

Abstract
Intratumoural hypoxia (low oxygen tension) is associated with aggressive disease and poor prognosis. Hypoxia-inducible factor-1 is a transcription factor activated by hypoxia that regulates the expression of genes that promote tumour cell survival, progression, metastasis, and resistance to chemo/radiotherapy. In addition to hypoxia, HIF-1 can be activated by growth factor-signalling pathways such as the mitogen-activated protein kinases- (MAPK-) and phosphatidylinositol-3-OH kinases- (PI3K-) signalling cascades. Mutations in these pathways are common in thyroid carcinoma and lead to enhanced HIF-1 expression and activity. Here, we summarise current data that highlights the potential role of both hypoxia and MAPK/PI3K-induced HIF-1 signalling in thyroid carcinoma progression, metastatic characteristics, and the potential role of HIF-1 in thyroid carcinoma response to radiotherapy. Direct or indirect targeting of HIF-1 using an MAPK or PI3K inhibitor in combination with radiotherapy may be a new potential therapeutic target to improve the therapeutic response of thyroid carcinoma to radiotherapy and reduce metastatic burden.
1. Introduction
The hypoxia-inducible factors (HIFs) are transcription factors that function under low oxygen tensions (hypoxia) and are, therefore, active in a number of diseases associated with low oxygen (O2) environments. These include ischemic disorders, atherosclerosis, and importantly cancer. HIF drives the survival and development of cancer cells by activating and repressing a multitude of genes that promote tumour cell survival, proliferation, invasion, and disease progression. As a result, hypoxia and HIF are associated with poor prognosis in many tumour types [1–3]. Hypoxia occurs in the majority of solid tumours, thus functional HIF is present in most tumour types indicating the importance of this signalling pathway in cancer. There is little known, however, about the role of HIF in thyroid carcinoma. Here, we summarise current literature that supports the potential significance of the HIF signalling pathway in progression and aggressiveness of thyroid carcinoma. Current data proposes that the HIF pathway may be a novel therapeutic target in reducing local tumour growth, metastatic burden, and resistance to chemo/radiotherapy.
2. Oxygen-Dependent Regulation of HIF-1
There are three known isoforms of HIF: HIF-1, 2, and 3. HIF-1 is expressed in all cells and is the most extensively researched, whereas the expression of the other isoforms is restricted to certain tissues. HIF-1 is a heterodimeric protein consisting of a constitutively expressed HIF-1β (also known as the ary lhydrocarbon receptor nuclear translocator; ARNT) subunit and an oxygen-labile HIF-1α subunit. Under conditions of low oxygen, HIF-1α is stabilised, heterodimerises with HIF-1β through the Per-ARNT-Sim (PAS) A and PAS B domains, and translocates to the nucleus. The complex then binds to the hypoxia-responsive element (HRE; consensus sequence G/ACGTG), in the promoter region of target genes via the basic helix-loop-helix (bHLH) DNA-binding domain and activates transcription. This process involves binding of the coactivators CREB-binding protein (CBP) and p300 [4, 5].
Under normoxia, HIF-1α is hydroxylated on proline residues 402 and/or 564 in the oxygen-dependent degradation domain (ODD). This process is carried out by specific oxygen-dependent enzymes known as proline hydroxylase domain proteins (PHDs). There are 3 PHDs: 1, 2, and 3. PHD2 is specifically involved in the hydroxylation of HIF-1α. The PHDs use O2 and 2-oxoglutarate (2-OG) as substrates. Upon hydroxylation, von Hippel-Lindau (VHL), a tumour-suppressor protein, binds HIF-1α and recruits the E3 ubiquitin ligase, leading to ubiquitination and proteosomal degradation of HIF-1α (Figure 1). Activation of asparaginyl hydroxylases such as factor inhibiting HIF-1 (FIH-1) represents an additional oxygen-dependent mechanism of inhibition of HIF-1α activity. FIH-1 hydroxylates asparagine-803 in the C-terminal transcriptional activation domain (C-TAD) of HIF-1α. This modification inhibits the interaction of C-TAD with the transcriptional co-activators CBP/p300, and thus inhibits the transcriptional activity of HIF-1 (Figure 1). Under hypoxia, the level of HIF-1α hydroxylation is reduced via inhibition of the PHD2 enzyme, resulting in stabilisation and accumulation of HIF-1α protein [4, 5]. An additional oxygen-sensitive mechanism of HIF-1 regulation is the generation of reactive oxygen species (ROS) from mitochondria. ROS inactivate PHD2 resulting in direct stabilisation of HIF-1α [6].
