419. doi: 10.1039/b307024g. [DOI] [PubMed] [Google Scholar]
- Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–1422. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources. Guide for the Care and Use of Laboratory Animals. Washington, D.C: National Academy Press; 1996. [Google Scholar]
- John CE, Jones SR. Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology. 2007;52:1596–1605. doi: 10.1016/j.neuropharm.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Furman CA, Zhang M, Guptaroy B, Gnegy ME. Rapid delivery of the dopamine transporter to the plasmalemmal membrane upon amphetamine stimulation. Neuropharmacology. 2005;49:750–758. doi: 10.1016/j.neuropharm.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Garris PA, Wightman RM. Different effects of cocaine and nomifensine on dopamine uptake in the caudate-putamen and nucleus accumbens. J Pharmacol Exp Ther. 1995;274:396–403. [PubMed] [Google Scholar]
- Jones SR, Joseph JD, Barak LS, Caron MG, Wightman RM. Dopamine neuronal transport kinetics and effects of amphetamine. J Neurochem. 1999;73:2406–2414. doi: 10.1046/j.1471-4159.1999.0732406.x. [DOI] [PubMed] [Google Scholar]
- Justice JB., Jr Quantitative microdialysis of neurotransmitters. J Neurosci Methods. 1993;48:263–276. doi: 10.1016/0165-0270(93)90097-b. [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Binda F, Khoshbouei H, Blakely RD, McMahon DG, Javitch JA, Galli A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc Natl Acad Sci U S A. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PH, Seviour PW, Iversen SD. Amphetamine and apomorphine responses in the rat following 6-OHDA lesions of the nucleus accumbens septi and corpus striatum. Brain Res. 1975;94:507–522. doi: 10.1016/0006-8993(75)90233-4. [DOI] [PubMed] [Google Scholar]
- Krueger BK. Kinetics and block of dopamine uptake in synaptosomes from rat caudate nucleus. J Neurochem. 1990;55:260–267. doi: 10.1111/j.1471-4159.1990.tb08847.x. [DOI] [PubMed] [Google Scholar]
- Kuczenski R. Effects of catecholamine releasing agents on synaptosomal dopamine biosynthesis: multiple pools of dopamine or multiple forms of tyrosine hydroxylase. Neuropharmacology. 1975;14:1–10. doi: 10.1016/0028-3908(75)90060-x. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Melega WP, Cho AK, Segal DS. Extracellular dopamine and amphetamine after systemic amphetamine administration: comparison to the behavioral response. J Pharmacol Exp Ther. 1997;282:591–596. [PubMed] [Google Scholar]
- Kuczenski R, Segal DS, Aizenstein ML. Amphetamine, cocaine, and fencamfamine: relationship between locomotor and stereotypy response profiles and caudate and accumbens dopamine dynamics. J Neurosci. 1991;11:2703–2712. doi: 10.1523/JNEUROSCI.11-09-02703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Differential effects of amphetamine and dopamine uptake blockers (cocaine, nomifensine) on caudate and accumbens dialysate dopamine and 3-methoxytyramine. J Pharmacol Exp Ther. 1992;262:1085–1094. [PubMed] [Google Scholar]
- Kuhr WG, Bigelow JC, Wightman RM. In vivo comparison of the regulation of releasable dopamine in the caudate nucleus and the nucleus accumbens of the rat brain. J Neurosci. 1986;6:974–982. doi: 10.1523/JNEUROSCI.06-04-00974.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhr WG, Ewing AG, Near JA, Wightman RM. Amphetamine attenuates the stimulated release of dopamine in vivo. J Pharmacol Exp Ther. 1985;232:388–394. [PubMed] [Google Scholar]
- Kulagina NV, Zigmond MJ, Michael AC. Glutamate regulates the spontaneous and evoked release of dopamine in the rat striatum. Neurosci. 2001;102:121–128. doi: 10.1016/s0306-4522(00)00480-2. [DOI] [PubMed] [Google Scholar]
- Lew R, Patel A, Vaughan RA, Wilson A, Kuhar MJ. Microheterogeneity of dopamine transporters in rat striatum and nucleus accumbens. Brain Res. 1992;584:266–271. doi: 10.1016/0006-8993(92)90905-o. [DOI] [PubMed] [Google Scholar]
- Lin Z, Uhl GR. Dopamine transporter mutants with cocaine resistance and normal dopamine uptake provide targets for cocaine antagonism. Mol Pharmacol. 2002;61:885–891. doi: 10.1124/mol.61.4.885. [DOI] [PubMed] [Google Scholar]
- May LJ, Kuhr WG, Wightman RM. Differentiation of dopamine overflow and uptake processes in the extracellular fluid of the rat caudate nucleus with fast-scan in vivo voltammetry. J Neurochem. 1988;51:1060–1069. doi: 10.1111/j.1471-4159.1988.tb03069.x. [DOI] [PubMed] [Google Scholar]
- Meiergerd SM, Schenk JO. Kinetic evaluation of the commonality between the site(s) of action of cocaine and some other structurally similar and dissimilar inhibitors of the striatal transporter for dopamine. J Neurochem. 1994;63:1683–1692. doi: 10.1046/j.1471-4159.1994.63051683.x. [DOI] [PubMed] [Google Scholar]
