

Abstract
Normal epithelial thyroid cells in culture are inhibited by TGF-β1. Instead, transformed thyroid cell lines are frequently resistant to its growth inhibitory effect. Loss of TGF-β responsiveness could be due to a reduced expression of TGF-β receptors, as shown in transformed rat thyroid cell lines and in human thyroid tumors, or to alterations of other genes controlling TGF-β signal transduction pathway. However, in thyroid neoplasia, a complex pattern of alterations occurring during transformation and progression has been identified. Functionally, TGF-β1 acts as a tumor suppressor in the early stage of transformation or as a tumor promoter in advanced cancer. This peculiar pleiotropic behaviour of TGF-β may result from cross-talk with signalling pathways mediated by other growth factors, among which EGF-like ligands play an important role. This paper reports evidences on TGF-β1 and EGF systems in thyroid tumors and on the cross-talk between these growth factors in thyroid cancer.
1. Introduction
Thyroid gland homeostasis is maintained through a fine regulation of thyrocyte growth and differentiation. This regulation occurs through complex interactions between thyroid-stimulating hormone (TSH) and other growth factors and cytokines [1]. Evidence supports the role of transforming growth factor-beta 1 (TGF-β1) and epidermal growth factor- (EGF-) like ligands in the regulation of thyroid proliferation and differentiation, given the numerous information focused on mechanism of signal transduction and cross-talk. It is emerging the concept that the basis behind the pleiotropic nature of TGF-β in the context of cell-type and tumorigenesis derived probably from differences in the mechanism of such cross-talk.
The TGF-β superfamily, which includes TGF-βs, activins, and bone morphogenetic proteins (BMPs), is a multifunctional dimeric proteins family that regulates growth, differentiation and extracellular matrix production in many different cell types [2]. In epithelial cells, TGF-β acts as a tumor suppressor by inhibiting cell growth or by regulating cellular differentiation or apoptosis.
EGF is the prototype of a large family of peptides that consists of about a dozen members and has an essential role in embryonic development as well as in inducing cell growth. Mutations in the kinase domain of EGFR/ErbB1 (epidermal growth factor receptor) and ErbB2 are responsible of ligand-independent activation of cytoplasmic signal transducers that regulate motility, adhesion, protection from apoptosis, and transformation [3].
TGF-β and EGF are physiological regulators of thyroid cell differentiation and proliferation. TGF-β is normally expressed and secreted by thyrocytes, acting as a potent inhibitor of thyroid cell growth [4]. EGF, instead, acts as a strong mitogen for follicular thyroid cells [5]. Any alterations of these two factors or their signalling pathways may play an important role in the stepwise transition towards malignancy, including the ability to become, at least partially, resistant to growth inhibition, to proliferate without dependence on growth factors, to replicate without limit, to invade, and to metastasize. TGF-β1 appears to have a dual effect in tumorigenesis. It can act as a tumor suppressor in the pretumor stage, and as a tumor promoter in late stage of tumorigenesis. It is likely that during tumorigenesis, as a result of genetic and/or epigenetic changes, the balance between those opposing functions of TGF-β1 changes resulting in a switch to tumor promotion; however, the precise mechanism for this switch remains to be clarified [6]. In this paper we focused our attention on the role of EGF and TGF-β in the control of proliferation and differentiation of thyroid cells.
2. Thyroid Cell Regulation by Growth Factors
Physiological regulation of thyroid cell growth and function involves a complex network of factors that act through endocrine, paracrine, or autocrine mechanisms. The proliferation and differentiation of thyroid epithelial cells are under the control of a positive systemic signal, TSH, and a negative locally produced signal, TGF-β. The main function of the thyroid is the formation, storage, and secretion of thyroid hormones tightly controlled by TSH, but requiring insulin/insulin-like growth factor I (IGF-I) [7]. The steps leading to the thyroid hormone formation include thyroglobulin (TG) synthesis and transport to the lumen of thyroid follicles, the iodide uptake by the sodium iodide symporter (NIS), iodination of TG, and coupling of TG iodotyrosine residues by the thyroperoxidase (TPO). In rat thyroid cells, the expression of the TG, TPO, NIS, and TSH receptor (TSHR) is under the control of thyroid-restricted transcription factors, such as thyroid transcription factor-1 (TTF-1) [8], that plays the most important role in the expression of all the genes, Pax-8, and TTF-2 [9, 10].
TSH, through the activation of its receptor, has been shown to stimulate more than one signal transduction pathway, the main of which being the adenylcyclase/cAMP (cyclic adenosine monophosphate) pathway. cAMP seems to account for the mitogenic effects of TSH in human thyroid cells, mediated by the activation of cAMP-dependent protein kinases [11]. TSH-induced cAMP also appears to play a central role in iodide uptake and metabolism in the dog follicular cells [12], in TG and TPO gene expression in rat follicular cells [13]. Although TSH is the major regulator of thyroid growth and functions, it has been shown that a number of growth factors affect the proliferation and function of thyroid epithelial cells. In fact, TSH effects can be potentiated by several growth factors such as insulin and IGF-I in rat thyroid cells in culture [14]. Insulin or IGF-I synergizes with TSH to induce thyroid cell growth and to maintain specialized cell functions [11]. There is evidence that TSHR and TG gene expression are regulated by insulin/IGF-I as well as TSH in a rat thyroid cell line (FRTL-5) [15, 16]. Moreover, an important regulator of thyroid growth that stimulates proliferation in vitro includes EGF.
3. Epidermal Growth Factor-Related Ligands and Their Receptors
EGF is the prototype of a large family of peptides structurally related by possessing an EGF-like domain that consists of 6 cysteine residues capable of forming three disulfide-bonded intramolecular loops. These ligands are expressed in the extracellular domain of transmembrane proteins and are generated by regulated proteolysis to yield growth factors that contain 49–85 amino acids. The components of the EGF-like growth factors family are functionally related on the basis of binding to the members of the tyrosine kinase ErbB family (EGFR/ErbB1, ErbB2, ErbB3, and ErbB4) and are divided into three groups: the first includes EGF, transforming growth factor-alpha (TGF-α) and amphiregulin, which all bind specifically to EGFR/ErbB1. A second group includes betacellulin, heparin-binding EGF (HB-EGF), and epiregulin binding to both EGFR/ErbB1 and ErbB4, while the third group comprises the neuregulin family, differentiated by their binding to ErbB3 and ErbB4 (NRG1 and NRG2) or only to ErbB4 (NRG3 and NRG4) [17]. All four human receptors share four extracellular domains with high structural homology, a single transmembrane spanning helix, and a cytoplasmic portion that contains a conserved but not equally functional tyrosine kinase domain. Only the EGFR/ErbB1 and ErbB4 are fully functional in terms of ligand binding and kinase activity. ErbB2 fails to bind any of the known ErbB ligands but contributes its potent kinase activity to all possible heterodimers. ErbB3 has an impaired kinase activity and relies on the kinase activity of its heterodimerization partners for activation. Heterodimers of ErbB2 and ErbB3 are the most potent ErbB pair in mitogenic signalling [17–19].
After ligand binding, ErbB receptors achieve activation by forming homo- or heterodimeric receptor complexes. The dimerization of ErbB receptors represents the most important mechanism that drives transformation. Dimeric receptors become catalytically active and are able to phosphorylate the cytoplasmic receptor domain that serves as docking site for a variety of signalling molecules whose recruitment leads to the activation of intracellular pathways controlling diverse genetic programs. The two major signalling pathways activated by ErbB receptors are the mitogen-activated protein kinases (MAPKs) pathway which stimulates proliferation, and Phosphatidylinositol 3-kinases/AKT (PI3K-AKT) pathway which promotes cell survival (Figure 1). The specific combination of ErbB receptors in the dimer defines the downstream signalling network as well as the intensity and the duration of the stimulation. Indeed, heterodimers that involve ErbB3 stimulates the activation of PI3K pathway [20].
Figure 1.
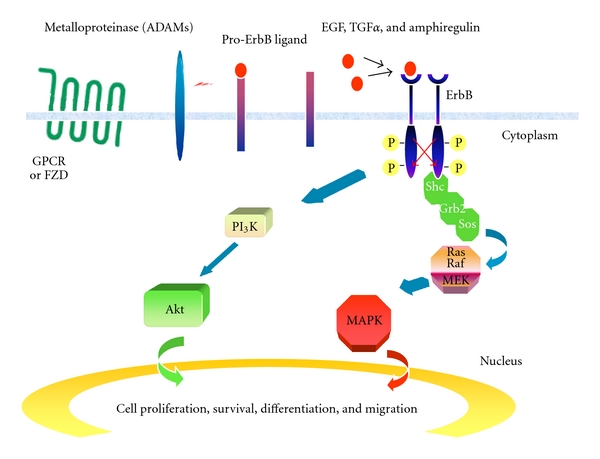
Mechanisms of action of ErbB receptor. In tumor cells, ErbB receptor tyrosine kinases are activated by autocrine or paracrine production of EGF family ligands. Autocrine ligand production results from the activation of G-protein coupled receptors (GPCRs) or frizzled (FZD) receptor which causes the metalloproteinases-mediated cleavage and release of pro-EGF-related ligands (ectodomain shedding). Binding of ligands to the extracellular domain of ErbB receptors leads to receptor dimerization, autophosphorylation, and activation of several downstream signalling pathways. In particular, tyrosine-phosphorylated ErbB receptors bind the adaptor proteins Shc and Grb2 leading to Sos recruitment and Ras/MAPK pathway activation. The PI3K/Akt pathway is stimulated through recruitment of the p85 adaptor subunit of PI3K to the receptor.
Amplification, overexpression and gene mutation of EGFR/ErbB1 and ErbB2 have been found in various human cancers [17, 21]. EGFR/ErbB1 is overexpressed in bladder, breast, head and neck, kidney, nonsmall cell lung, and prostate cancers [22]. Three truncated forms of the EGFR/ErbB1 have been described [23] among which the EGFRvIII (variant III) lacks the majority of the ectodomain and does not bind EGF. This variant is the most common in glioblastoma multiforme and also occurs in lung, breast, ovarian, and prostate cancer [24]. ErbB2 protein is overexpressed in breast cancer due to gene amplification in 15%–30% of invasive ductal breast cancers and overexpression correlates with poor prognosis and disease progression [25]. A number of studies have shown mutations in the kinase domain of EGFR/ErbB1 and ErbB2 [3, 21]. Intragenic somatic mutations in the ErbB2 gene were reported in 5% of nonsmall cell lung cancer, 5% of gastric carcinomas, 3% of colorectal carcinomas, and 5% of breast carcinomas [26–28]. Accumulating evidence has suggested that also ErbB3 plays a critical role in cancer. Overexpression of ErbB3 often accompanies EGFR/ErbB1 or ErbB2 overexpression and has been frequently detected in a variety of cancers, including those of the breast [29], colon [30], stomach [31], ovary [32], and pancreas [33]. In ErbB2-driven cancers, ErbB3 functions as an intimate signalling partner that promotes the transforming potency of ErbB2, usually by activating the PI3K/AKT pathway [34]. ErbB4 receptor is made in at least four different full-length isoforms as a consequence of alternative mRNA splicing [35]. It has both oncogenic and tumor suppressive functions. Supporting a role in promoting growth, overexpression of ErbB4 enhances growth of human breast cancer cells [36] and transforms mouse mammary epithelial cells to form tumors in vitro and in vivo [37]. Supporting a suppressive role for mammary tumor growth activation of ErbB4 in breast cancer cells has been associated with cell-cycle arrest, differentiation, and apoptosis in vitro [38].
4. Epidermal Growth Factor-Related Ligands and Their Receptors in Thyroid
EGF is synthesized by thyroid gland and is able to induce thyroid cell proliferation in several species together with the loss of thyroid specific functions [5]. Moreover, EGF enhances the migration and invasiveness of papillary thyroid cancer [5, 39–41]. Accordingly, in vitro growth was inhibited when either neutralizing anti-TGF-α or anti-EGFR/ErbB1 antibodies were applied to thyroid carcinoma cell lines [42]. A correlation between the staining intensity of EGF and recurrence has been found statistically significant in a set of human papillary thyroid tumors, indicating EGF as a predictor of papillary thyroid carcinoma aggressiveness [43]. Follicular epithelial thyroid cells expressed weakly TGF-α protein, whereas 66% of hyperplastic thyroid nodules, 100% of thyroid adenocarcinomas, and all cases of papillary, follicular, and medullary carcinomas displayed intense staining for TGF-α. A parallel pattern of staining was observed for the EGFR/ErbB1 in these tissues, suggesting the potential for an in vivo autocrine loop [44]. Papillary carcinomas and their lymph node metastases coexpressed EGFR/ErbB1 and EbB2 mRNA transcripts at a higher level than in normal thyroid tissues [45]. Moreover, in the same histotype of cancer, it has also been demonstrated the coexpression of TGF-α and EGFR/ErbB1 mRNA transcripts at higher levels than in non-neoplastic thyroid tissues [46].
Rat thyroid epithelial cells transformed with K-ras oncogene were found to express high levels of ErbB4 receptor and Neuregulin-1 (HRG/NDF/NRG1) ligand compared to rat thyroid epithelial control cells. Treatment of K-ras transformed thyroid cells with neutralizing antibody against NRG1 reduced by 50% cell proliferation, demonstrating the presence of an active NRG1 protein secreted in the supernatant by the cells. These data indicate that in K-ras rat thyroid epithelial cells, the growth factor NRG1 signals through the heterodimer ErbB2/ErbB4 receptors in an autocrine fashion [47]. Nevertheless, in human papillary carcinomas the protein overexpression and nuclear localization of the NRG1 precursor isoform compared to normal thyroid tissues was not associated with the expression of ErbB receptors, while the expression of neuregulin-β3 (Hrgβ3) was significantly correlated to ErbB3 protein expression, indicating this receptor as the cognate one [48].
EGFR/ErbB1 was higher expressed in anaplastic and papillary thyroid cancers than in normal thyroid tissues. In particular, Bergstrom et al. [49] have shown that EGFR/ErbB1 was expressed in all six anaplastic thyroid carcinomas examined and it was constitutive phosphorylated in 3 of the 6 cell lines tested. Song [50] has demonstrated that EGFR/ErbB1 was expressed in 90% of papillary carcinomas analyzed. Moreover, the role of EGFR/ErbB1 in stimulating the growth of thyroid tumors has been highlighted by the capability of Gefitinib, a small molecule inhibitor of EGFR/ErbB1, to reduce the growth of papillary, follicular, and anaplastic thyroid cancer cells [51, 52]. By immunohistochemical analyses, cytoplasmic immunopositivity of EGFR/ErbB1 was observed in papillary carcinomas, while no nuclear positivity for EGF and EGFR/ErbB1 was demonstrated. A study by Akslen et al. significantly associated cytoplasmatic EGFR/ErbB1 with the increased risk of recurrent tumor. Nuclear positivity for EGF and EGFR/ErbB1 was demonstrated to be a feature of both follicular adenomas and follicular carcinomas [53, 54].
The role of the ErbB2 proto-oncogene in thyroid carcinoma has been controversially discussed. It has been reported that high levels of ErbB2 mRNA expression correlated with lymph node metastasis in papillary carcinoma [45]. Studies by Utrilla et al. [55] and Haugen et al. [56] showed that there are marked differences in the pattern of ErbB2 immunoreactivity depending on the tumor type. The investigators demonstrated positivity for papillary carcinomas and negativity for follicular adenomas and follicular carcinomas, but they showed controversial results about medullary carcinomas. Moreover, ErbB2 was not detected in papillary carcinomas by immunohistochemistry [57]. Some studies investigated ErbB2 correlation with prognosis. Sugg et al. [58] found ErbB2 staining correlation with degree of differentiation, while Gumurdulu et al. [59] demonstrated that further investigations on ErbB2 role in thyroid tumors are required for determination of prognosis. Other studies have associated cytoplasmatic reactivity with patient's sex in tumor [60] or with development of metastasis [61].
Few studies analyzed all ErbB family members and their implication in thyroid tumors. Wiseman et al. [62] showed that EGFR/ErbB1, ErbB2, ErbB3, and ErbB4 were expressed in 76%, 2%, 57%, and 73% of differentiated thyroid carcinomas analyzed (90 cases of papillary and 6 cases of follicular carcinomas examined), respectively. Moreover, EGFR/ErbB1 and ErbB3 showed significantly increased expression, while ErbB4 showed significantly decreased expression in these tumors compared with benign thyroid lesions. ErbB3 expression correlated with the presence of lymph node metastases, tumor type, and higher N stage; the expression of ErbB4 correlated with lower T stage. Kato et al. [63] demonstrated that the transcription level of ErbB2 and ErbB3 genes was increased in papillary carcinomas compared to normal tissues, suggesting that the expression of an ErbB2-ErbB3 heterodimer may correlate with the aggressiveness of a thyroid tumor. Interestingly, coexisting protein overexpression of EGFR/ErbB1, ErbB2, ErbB3, and ErbB4 was demonstrated in 64% of papillary thyroid carcinomas providing numerous possibilities for functional receptor interactions [64].
5. TGF-β Effects on Thyroid Cell Physiology
TGF-β is synthesized as an inactive precursor that can be activated by different proteases produced by thyrocytes. Its expression is upregulated during TSH-induced thyroid hyperplasia in rats, suggesting that an increased local expression of TGF-β1, during thyroid hyperplasia, may contribute to the temporal stabilization of goiter size [65]. TGF-β signalling is propagated via cell surface serine/threonine kinases, TGF-β type I receptor (TβRI) and TGF-β type II receptor (TβRII). Both receptors are expressed on thyrocytes at equimolar amount [66] and, upon ligand binding by type II receptor, TβRI is recruited to an heteromeric complex and phosphorylated by TβRII, thus activating its serine/threonine kinase in order to phosphorylate the transcription factors R-Smads (Smad2 and Smad3). The phosphorylated Smad2 or Smad3 associate with the common partner Co-Smad, Smad4, forming complexes that accumulate in the nucleus, where they regulate target genes expression either by interacting with other transcription factors and coactivators or corepressors or by directly binding to defined sequences in the promoter [67]. In contrast to R-Smads and Co-Smad, the Inhibitory Smads (I-Smads), including Smad6 and Smad7, bind to TβRI and compete with R-Smads for activation by TβRI resulting in the inhibition of TGF-β signalling [68, 69]. A Smad ubiquitin regulatory factor 1 (Smurf1), being a HECT-type E3 (Homologous to the E6-AP C Terminus) ubiquitin ligase, interacts with inhibitory Smad7 and induces cytoplasmic localization of Smad7. Smurf1, then, associates with TβRI and enhances the turnover of this receptor [70] (Figure 2).
Figure 2.