- Miller HH, Shore PA, Clarke DE. In vivo monoamine oxidase inhibition by D-amphetamine. Biochem Pharmacol. 1980;10:1347–1354. doi: 10.1016/0006-2952(80)90429-3. [DOI] [PubMed] [Google Scholar]
- Miller AD, Forster GL, Metcalf KM, Blaha CD. Excitotoxic lesions of the pedunculopontine differentially mediate morphine- and d-amphetamine-evoked striatal dopamine efflux and behaviors. Neuroscience. 2002;111:351–362. doi: 10.1016/s0306-4522(01)00595-4. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Gong LW, Khanna B, Sulzer D, Lindau M. Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J Neurosci. 2003;23:5835–5845. doi: 10.1523/JNEUROSCI.23-13-05835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J, Mooslehner KA, Chan PM, Emson PC, Stamford JA. Presynaptic control of striatal dopamine neurotransmission in adult vesicular monoamine transporter 2 (VMAT2) mutant mice. J Neurochem. 2003;85:898–910. doi: 10.1046/j.1471-4159.2003.01732.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1986. [Google Scholar]
- Richelson E, Pfenning M. Blockade by antidepressants and related compounds of biogenic amine uptake into rat brain synaptosomes: most antidepressants selectively block norepinephrine uptake. Eur J Pharmacol. 1984;104:277–286. doi: 10.1016/0014-2999(84)90403-5. [DOI] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Robinson DL, Howard EC, McConnell S, Gonzales RA, Wightman RM. Disparity between tonic and phasic ethanol-induced dopamine increases in the nucleus accumbens of rats. Alcohol Clin Exp Res. 2009;33:1187–1196. doi: 10.1111/j.1530-0277.2009.00942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS Institute Inc. SAS/STAT® 9.1 User's Guide. Cary, NC: SAS Institute Inc; 2004. [Google Scholar]
- Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, Leeb-Lundberg LM, Carvelli L, Javitch JA, Galli A. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci U S A. 2000;97:6850–6855. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Behavioral dopamine signals. Trends Neurosci. 2007;30:203–210. doi: 10.1016/j.tins.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Roitman MF, Phillips PE, Carelli RM, Wightman RM. Rapid dopamine signaling in the nucleus accumbens during contingent and noncontingent cocaine administration. Neuropsychopharmacology. 2005;30:853–863. doi: 10.1038/sj.npp.1300619. [DOI] [PubMed] [Google Scholar]
- Suaud-Chagny MF, Buda M, Gonon FG. Pharmacology of electrically evoked dopamine release studied in the rat olfactory tubercle by in vivo electrochemistry. Eur J Pharmacol. 1989;164:273–283. doi: 10.1016/0014-2999(89)90468-8. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Martin LP, Anderson DR. GABAA receptor-mediated inhibition of rat substantia nigra dopaminergic neurons by pars reticulata projection neurons. J Neurosci. 1995;15:3092–3103. doi: 10.1523/JNEUROSCI.15-04-03092.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Sparta DR, Hopf FW, Bowers MS, Melis M, Bonci A. Strain specific synaptic modifications on ventral tegmental area dopamine neurons after ethanol exposure. Biol Psychiatry. 2009;65:646–653. doi: 10.1016/j.biopsych.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann DJ, Basse-Tomusk A, Wilson RL, Rebec GV, Wightman RM. Interference by DOPAC and ascorbate during attempts to measure drug-induced changes in neostriatal dopamine with Nafion-coated, carbon-fiber electrodes. J Neurosci Methods. 1990;35:9–18. doi: 10.1016/0165-0270(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Amatore C, Engstrom RC, Hale PD, Kristensen EW, Kuhr WG, May LJ. Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience. 1988;25:513–523. doi: 10.1016/0306-4522(88)90255-2. [DOI] [PubMed] [Google Scholar]
- Wu Q, Reith ME, Kuhar MJ, Carroll FI, Garris PA. Preferential increases in nucleus accumbens dopamine after systemic cocaine administration are caused by unique characteristics of dopamine neurotransmission. J Neurosci. 2001a;21:6338–6347. doi: 10.1523/JNEUROSCI.21-16-06338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Reith ME, Wightman RM, Kawagoe KT, Garris PA. Determination of release and uptake parameters from electrically evoked dopamine dynamics measured by real-time voltammetry. J Neurosci Methods. 2001b;112:119–133. doi: 10.1016/s0165-0270(01)00459-9. [DOI] [PubMed] [Google Scholar]
- Wu Y, Pearl SM, Zigmond MJ, Michael AC. Inhibitory Glutamatergic regulation of evoked dopamine release in striatum. Neurosci. 2000;96:65–72. doi: 10.1016/s0306-4522(99)00539-4. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Pehek EA. A neurochemical heterogeneity of the rat striatum as measured by in vivo electrochemistry and microdialysis. Brain Res. 1990;506:236–242. doi: 10.1016/0006-8993(90)91256-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.