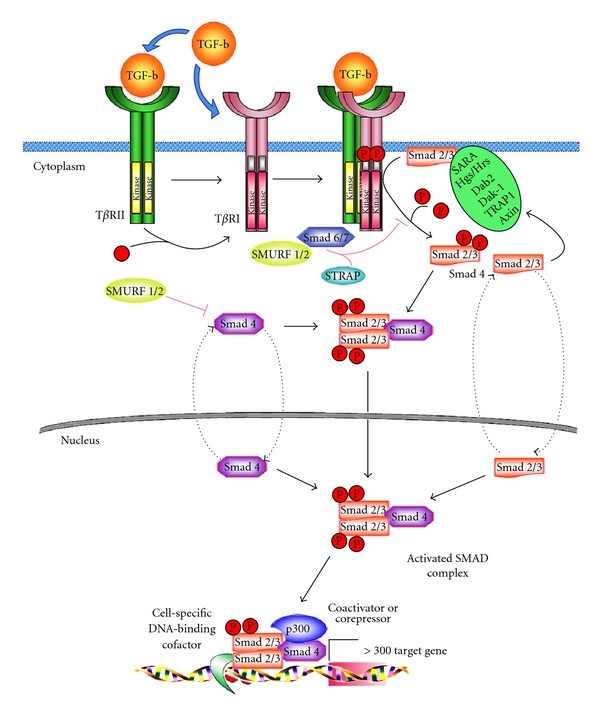
Intracellular signal transduction of TGF-β signalling. TGF-β ligand binds to TβRI and TβRII to constitute active heterodimers that possess serin-threonin kinase activity. Activated TβRI phosphorylates Smad2 and Smad3, which in turn form a complex with Smad4. Smad2 or Smad3/Smad4 complex translocates into the nucleus where interacts with other DNA-binding transcription factors, coactivators and corepressors, regulating the transcription of target genes.
TGF-β is the important negative regulator of thyrocyte: it antagonizes the mitogenic effects of the main growth factors in cultured cells of human [65, 71], dog [72], pig [73], and rat origin [4, 74]. TGF-β delays progression during the mid–late G1 phase by directly controlling cyclin D1-3 levels and preventing the relocalization of p27/kip1 inhibitor from cyclin E/cdk2 to cyclin D3/cdk4 complexes [72, 74]. TGF-β has been shown to downregulate the expression of thyroid-specific genes in the majority of species. The iodide trapping is inhibited in rat thyroid cells by blocking the protein kinase A (PKA) pathway, but not the PKA-induced DNA synthesis [75]. Moreover, we have demonstrated that the overexpression of ErbB2 in FRTL-5 cells, responsible of the resistance to the inhibitory action of TGF-β on cell proliferation, does not affect the inhibition of iodide uptake caused by TGF-β [66]. This effect was likely due in part to the inhibition of the expression of NIS mRNA and its protein [76, 77] and in part to the inhibition of the expression and activity of the Na+/I- ATPase, an enzyme that plays a key role in iodide uptake [78]. Similar responses have also been observed in porcine cell cultures, where TGF-β inhibits iodide uptake and metabolism, cAMP formation, and T4 release [41]. In human thyroid primary cultures, TGF-β inhibits most effects of cAMP on gene expression [79]. The rat thyroid cell lines represent a good model to study the TGF-β action in regulating the TG expression respect to porcine thyroid follicular cells where, TGF-β has no effect [80]. In the FRTL-5 rat thyroid epithelial cell line, the addition of TGF-β inhibits Pax8 mRNA causing a decreased formation of Pax8/DNA complexes, both with or without the addition of TSH that is responsible of the TGFβ1-induced suppression of TG gene expression [81]. In the same cells, it has been demonstrated that the inhibition of TG biosynthesis and TSHR expression by TGF-β1 could be counterbalanced by blocking the nuclear translocation of Smad2 and Smad4 [82]. Instead, Smad3 has a key role in the reduction of NIS expression, because its physical interaction with Pax8 in turn diminishes Pax8 binding to DNA sequence involved in the regulation of NIS [83].
In porcine thyroid cells cultured in suspension, TGF-β counteracts TSH positive effect on folliculogenesis causing an inversion of cell polarity [84]. The ability of TGF-β to modulate cytoskeleton organization and extracellular matrix protein distribution has been demonstrated not only in porcine thyroid cells, where it stimulates the expression of plasminogen activator inhibitor-1 (PAI-1), clusterin, trombospondin-1 [80, 85, 86], but also in rat thyroid cells [87]. Finally, in the same cellular model TGF-β1 inhibits the major histocompatibility complex (MHC) class I by regulating two elements at the transcription start site of the flanking region and increases the downstream regulatory element (DRE) binding of an ubiquitously expressed Y-box protein, termed TSEP-1 (TSHR suppressor element-binding Protein-1), an important suppressor of the TSHR and of MHC class I and class II expression [88]. TGF-β1 stimulates monocyte chemoattractant Protein-1 (MCP-1) and colony-stimulating factor (CSF) [89], as well as endothelin and its receptors in human thyroid follicular cells [90].
Therefore, it can be concluded that TGF-β exerts an important effect on thyroid cells in all the species tested, inhibiting the proliferation and function and modulating extracellular matrix (ECM) formation.
6. Role of TGF-β in Thyroid Cancer
The inhibition of cellular proliferation is one of the primary action of the TGF-β signalling. This factor is involved in the regulation of other numerous cellular functions as embryogenesis, differentiation, apoptosis, angiogenesis, immunosuppression, and wound-healing process [91]. Given the multifunctional role of TGF-β, any aberration of its normal signalling cascade may have wide-ranging pathologic consequences. Yet, paradoxically, TGF-β also modulates processes such as cell invasion and microenvironment modification that cancer cells may exploit to their advantage. Consequently, the output of a TGF-β response is highly contextual throughout development, across different tissues, and also in cancer [92].
Thyroid cancer incidence has significantly increased during the past decades [93], and it has become one of the ten leading cancer types in females being more frequent than ovarian, urinary, bladder, or pancreas cancer [94]. Although the majority of thyroid cancers have an excellent prognosis, there are a small percentage of cases that show an extensive local invasion and distant metastases, which frequently do not respond to standard treatments and have a worsened prognosis. The genetic basis for the initiation and development of the common type of thyroid cancer, papillary thyroid carcinoma (PTC), is well characterized. It has been demonstrated that the activation of oncogenes like RAS, BRAF, RET/PTC, and PI3K/AKT plays an important role in thyroid tumorigenesis [95]. However, it is also interesting to underscore the differences among the tumors arising from the different mutations. Studies in vitro and in vivo have clearly shown that other oncoproteins, like EGF and TGF-β, exert their own oncogenic drive, conferring a distinct biological behaviour on thyroid tumors. The homoeostasis of growth in differentiated epithelia reflects a critical balance between the promotion and suppression of cell division. The thyroid gland is among the most common sites of epithelial hyperplasia, affecting up to 15% of the adult population, typically presenting as “sporadic” or multinodular nontoxic Goiter (MNTG), the hyperplastic gland usually contains well-defined nodules of varying size, surrounded by a normal epithelium. A wide range of studies has revealed evidence for an involvement of several autocrine growth stimulators and their receptors in the progression of MNTG. Prominent among these is TGF-β, and a lack of response to TGF-β inhibitory action in thyroid cells may be responsible for some cases of MNTG in human cells [71]. In rats, thyroid hyperplasia induced by iodide deficiency and goitrogen is accompanied by an increase in TGF-β1 expression and the arrest of goiter growth after 4 weeks. This surprising result is thought to reflect a critical role of TGF-β1 in stabilizing goiter mass [65, 96]. This would suggest that the increase in TGF-β1 levels observed during goiter induction might be a mechanism to counteract the goitrogenic effect of endogenous TSH [65, 97]. Instead, in a group of patients with either papillary adenomas (n = 14) or carcinomas (n = 14), not significant changes in blood levels of TGF-β1 have been observed compared to normal controls [98].
Increased expression of TGF-β, NFkB (nuclear factor of κB), and CDC42, compared to the normal thyroid tissue, has been demonstrated in a group of human papillary thyroid carcinomas, analyzed by oligonucleotide microarray of microscopically dissected intratumoral samples from central and invasive regions. These data together with reduced levels of mRNAs encoding proteins involved in cell-cell adhesion and communication and an overexpression of vimentin strongly support the hypothesis that the TGF-β, responsible of epithelial mesenchymal transition (EMT) induction, increases the tumor invasiveness in papillary thyroid carcinomas [95].
Perturbations of TGF-β signalling are central to tumorigenesis and tumor progression. TβRII is commonly inactivated through mutation and loss of heterozygosity (LOH) in several types of carcinoma [99]. Lazzereschi et al. [100], have obtained similar results in a series of human thyroid tumors, from benign lesions (adenomas) to neoplastic lesions of increasing aggressiveness (papillary and follicular carcinomas) up to the extremely aggressive anaplastic tumors. Northern blot analyses demonstrated a statistically significant reduction (about 2-3-fold less) of TβRII expression in papillary thyroid carcinomas in comparison with the respective healthy tissues. Immunostaining of the formalin-fixed sections with specific anti-TβRII antibodies substantiates these data, clearly demonstrating that the greatest reduction in TβRII immunoreactivity was found in the highly malignant, undifferentiated anaplastic thyroid tumors. A comparative study performed in different types of epithelial thyroid carcinomas of patients from different regions of the world demonstrated a strong decrease of TβRII expression in follicular cancer of patients from China, Japan, and USA (50%, 55%, and 90% respectively). In papillary thyroid carcinoma TβRII was decreased in 75%, 77% and 96% of patients from Japan, China, and USA, respectively. Finally, in undifferentiated cancer, the reduction of TβRII expression was observable in 83% of patients from USA and in 100% of patients from China [101].
Inactivating mutations in Smad2 and 4 are frequently found in some cancers [102]. In a group of 20 follicular thyroid neoplasms, classified as 11 adenomas and 9 minimally invasive follicular carcinomas, according to current pathological criteria, Smad2 expression investigated by immunohistochemistry has been lost, while TβRII expression was lost in 78%. These data indicate that the downregulation of TβRII remains the major consistent abnormality in thyroid carcinomas that may be used to differentiate minimally invasive carcinomas from adenomas, while the downregulation of Smad2 could be another mechanism by which carcinomas become independent from TGFβ–mediated growth inhibition [103].
The tumor suppressor role of TβRII has been demonstrated in vitro in a model of K-ras transformed rat thyroid cells, where the overexpression of TβRII induces not only a partial reversion of malignant phenotype restoring the sensitivity to TGF-β, but also a significant reduction in spontaneous and lung artificial metastases when transplanted in athymic nude mice [104]. In this model, the overexpression of TβRII is essential to reduce the invasive potential and to modulate the adhesive and migratory cell behaviours by controlling the integrin functions rather than integrin receptor expression [105]. The inhibitory action of TGF-β1 on cellular migration, invasion, and adhesion is present in a set of human PTC and follicular thyroid carcinoma (FTC) cell lines, while inhibition of TGFβ-induced cell growth is maintained only in FTC cell lines [106]. Other authors demonstrated that in human papillary thyroid carcinoma cell line TPC-1, these effects are probably due to the lower level of Smads 2, 3, and 4 associated to an increase in Smad7 expression [107]. Otherwise, the altered expression of TGF-β pathway proteins not always is responsible of resistance to the TGF-β action. A human anaplastic carcinoma cell line, despite the activity of receptors signalling, Smad2 phosphorylation and nuclear translocation of Smad2/4 complexes, is strongly resistant to the TGF-β action, suggesting that other signalling mechanisms might be related to the escape from TGF-β sensitivity [108].
Sensitivity to TGF-β is impaired in thyroid tumors and escape from TGF-β action is actively selected during thyroid tumor development. Destruction of the TGF-β signalling at the level of Smad genes is common in human carcinomas, the deficiencies of Smad4 have been hypothesized to underlie TGF-β resistance of tumor cells and to strongly accelerate the malignant progression of neoplastic lesions initiated by other oncogenic stimuli [92]. Lazzereschi et al. [109] showed, for the first time, the mutational and the expression status of Smad4 in a consistent number of thyroid tumors of different histotypes, demonstrating the high frequency of Smad4 abnormalities (27%) in thyroid tumors, comparable only with Smad4 mutation frequency in tumors arising from the gastrointestinal tract, both sporadic and inherited. The high frequency of alterations in Smad4 sequence led the authors to propose that these changes may constitute a nearly and frequent event in thyroid tumors natural history. Smad4 inactivation in tumors is generally a late event linked to progression to overt carcinoma. In human papillary thyroid cell lines, TPC-1 and BCPAP, it has been recently demonstrated a strong reduction in the level of SMAD4 protein, which is responsible for an alteration of TGF-β signalling and for some of the TGF-β-mediated biological effects. The overexpression of Smad4, restoring TGF-β signal transduction, determines a significant increase of antiproliferative response to TGF-β, reduces the invasive behaviour of these cells as well as is responsible of a significant increase of E-cadherin expression, indicating that the level of SMAD4 is a critical regulator of these processes. To remark the important role of SMAD4 in thyroid carcinogenesis contributes also the finding obtained by immunohistochemistry that 7 out of 23 (30%) PTC tumor samples, including 1 case of follicular variant of PTCs, present a weak and focal intensity of SMAD4 staining compared to normal tissue from the opposite lobe [110].
The stability of TβRI represents an important regulatory mechanism for TGF-β signalling both in cell culture studies and in vivo models. TGF-β receptors are ubiquitinated and degraded through the action of several cooperating protein complexes containing E3 ligases as well as other important regulators of protein degradation. The I-Smads regulate many of these complexes, orchestrating both ubiquitination and de-ubiquitination [111]. The levels of Smurf1 and Smad7 are overexpressed in the anaplastic thyroid carcinoma cell line [112], and an increase of SMAD7 expression has been found in a group of papillary and follicular carcinomas with respect to benign pathologies, indicating SMAD7 as another SMAD involved in thyroid tumorigenesis [113].
It is known that TGF-β's role in human cancer appears both complex and context depended. Depending on the tumor type and the stage of tumor progression, it can exercite strong tumor suppressive or tumor-promoting functions. More recently, it has been demonstrated that also in thyroid cells, as well as in the skin tumors, or in metastatic colon cancer [114], TGF-β can acts as tumor-promoting factor. The expression of BRAFV660E, in normal rat thyrocytes and in fifty cases of human PTC determines a reduction of NIS expression and an increase of TGF-β secretion, suggesting an hyperactivation of TGF-β signalling, responsible of the pro-tumorigenic activity [115].
The aberrant microRNA (miR) expression, involved in the cell growth suppressive function, has been demonstrated in a large number of follicular thyroid neoplasias [116]. More recently, a new important function of TGF-β, involving the regulation of expression of miR levels (miR-200 and miR-30), has been discovered in human anaplastic thyroid carcinoma (ATC). ATCs represent a more aggressive type of thyroid cancer arising from mesenchymal de-/transdifferentiation of epithelial thyroid cells that rapidly invade the adjacent tissue. The main function of miR-200 and miR-30 is to negatively regulate the EMT process in follicular cells. The low levels of miR-200 and miR-30 in ATCs respect to that observed in thyroid normal tissues, in PTCs or in FTCs, strongly suggest that the invasive potential of ATC is due to enhancement of EMT process. In addition, it has been demonstrated that the reduction of miR-200 and miR-30 in these carcinomas is caused by a strong activation of TGF-β signalling due to an upregulation of TβRI and Smad2. Therefore, the authors not only propose a novel molecular panel to identify ATCs, but also they suggest the inhibition of TGF-β signalling represent a new likely approach for the treatment of these carcinomas [117].
7. Growth Factors Cross-Talk in Thyroid: Role of TGF-β and EGF Systems in the Regulation of Thyroid Growth
Complex and apparently redundant interactions between hormones and growth factors regulate thyroid cell proliferation and differentiation. However, information about the cross-talk between different growth factors that regulate thyroid cell growth is limited. Ariga et al. [118] studied the signalling pathway through which the synergistic actions between IGF-I and TSH are mediated in FRTL-5 cells. Also, TGF-β1 and IGF-I appear to interact and have opposite effects on the growth of rat thyroid cells [75]. In particular, TGF-β1 attenuates IGF-I-stimulated MAPK phosphorylation through inhibition of IRS-1 (insulin receptor substrate-1) tyrosine phosphorylation, IRS-1/Grb2/Sos complex formation and CrkII tyrosine phosphorylation thus leading to the suppression of FRTL-5 cell growth [119]. ErbB ligands and ErbB receptors provide a complex and multilayered network of signalling that is deregulated in many human tumors. However, several are the causes and mechanisms of uncontrolled signalling by ErbB receptors suggesting that differences exist within the ErbB family in the mechanism of regulation but also in the cross-talk with other growth factors.
TGF-β is involved in two opposing activities: it is able to function as a growth inhibitor at early stages of carcinogenesis and as a growth promoter at later stages of neoplasia when tumor cells that have developed the capability to bypass the tumor inhibitor function of TGF-β, paradoxically, may use it for tumor progression by means of multiple mechanisms. Overexpression of TGF-β ligands has been reported in most cancers [120] and correlates with markers of a more metastatic phenotype and/or a poor patient outcome. This dual role of TGF-β is believed to result from molecular cross-talk with a complex network of signalling pathways involving either direct effects on tumor cells or paracrine effects on other cells [121]. Several reports provided evidence that TGF-β can collaborate with EGF/ErbB receptors system. Indeed, it has been shown that Smads proteins may cross-talk with mitogenic growth-factor signalling through receptor tyrosine kinase-induced MEK/MAPK protein kinases both in a synergistic (MEK1-induced Smad2 phosphorylation) [122] or an antagonistic interplay (MAPK-induced Smad1 phosphorylation) [123, 124]. In tumors, ErbB receptors and their ligands promote growth and confer apoptosis resistance, thus overcoming TGF-β1 growth inhibition and apoptotic effect. Indeed, in rat hepatocytes an autocrine loop of TGF-β in cells that have undergone EMT, induces an upregulation of EGFR/ErbB1 ligands by promoting their shedding through the activation of ADAM17 (a disintegrin and metalloprotease 17) and thus allowing some cells to escape from TGFβ-induced pro-apoptotic effect [125, 126]. A further mechanistic insight on the conversion of the function of TGF-β from tumor suppressor to tumor promoter has been provided by the evidence that the EGF signalling pathway may enhance TGF-β responses. EGF increases the stability of TβRII thus preventing full loss of TβRII expression in late stage of cancers, and thereby permits some of the direct oncogenic behaviour of TGF-β during tumor progression [127]. Uttamsingh et al. [128] showed that in conjunction with EGF, TGF-β1 helps to augment migration, invasion and anchorage-independent growth of intestinal epithelial cells, in agreement with reports indicating that activation of TGF-β signalling promotes pulmonary metastasis of mammary tumors in neu transgenic mice [129]. Moreover, the elevated and prolonged activation of ERK/MAPK and its requirement for EGF and TGFβ1-induced EMT and migration/invasion of intestinal cells is in agreement with that between ErbB2 and TGF-β1 in mammary epithelial cells [130]. In fibroblasts, TGF-β induces the upregulation of ErbBs ligands and activation of cognate receptors via the canonical Smad pathway, thus allowing the induction of fibroblast cell morphologic transformation and anchorage-independent growth [131].
Contrasting to this plethora of data on cross-talk between EGF and TGF-β1 signals, there are very few results in the literature relating the interconnection between EGF and TGF-β signalling pathways in thyroid cancer cells.
Rat thyroid epithelial cells overexpressing the ErbB2 proto-oncogene are not transformed in vitro but no longer depend on TSH for cell growth and become resistant to the growth inhibitory effects of TGF-β1, thus suggesting that ErbB2 proto-oncogene, when overexpressed, is able to interfere and cross-talk with growth factors that control in a positive and negative manner the thyroid cell proliferation [132]. Using collagen gel-cultured porcine follicles, Nilsson et al. [133] demonstrated that the morphoregulatory effects of EGF are highly influenced by TGF-β1. In particular, TGF-β1 inhibits EGF-induced thyrocytes proliferation, but it synergizes with EGF in the stimulation of cell migration. This latter effect could be due to the known role of TGF-β1 to regulate the cell-matrix interactions, by stimulating the synthesis of extracellular matrix components, inhibiting proteases and inducing changes of integrin synthesis. However, contrasting to these results, TGF-β1 inhibited both EGF-induced mitogenesis and motogenesis in rabbit corneal epithelial cells [134] indicating that the modulation by TGF-β1 of EGF responses differs among epithelial cell types.
Moreover, in a follicular thyroid cancer cell line lacking endogenous TSHR, EGF, and TGF-β have been shown to enhance VEGF (vascular endothelial growth factor) secretion. Since the loss of the TSHR is characteristic of anaplastic thyroid cancer, which usually exhibits significantly increased VEGF expression and a high degree of angiogenesis compared with differentiated thyroid cancer, the finding of VEGF stimulation by EGF and TGF-β, highlights the important role of these growth factors in thyroid tumor progression and aggressiveness [135]. Preliminary data from our laboratory demonstrate that cotreatment with EGF and TGF-β1 results in opposite effects in human thyroid cancer cell lines. Indeed, TGF-β1 inhibits EGF-mediated migration in invasion/wound healing assay, while a synergistic effect between TGF-β1 and EGF is observed in anchorage-independent growth assay. These findings demonstrate that cell invasion and anchorage-independent growth capability involve different factors and molecular mechanisms (Mincione G. et al., unpublished results).
8. Conclusions
The findings described in this paper support the hypothesis that a network formed by the EGF/ErbBs system and TGF-β pathway is involved in the pathogenesis and progression of thyroid tumors. Further understanding of the complexity of cross-talk between these pathways in thyroid disease related to gain of function of ErbB, inactivation of growth suppression function, or activation of tumor promoter activity of TGF-β1 will offer a broader spectrum of points of intervention and will lead to continued advances in thyroid cancer treatment.
References
- 1.Vassart G, Dumont JE. The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocrine Reviews. 1992;13(3):596–611. doi: 10.1210/edrv-13-3-596. [DOI] [PubMed] [Google Scholar]
- 2.Kingsley DM. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes and Development. 1994;8(2):133–146. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- 3.Wang SE, Narasanna A, Perez-Torres M, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10(1):25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 4.Colletta G, Cirafici AM, Di Carlo A. Dual effect of transforming growth factor beta on rat thyroid cells: inhibition of thyrotropin-induced proliferation and reduction of thyroid-specific differentiation markers. Cancer Research. 1989;49(13):3457–3462. [PubMed] [Google Scholar]
- 5.Asmis LM, Gerber H, Kaempf J, Studer H. Epidermal growth factor stimulates cell proliferation and inhibits iodide uptake of FRTL-5 cells in vitro. Journal of Endocrinology. 1995;145(3):513–520. doi: 10.1677/joe.0.1450513. [DOI] [PubMed] [Google Scholar]
- 6.Massaguè J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103(2):295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 7.Dumont JE, Maenhaut C, Lamy F. Control of thyroid cell proliferation and goitrogenesis. Trends in Endocrinology and Metabolism. 1992;3(1):12–17. doi: 10.1016/1043-2760(92)90086-g. [DOI] [PubMed] [Google Scholar]
- 8.Musti AM, Ursini VM, Avvedimento EV, Zimarino V, Lauro RD. A cell type specific factor recognizes the rat thyroglobulin promoter. Nucleic Acids Research. 1987;15(20):8149–8166. doi: 10.1093/nar/15.20.8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zannini M, Avantaggiato V, Biffali E, et al. TTF-2, a new forkhead protein, shows a temporal expression in the developing thyroid which is consistent with a role in controlling the onset of differentiation. EMBO Journal. 1997;16(11):3185–3197. doi: 10.1093/emboj/16.11.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zannini M, Francis-Lang H, Plachov D, Di Lauro R. Pax-8, a paired domain-containing protein, binds to a sequence overlapping the recognition site of a homeodomain and activates transcription from two thyroid-specific promoters. Molecular and Cellular Biology. 1992;12(9):4230–4241. doi: 10.1128/mcb.12.9.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roger PP, Taton M, Van Sande J, Dumont JE. Mitogenic effects of thyrotropin and adenosine 3’,5’-monophosphate in differentiated normal human thyroid cells in vitro. Journal of Clinical Endocrinology and Metabolism. 1988;66(6):1158–1165. doi: 10.1210/jcem-66-6-1158. [DOI] [PubMed] [Google Scholar]
- 12.Filetti S, Rapoport B. Autoregulation by iodine of thyroid protein synthesis: influence of iodine on amino acid transport in cultured thyroid cells. Endocrinology. 1984;114(4):1379–1385. doi: 10.1210/endo-114-4-1379. [DOI] [PubMed] [Google Scholar]
- 13.Van Heuverswyn B, Leriche A, Van Sande J, Dumont JE, Vassart G. Transcriptional control of thyroglobulin gene expression by cyclic AMP. FEBS Letters. 1985;188(2):192–196. doi: 10.1016/0014-5793(85)80370-7. [DOI] [PubMed] [Google Scholar]
- 14.Tramontano D, Cushing GW, Moses AC, Ingbar SH. Insulin-like growth factor-I stimulates the growth of rat thyroid cells in culture and synergizes the stimulation of DNA synthesis induced by TSH and Graves’-IgG. Endocrinology. 1986;119(2):940–942. doi: 10.1210/endo-119-2-940. [DOI] [PubMed] [Google Scholar]
- 15.Santisteban P, Kohn LD, Di Lauro R. Thyroglobulin gene expression is regulated by insulin and insulin-like growth factor I, as well as thyrotropin, in FRTL-5 thyroid cells. Journal of Biological Chemistry. 1987;262(9):4048–4052. [PubMed] [Google Scholar]
- 16.Saji M, Akamizu T, Sanchez M, et al. Regulation of thyrotropin receptor gene expression in rat FRTL-5 thyroid cells. Endocrinology. 1992;130(1):520–533. doi: 10.1210/endo.130.1.1309347. [DOI] [PubMed] [Google Scholar]
- 17.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2001;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 18.Pinkas-Kramarski R, Soussan L, Waterman H, et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO Journal. 1996;15(10):2452–2467. [PMC free article] [PubMed] [Google Scholar]
- 19.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO Journal. 1997;16(7):1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alimandi M, Romano A, Curia MC, et al. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10(9):1813–1821. [PubMed] [Google Scholar]
- 21.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nature Reviews Cancer. 2007;7(3):169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 22.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Critical Reviews in Oncology/Hematology. 1995;19(3):183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 23.Zandi R, Larsen AB, Andersen P, Stockhausen M-T, Poulsen HS. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cellular Signalling. 2007;19(10):2013–2023. doi: 10.1016/j.cellsig.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 24.Moscatello DK, Holgado-Madruga M, Godwin AK, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Research. 1995;55(23):5536–5539. [PubMed] [Google Scholar]
- 25.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 26.Lee JW, Soung YH, Seo SH, et al. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clinical Cancer Research. 2006;12(1):57–61. doi: 10.1158/1078-0432.CCR-05-0976. [DOI] [PubMed] [Google Scholar]
- 27.Stephens P, Hunter C, Bignell G, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431(7008):525–526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 28.Shigematsu H, Takahashi T, Nomura M, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Research. 2005;65(5):1642–1646. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 29.Naidu R, Yadav M, Nair S, Kutty MK. Expression of c-erbB3 protein in primary breast carcinomas. British Journal of Cancer. 1998;78(10):1385–1390. doi: 10.1038/bjc.1998.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maurer CA, Friess H, Kretschmann B, et al. Increased expression of erbB3 in colorectal cancer is associated with concomitant increase in the level of erbB2. Human Pathology. 1998;29(8):771–777. doi: 10.1016/s0046-8177(98)90444-0. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi M, Iwamatsu A, Shinohara-Kanda A, Ihara S, Fukui Y. Activation of ErbB3-PI3-kinase pathway is correlated with malignant phenotypes of adenocarcinomas. Oncogene. 2003;22(9):1294–1301. doi: 10.1038/sj.onc.1206256. [DOI] [PubMed] [Google Scholar]
- 32.Rajkumar T, Stamp GWH, Hughes CM, Gullick WJ. c-erbB3 protein expression in ovarian cancer. Journal of Clinical Pathology. 1996;49(4):M199–M202. doi: 10.1136/mp.49.4.m199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friess H, Wang L, Zhu Z, et al. Growth factor receptors are differentially expressed in cancers of the papilla of vater and pancreas. Annals of Surgery. 1999;230(6):767–775. doi: 10.1097/00000658-199912000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, III, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(15):8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Junttila TT, Sundvall M, Määttä JA, Elenius K. ErbB4 and its isoforms: selective regulation of growth factor responses by naturally occurring receptor variants. Trends in Cardiovascular Medicine. 2000;10(7):304–310. doi: 10.1016/s1050-1738(01)00065-2. [DOI] [PubMed] [Google Scholar]
- 36.Määttä JA, Sundvall M, Junttila TT, et al. Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Molecular Biology of the Cell. 2006;17(1):67–79. doi: 10.1091/mbc.E05-05-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch CC, Vargo-Gogola T, Martin MD, Fingleton B, Crawford HC, Matrisian LM. Matrix metalloproteinase 7 mediates mammary epithelial cell tumorigenesis through the ErbB4 receptor. Cancer Research. 2007;67(14):6760–6767. doi: 10.1158/0008-5472.CAN-07-0026. [DOI] [PubMed] [Google Scholar]
- 38.Muraoka-Cook RS, Caskey LS, Sandahl MA, et al. Heregulin-dependent delay in mitotic progression requires HER4 and BRCA1. Molecular and Cellular Biology. 2006;26(17):6412–6424. doi: 10.1128/MCB.01950-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoelting T, Siperstein AE, Clark OH, Duh Q-Y. Epidermal growth factor enhances proliferation, migration, and invasion of follicular and papillary thyroid cancer in vitro and in vivo. Journal of Clinical Endocrinology and Metabolism. 1994;79(2):401–408. doi: 10.1210/jcem.79.2.8045955. [DOI] [PubMed] [Google Scholar]
- 40.van der Laan BF, Freeman JL, Asa SL. Expression of growth factors and growth factor receptors in normal and tumorous human thyroid tissues. Thyroid. 1995;5(1):67–73. doi: 10.1089/thy.1995.5.67. [DOI] [PubMed] [Google Scholar]
- 41.Bechtner G, Schopohl D, Rafferzeder M, Gärtner R, Welsch U. Stimulation of thyroid cell proliferation by epidermal growth factor is different from cell growth induced by thyrotropin or insulin-like growth factor I. European Journal of Endocrinology. 1996;134(5):639–648. doi: 10.1530/eje.0.1340639. [DOI] [PubMed] [Google Scholar]
- 42.Gabler B, Aicher T, Heiss P, Senekowitsch-Schmidtke R. Growth inhibition of human papillary thyroid carcinoma cells and multicellular spheroids by anti-EGF-receptor antibody. Anticancer Research. 1997;17(4):3157–3159. [PubMed] [Google Scholar]
- 43.Mizukami Y, Nonomura A, Hashimoto T, et al. Immunohistochemical demonstration of epidermal growth factor and c-myc oncogene product in normal, benign and malignant thyroid tissues. Histopathology. 1991;18(1):11–18. doi: 10.1111/j.1365-2559.1991.tb00808.x. [DOI] [PubMed] [Google Scholar]
- 44.Gorgoulis V, Aninos D, Priftis C, et al. Expression of epidermal growth factor, transforming growth factor-alpha and epidermal growth factor receptor in thyroid tumors. In Vivo. 1992;6(3):291–296. [PubMed] [Google Scholar]
- 45.Aasland R, Lillehaug JR, Male R, Josendal O, Varhaug JE, Kleppe K. Expression of oncogenes in thyroid tumours: coexpression of c-erbB2/neu and c-erbB. British Journal of Cancer. 1988;57(4):358–363. doi: 10.1038/bjc.1988.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haugen DR, Akslen LA, Verhaug JE, Lillehaug JR. Demonstration of a TGF-alpha-EGF-receptor autocrine loop and c-myc protein over-expression in papillary thyroid carcinomas. International Journal of Cancer. 1993;55(1):37–43. doi: 10.1002/ijc.2910550108. [DOI] [PubMed] [Google Scholar]
- 47.Mincione G, Piccirelli A, Lazzereschi D, Salomon DS, Colletta G. Heregulin-dependent autocrine loop regulates growth of K-ras but not erbB-2 transformed rat thyroid epithelial cells. Journal of Cellular Physiology. 1998;176(2):383–391. doi: 10.1002/(SICI)1097-4652(199808)176:2<383::AID-JCP17>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 48.Fluge O, Akslen LA, Haugen DRF, Varhaug JE, Lillehaug JR. Expression of heregulins and associations with the ErbB family of tyrosine kinase receptors in papillary thyroid carcinomas. International Journal of Cancer. 2000;87(6):763–770. doi: 10.1002/1097-0215(20000915)87:6<763::aid-ijc1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 49.Bergstrom JD, Westermark B, Heldin N-E. Epidermal growth factor receptor signaling activates met in human anaplastic thyroid carcinoma cells. Experimental Cell Research. 2000;259(1):293–299. doi: 10.1006/excr.2000.4967. [DOI] [PubMed] [Google Scholar]
- 50.Song B. Immunohistochemical demonstration of epidermal growth factor receptor and ceruloplasmin in thyroid diseases. Acta Pathologica Japonica. 1991;41(5):336–343. doi: 10.1111/j.1440-1827.1991.tb01656.x. [DOI] [PubMed] [Google Scholar]
- 51.Schiff BA, McMurphy AB, Jasser SA, et al. Epidermal growth factor receptor (EGFR) is overexpressed in anaplastic thyroid cancer, and the EGFR inhibitor gefitinib inhibits the growth of anaplastic thyroid cancer. Clinical Cancer Research. 2004;10(24):8594–8602. doi: 10.1158/1078-0432.CCR-04-0690. [DOI] [PubMed] [Google Scholar]
- 52.Hoffmann S, Gläser S, Wunderlich A, et al. Targeting the EGF/VEGF-R system by tyrosine-kinase inhibitors-a novel antiproliferative/antiangiogenic strategy in thyroid cancer. Langenbeck’s Archives of Surgery. 2006;391(6):589–596. doi: 10.1007/s00423-006-0104-y. [DOI] [PubMed] [Google Scholar]
- 53.Akslen LA, Myking AO, Salvesen H, Varhaug JE. Prognostic impact of EGF-receptor in papillary thyroid carcinoma. British Journal of Cancer. 1993;68(4):808–812. doi: 10.1038/bjc.1993.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marti U, Ruchti C, Kämpf J, et al. Nuclear localization of epidermal growth factor and epidermal growth factor receptors in human thyroid tissues. Thyroid. 2001;11(2):137–145. doi: 10.1089/105072501300042785. [DOI] [PubMed] [Google Scholar]
- 55.Utrilla JC, Martin-Lacave I, San Martin MV, Fernandex-Santos JM, Galera-Davidson H. Expression of c-erbB-2 oncoprotein in human thyroid tumours. Histopathology. 1999;34(1):60–65. doi: 10.1046/j.1365-2559.1999.00563.x. [DOI] [PubMed] [Google Scholar]
- 56.Haugen DRF, Akslen LA, Varhaug JE, Lillehaug JR. Expression of c-erbB-2 protein in papillary thyroid carcinomas. British Journal of Cancer. 1992;65(6):832–837. doi: 10.1038/bjc.1992.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murakawa T, Tsuda H, Tanimoto T, Tanabe T, Kitahara S, Matsubara O. Expression of KIT, EGFR, HER-2 and tyrosine phosphorylation in undifferentiated thyroid carcinoma: implication for a new therapeutic approach. Pathology International. 2005;55(12):757–765. doi: 10.1111/j.1440-1827.2005.01902.x. [DOI] [PubMed] [Google Scholar]
- 58.Sugg SL, Ezzat S, Zheng L, Rosen IB, Freeman JL, Asa SL. Cytoplasmic staining of erbB-2 but not mRNA levels correlates with differentiation in human thyroid neoplasia. Clinical Endocrinology. 1998;49(5):629–637. doi: 10.1046/j.1365-2265.1998.00580.x. [DOI] [PubMed] [Google Scholar]
- 59.Gumurdulu D, Uguz A, Erdogan S, Tuncer I, Demircan O. Expression of c-erbB-2 oncoprotein in different types of thyroid tumors: an immunohistochemical study. Endocrine Research. 2003;29(4):465–472. doi: 10.1081/erc-120026952. [DOI] [PubMed] [Google Scholar]
- 60.Akslen LA, Varhaug JE. Oncoproteins and tumor progression in papillary thyroid carcinoma: presence of epidermal growth factor receptor, c-erbB-2 protein, estrogen receptor related protein, p21-ras protein, and proliferation indicators in relation to tumor recurrences and patient survival. Cancer. 1995;76(9):1643–1654. doi: 10.1002/1097-0142(19951101)76:9<1643::aid-cncr2820760922>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 61.Kremser R, Obrist P, Spizzo G, et al. Her2/neu overexpression in differentiated thyroid carcinomas predicts metastatic disease. Virchows Archiv. 2003;442(4):322–328. doi: 10.1007/s00428-003-0769-3. [DOI] [PubMed] [Google Scholar]
- 62.Wiseman SM, Griffith OL, Melck A, et al. Evaluation of type 1 growth factor receptor family expression in benign and malignant thyroid lesions. The American Journal of Surgery. 2008;195(5):667–673. doi: 10.1016/j.amjsurg.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 63.Kato S, Kobayashi T, Yamada K, et al. Expression of erbB receptors mRNA in thyroid tissues. Biochimica et Biophysica Acta. 2004;1673(3):194–200. doi: 10.1016/j.bbagen.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 64.Haugen DRF, Akslen LA, Varhaug JE, Lillehaug JR. Expression of c-erbB-3 and c-erbB-4 proteins in papillary thyroid carcinomas. Cancer Research. 1996;56(6):1184–1188. [PubMed] [Google Scholar]
- 65.Logan A, Smith C, Becks GP, Gonzalez AM, Phillips ID, Hill DJ. Enhanced expression of transforming growth factor-β1 during thyroid hyperplasia in rats. Journal of Endocrinology. 1994;141(1):45–57. doi: 10.1677/joe.0.1410045. [DOI] [PubMed] [Google Scholar]
- 66.Coppa A, Mincione G, Mammarella S, Ranieri A, Colletta G. Epithelial rat thyroid cell clones, escaping from transforming growth factor beta negative growth control, are still inhibited by this factor in the ability to trap iodide. Cell Growth and Differentiation. 1995;6(3):281–290. [PubMed] [Google Scholar]
- 67.Attisano L, Wrana JL. Smads as transcriptional co-modulators. Current Opinion in Cell Biology. 2000;12(2):235–243. doi: 10.1016/s0955-0674(99)00081-2. [DOI] [PubMed] [Google Scholar]
- 68.Imamura T, Takase M, Nishihara A, et al. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389(6651):622–626. doi: 10.1038/39355. [DOI] [PubMed] [Google Scholar]
- 69.Nakao A, Afrakhte M, Morén A, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGFbeta signalling. Nature. 1997;389(6651):631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki C, Murakami G, Fukuchi M, et al. Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane. Journal of Biological Chemistry. 2002;277(42):39919–39925. doi: 10.1074/jbc.M201901200. [DOI] [PubMed] [Google Scholar]
- 71.Grubeck-Loebenstein B, Buchan G, Sadeghi R, et al. Transforming growth factor beta regulates thyroid growth. Role in the pathogenesis of nontoxic goiter. Journal of Clinical Investigation. 1989;83(3):764–770. doi: 10.1172/JCI113955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Depoortere F, Pirson I, Bartek J, Dumont JE, Roger PP. Transforming growth factor beta(1) selectively inhibits the cyclic AMP-dependent proliferation of primary thyroid epithelial cells by preventing the association of cyclin D3-cdk4 with nuclear p27(kip1) Molecular Biology of the Cell. 2000;11(3):1061–1076. doi: 10.1091/mbc.11.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Franzén A, Piek E, Westermark B, ten Dijke P, Heldin N-E. Expression of transforming growth factor-beta1, activin A, and their receptors in thyroid follicle cells: negative regulation of thyrocyte growth and function. Endocrinology. 1999;140(9):4300–4310. doi: 10.1210/endo.140.9.6961. [DOI] [PubMed] [Google Scholar]
- 74.Carneiro C, Alvarez CV, Zalvide J, Vidal A, Domínguez F. TGF-beta1 actions on FRTL-5 cells provide a model for the physiological regulation of thyroid growth. Oncogene. 1998;16(11):1455–1465. doi: 10.1038/sj.onc.1201662. [DOI] [PubMed] [Google Scholar]
- 75.Pang X-P, Park M, Hershman JM. Transforming growth factor-beta blocks protein kinase-A-mediated iodide transport and protein kinase-C-mediated DNA synthesis in FRTL-5 rat thyroid cells. Endocrinology. 1992;131(1):45–50. doi: 10.1210/endo.131.1.1612026. [DOI] [PubMed] [Google Scholar]
- 76.Kawaguchi A, Ikeda M, Endo T, Kogai T, Miyazaki A, Onaya T. Transforming growth factor-beta1 suppresses thyrotropin-induced Na+/I− symporter messenger RNA and protein levels in FRTL-5 rat thyroid cells. Thyroid. 1997;7(5):789–794. doi: 10.1089/thy.1997.7.789. [DOI] [PubMed] [Google Scholar]
- 77.Pekary AE, Hershman JM. Tumor necrosis factor, ceramide, transforming growth factor-beta1, and aging reduce Na+/I− symporter messenger ribonucleic acid levels in FRTL-5 cells. Endocrinology. 1998;139(2):703–712. doi: 10.1210/endo.139.2.5760. [DOI] [PubMed] [Google Scholar]
- 78.Pekary AE, Levin SR, Johnson DG, Berg L, Hershman JM. Tumor necrosis factor-alpha (TNF-alpha) and transforming growth factor-beta 1 (TGF-beta 1) inhibit the expression and activity of Na + /K( + )-ATPase in FRTL-5 rat thyroid cells. Journal of Interferon and Cytokine Research. 1997;17(4):185–195. doi: 10.1089/jir.1997.17.185. [DOI] [PubMed] [Google Scholar]
- 79.Taton M, Lamy F, Roger PP, Dumont JE. General inhibition by transforming growth factor beta 1 of thyrotropin and cAMP responses in human thyroid cells in primary culture. Molecular and Cellular Endocrinology. 1993;95(1-2):13–21. doi: 10.1016/0303-7207(93)90024-e. [DOI] [PubMed] [Google Scholar]
- 80.Claisse D, Martiny I, Chaqour B, et al. Influence of transforming growth factor beta1 (TGF-beta1) on the behaviour of porcine thyroid epithelial cells in primary culture through thrombospondin-1 synthesis. Molecular and Cellular Endocrinology. 1999;112, part 9:1405–1416. doi: 10.1242/jcs.112.9.1405. [DOI] [PubMed] [Google Scholar]
- 81.Kang H-C, Ohmori M, Harii N, Endo T, Onaya T. Pax-8 is essential for regulation of the thyroglobulin gene by transforming growth factor-beta1. Endocrinology. 2001;142(1):267–275. doi: 10.1210/endo.142.1.7918. [DOI] [PubMed] [Google Scholar]
- 82.Nicolussi A, D’Inzeo S, Santulli M, Colletta G, Coppa A. TGF-beta control of rat thyroid follicular cells differentiation. Molecular and Cellular Endocrinology. 2003;207(1-2):1–11. doi: 10.1016/s0303-7207(03)00238-7. [DOI] [PubMed] [Google Scholar]
- 83.Costamagna E, García B, Santisteban P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. Journal of Biological Chemistry. 2004;279(5):3439–3446. doi: 10.1074/jbc.M307138200. [DOI] [PubMed] [Google Scholar]
- 84.Delorme N, Remond C, Sartelet H, et al. TGFbeta1 effects on functional activity of porcine thyroid cells cultured in suspension. Journal of Endocrinology. 2002;173(2):345–355. doi: 10.1677/joe.0.1730345. [DOI] [PubMed] [Google Scholar]
- 85.Kotlarz G, Wegrowski Y, Martiny L, Declerck PJ, Bellon G. Enhanced expression of plasminogen activator inhibitor-1 by dedifferentiated thyrocytes. Biochemical and Biophysical Research Communications. 2002;295(3):737–743. doi: 10.1016/s0006-291x(02)00712-x. [DOI] [PubMed] [Google Scholar]
- 86.Wegrowski Y, Perreau C, Martiny L, Haye B, Maquart FX, Bellon G. Transforming growth factor beta-1 up-regulates clusterin synthesis in thyroid epithelial cells. Experimental Cell Research. 1999;247(2):475–483. doi: 10.1006/excr.1998.4378. [DOI] [PubMed] [Google Scholar]
- 87.Garbi C, Colletta G, Cirafici AM, Marchisio PC, Nitsch L. Transforming growth factor-beta induces cytoskeleton and extracellular matrix modifications in FRTL-5 thyroid epithelial cells. European Journal of Cell Biology. 1990;53(2):281–289. [PubMed] [Google Scholar]
- 88.Napolitano G, Montani V, Giuliani C, et al. Transforming growth factor-beta1 down-regulation of major histocompatibility complex class I in thyrocytes: coordinate regulation of two separate elements by thyroid-specific as well as ubiquitous transcription factors. Molecular Endocrinology. 2000;14(4):486–505. doi: 10.1210/mend.14.4.0454. [DOI] [PubMed] [Google Scholar]
- 89.Matsumura M, Banba N, Motohashi S, Hattori Y. Interleukin-6 and transforming growth factor-beta regulate the expression of monocyte chemoattractant protein-1 and colony-stimulating factors in human thyroid follicular cells. Life Sciences. 1999;65(12):PL129–PL135. doi: 10.1016/s0024-3205(99)00368-9. [DOI] [PubMed] [Google Scholar]
- 90.Tseng Y-CL, Lahiri S, Jackson S, Burman KD, Wartofsky L. Endothelin binding to receptors and endothelin production by human thyroid follicular cells: effects of transforming growth factor-beta and thyrotropin. Journal of Clinical Endocrinology and Metabolism. 1993;76(1):156–161. doi: 10.1210/jcem.76.1.8421082. [DOI] [PubMed] [Google Scholar]
- 91.Truty MJ, Urrutia R. Basics of TGF-beta and pancreatic cancer. Pancreatology. 2007;7(5-6):423–435. doi: 10.1159/000108959. [DOI] [PubMed] [Google Scholar]
- 92.Massagué J. TGFβ in cancer. Cell. 2008;134(2):215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. Journal of the American Medical Association. 2006;295(18):2164–2167. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- 94.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA: A Cancer Journal for Clinicians. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 95.Vasko V, Espinosa AV, Scouten W, et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(8):2803–2808. doi: 10.1073/pnas.0610733104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bidey SP, Hill DJ, Eggo MC. Growth factors and goitrogenesis. Journal of Endocrinology. 1999;160(3):321–332. doi: 10.1677/joe.0.1600321. [DOI] [PubMed] [Google Scholar]
- 97.Pisarev MA, Thomasz L, Juvenal GJ. Role of transforming growth factor beta in the regulation of thyroid function and growth. Thyroid. 2009;19(8):881–892. doi: 10.1089/thy.2007.0303. [DOI] [PubMed] [Google Scholar]
- 98.Vesely D, Astl J, Lastuvka P, Matucha P, Sterzl I, Betka J. Serum levels of IGF-I, HGF, TGFbeta1, bFGF and VEGF in thyroid gland tumors. Physiological Research. 2004;53(1):83–89. [PubMed] [Google Scholar]
- 99.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine and Growth Factor Reviews. 2006;17(1-2):41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 100.Lazzereschi D, Ranieri A, Mincione G, Taccogna S, Nardi F, Colletta G. Human malignant thyroid tumors displayed reduced levels of transforming growth factor beta receptor type II messenger RNA and protein. Cancer Research. 1997;57(10):2071–2076. [PubMed] [Google Scholar]
- 101.Imamura Y, Jin L, Grande JP, et al. Analysis of TGF-β and TGF-β-RII in thyroid neoplasms from the United States, Japan, and China. Endocrine Pathology. 1998;9(3):209–216. doi: 10.1007/BF02739960. [DOI] [PubMed] [Google Scholar]
- 102.Seoane J. Escaping from the TGFbeta anti-proliferative control. Carcinogenesis. 2006;27(11):2148–2156. doi: 10.1093/carcin/bgl068. [DOI] [PubMed] [Google Scholar]
- 103.West J, Munoz-Antonia T, Johnson JG, Klotch D, Muro-Cacho CA. Transforming growth factor-beta type II receptor and Smad proteins in follicular thyroid tumors. Laryngoscope. 2000;110(8):1323–1327. doi: 10.1097/00005537-200008000-00019. [DOI] [PubMed] [Google Scholar]
- 104.Turco A, Coppa A, Aloe S, et al. Overexpression of transforming growth factor beta-type II receptor reduces tumorigenicity and metastastic potential of K-ras-transformed thyroid cells. International Journal of Cancer. 1999;80(1):85–91. doi: 10.1002/(sici)1097-0215(19990105)80:1<85::aid-ijc17>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 105.Nicolussi A, D’Inzeo S, Gismondi A, Coppa A. Reduction of invasive potential in K-ras-transformed thyroid cells by restoring of TGF-β pathway. Clinical and Experimental Metastasis. 2006;23(5-6):237–248. doi: 10.1007/s10585-006-9023-0. [DOI] [PubMed] [Google Scholar]
- 106.Hölting T, Zielke A, Siperstein AE, Clark OH, Duh Q-Y. Transforming growth factor-beta 1 is a negative regulator for differentiated thyroid cancer: studies of growth, migration, invasion, and adhesion of cultured follicular and papillary thyroid cancer cell lines. Journal of Clinical Endocrinology and Metabolism. 1994;79(3):806–813. doi: 10.1210/jcem.79.3.8077365. [DOI] [PubMed] [Google Scholar]
- 107.Matsuo SE, Leoni SG, Colquhoun A, Kimura ET. Transforming growth factor-beta1 and activin A generate antiproliferative signaling in thyroid cancer cells. Journal of Endocrinology. 2006;190(1):141–150. doi: 10.1677/joe.1.06713. [DOI] [PubMed] [Google Scholar]
- 108.Heldin NE, Bergström D, Hermansson A, et al. Lack of responsiveness to TGF-beta1 in a thyroid carcinoma cell line with functional type I and type II TGF-beta receptors and Smad proteins, suggests a novel mechanism for TGF-beta insensitivity in carcinoma cells. Molecular and Cellular Endocrinology. 1999;153(1-2):79–90. doi: 10.1016/s0303-7207(99)00086-6. [DOI] [PubMed] [Google Scholar]
- 109.Lazzereschi D, Nardi F, Turco A, et al. A complex pattern of mutations and abnormal splicing of Smad4 is present in thyroid tumours. Oncogene. 2005;24(34):5344–5354. doi: 10.1038/sj.onc.1208603. [DOI] [PubMed] [Google Scholar]
- 110.D’Inzeo S, Nicolussi A, Ricci A, et al. Role of reduced expression of SMAD4 in papillary thyroid carcinoma. Journal of Molecular Endocrinology. 2010;45(4):229–244. doi: 10.1677/JME-10-0044. [DOI] [PubMed] [Google Scholar]
- 111.Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136(22):3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 112.Cerutti JM, Ebina KN, Matsuo SE, Martins L, Maciel RMB, Kimura ET. Expression of Smad4 and Smad7 in human thyroid follicular carcinoma cell lines. Journal of Endocrinological Investigation. 2003;26(6):516–521. doi: 10.1007/BF03345213. [DOI] [PubMed] [Google Scholar]
- 113.Matsuo SE, Fiore APZ, Siguematu SM, et al. Expression of SMAD proteins, TGF-beta/activin signaling mediators, in human thyroid tissues. Arquivos Brasileiros de Endocrinologia & Metabologia. 2010;54(4):406–412. doi: 10.1590/s0004-27302010000400010. [DOI] [PubMed] [Google Scholar]
- 114.Padua D, Massagué J. Roles of TGFbeta in metastasis. Cell Research. 2009;19(1):89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 115.Riesco-Eizaguirre G, Rodríguez I, De La Vieja A, et al. The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Research. 2009;69(21):8317–8325. doi: 10.1158/0008-5472.CAN-09-1248. [DOI] [PubMed] [Google Scholar]
- 116.Visone R, Pallante P, Vecchione A, et al. Specific microRNAs are downregulated in human thyroid anaplastic carcinomas. Oncogene. 2007;26(54):7590–7595. doi: 10.1038/sj.onc.1210564. [DOI] [PubMed] [Google Scholar]
- 117.Braun J, Hoang-Vu C, Dralle H, Hüttelmaier S. Downregulation of microRNAs directs the EMT and invasive potential of anaplastic thyroid carcinomas. Oncogene. 2010;29(29):4237–4244. doi: 10.1038/onc.2010.169. [DOI] [PubMed] [Google Scholar]
- 118.Ariga M, Nedachi T, Akahori M, et al. Signalling pathways of insulin-like growth factor-I that are augmented by cAMP in FRTL-5 cells. Biochemical Journal. 2000;348, part 2:409–416. [PMC free article] [PubMed] [Google Scholar]
- 119.Mincione G, Esposito DL, Di Marcantonio MC, Piccirelli A, Cama A, Colletta G. TGF-β1 modulation of IGF-I signaling pathway in rat thyroid epithelial cells. Experimental Cell Research. 2003;287(2):411–423. doi: 10.1016/s0014-4827(03)00155-1. [DOI] [PubMed] [Google Scholar]
- 120.Roberts AB, Wakefield LM. The two faces of transforming growth factor β in carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(15):8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Torre-Amione G, Beauchamp RD, Koeppen H, et al. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(4):1486–1490. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.De Caestecker MP, Parks WT, Frank CJ, et al. Smad2 transduces common signals from receptor serinethreonine and tyrosine kinases. Genes and Development. 1998;12(11):1587–1592. doi: 10.1101/gad.12.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kretzschmar M, Doody J, Massagué J. Opposing BMP and EGF signalling pathways converge on the TGF-β family mediator Smad1. Nature. 1997;389(6651):618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- 124.Kretzschmar M, Doody J, Timokhina I, Massagué J. A mechanism of repression of TGFfβ/Smad signaling by oncogenic Ras. Genes and Development. 1999;13(7):804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murillo MM, del Castillo G, Sánchez A, Fernández M, Fabregat I. Involvement of EGF receptor and c-Src in the survival signals induced by TGF-β1 in hepatocytes. Oncogene. 2005;24(28):4580–4587. doi: 10.1038/sj.onc.1208664. [DOI] [PubMed] [Google Scholar]
- 126.del Castillo G, Murillo MM, Alvarez-Barrientos A, et al. Autocrine production of TGF-β confers resistance to apoptosis after an epithelial-mesenchymal transition process in hepatocytes: role of EGF receptor ligands. Experimental Cell Research. 2006;312(15):2860–2871. doi: 10.1016/j.yexcr.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 127.Song K, Krebs TL, Danielpour D. Novel permissive role of epidermal growth factor in transforming growth factor β (TGF-β) signaling and growth suppression. Mediation by stabilization of TGF-β receptor type II. Journal of Biological Chemistry. 2006;281(12):7765–7774. doi: 10.1074/jbc.M511781200. [DOI] [PubMed] [Google Scholar]
- 128.Uttamsingh S, Bao X, Nguyen KT. Synergistic effect between EGF and TGF-β1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene. 2008;27(18):2626–2634. doi: 10.1038/sj.onc.1210915. [DOI] [PubMed] [Google Scholar]
- 129.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massagué J. Transforming growth factor β signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Seton-Rogers SE, Brugge JS. ErbB2 and TGF-beta: a cooperative role in mammary tumor progression? Cell Cycle. 2004;3(5):597–600. [PubMed] [Google Scholar]
- 131.Andrianifahanana M, Wilkes MC, Repellin CE, et al. ERBB receptor activation is required for profibrotic responses to transforming growth factor β. Cancer Research. 2010;70(19):7421–7430. doi: 10.1158/0008-5472.CAN-10-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mincione G, Cirafici M, Lazzereschi D, Pepe S, Ciardiello F, Colletta G. Loss of thyrotropin regulation and transforming growth factor beta-induced growth arrest in erbB-2 overexpressing rat thyroid cells. Cancer Research. 1993;53(22):5548–5553. [PubMed] [Google Scholar]
- 133.Nilsson M, Dahlman T, Westermark B, Westermark K. Transforming growth factor-β promotes epidermal growth factor-induced thyroid cell migration and follicle neoformation in collagen gel separable from cell proliferation. Experimental Cell Research. 1995;220(2):257–265. doi: 10.1006/excr.1995.1314. [DOI] [PubMed] [Google Scholar]
- 134.Mishima H, Nakamura M, Murakami J, Nishida T, Otori T. Transforming growth factor-beta modulates effects of epidermal growth factor on corneal epithelial cells. Current Eye Research. 1992;11(7):691–696. doi: 10.3109/02713689209000742. [DOI] [PubMed] [Google Scholar]
- 135.Hoffmann S, Hofbauer LC, Scharrenbach V, et al. Thyrotropin (TSH)-induced production of vascular endothelial growth factor in thyroid cancer cells in vitro: evaluation of TSH signal transduction and of angiogenesis-stimulating growth factors. Journal of Clinical Endocrinology & Metabolism. 2004;89(12):6139–6145. doi: 10.1210/jc.2004-1260. [DOI] [PubMed] [Google Scholar]