Figure 1.

Structure of HIF-1α and the oxygen-dependent regulation of HIF-1α protein stabilisation and activation: The N-terminal regions contain the basic helix-loop-helix (bHLH) domain involved in DNA-binding and the Per-Arnt-Sim (PAS) A and B domains required for heterodimerisation with HIF-1β. The oxygen-dependent degradation domain (ODD) is where PHD2 hydroxylates proline residues P402 and P562, which enables binding of VHL and proteosomal degradation. The terminal transcriptional activation domains (TADs) are responsible for transactivation of target genes. The N-TAD is located towards the N-terminus, with the C-TAD located at the extreme C-terminus. Factor inhibiting HIF-1 (FIH-1) hydroxylates asparagine H803 in the C-TAD, preventing the binding of coactivators CBP/p300, thus inhibiting activation of HIF-1α. Both hydroxylation processes use 2-oxoglutarate (2-OG) and O2 as substrates and produce succinate and carbon dioxide (CO2) as byproducts.
HIF-2α is likewise regulated by oxygen-dependent hydroxylation and dimerises with HIF-1β to form the functional HIF-2 complex [4, 7]. Both isoforms are similar in structure and function but have differences, particularly in the N-TADs [8]. This suggests that both isoforms may differ in the activation of target genes and the recruitment of coactivators. There are also fewer HIF-2α-regulated genes compared to HIF-1α. For example, in MCF-7 breast carcinoma cells, 80% of hypoxia-regulated genes were dependent on HIF-1α. A small group were dependent on HIF-2α, and the regulation of these genes was due to the interaction of HIF-2α with the transcription factor Elk-1 [9]. This interaction with Elk-1 is unique to HIF-2α.
Although HIF-1α and -2α show some overlap of target genes, the proteins do have distinct downstream targets. HIF-1 predominantly regulates the expression of genes encoding glycolytic proteins such as lactate dehydrogenase-A (LDH-A) and carbonic anhydrase-9 (CA-9), whereas in certain tissues expressing both HIF-1 and -2α, expression of genes such as vascular endothelial growth factor (VEGF) and erythropoietin (EPO) is mainly regulated by HIF-2α [7, 10, 11]. In support of this, high expression of HIF-2α but not -1α has been found in well-vascularised areas of neuroblastoma and is associated with aggressiveness [12]. Although HIF-1α is accepted as the most important of the HIFs, there is increasing evidence suggesting that HIF-2α may be of equal significance. The expression of HIF-2α is both tissue and cell-type specific, and the regulation of target genes differs depending on tissue type, tumour type, and coexpression with HIF-1α. A better understanding of how these factors lead to cell-specific differences in HIF-dependent gene regulation may help in the development of more effective therapeutics for diseases highly dependent on hypoxia. The HIF-3α isoform is also hypoxia regulated in a HIF-1-dependent manner and is an inhibitor of HIF-1 function [13].
3. Oxygen Independent Mechanisms of HIF Activation
3.1. Mutations in VHL
VHL targets all the HIF-αs for rapid proteosomal degradation and, as a result, plays a central role in molecular oxygen sensing [14]. Studies into the phenotype of VHL knockout (KO) mice provide direct evidence for the physiological relevance of the HIF pathway and VHL-regulated expression of HIF-1α in both normal foetal development and cancer progression. Genetic knock-out of VHL in the murine germ line results in embryonic lethality in mid-gestation due to abnormal vasculature formation thought to be HIF dependent [15]. In certain hereditary cancers such as clear cell renal carcinomas, Chuvash polycythemia, pheochromocytoma of the central nervous system or hemangioblastoma of the retina, normoxic degradation of HIF are impaired due to mutations in VHL. These tumours are usually well vascularised, a characteristic largely attributed to deregulated HIF-1α signalling [16, 17].
3.2. Metabolic Signalling Pathways
Metabolic signalling pathways such as the tricarboxylic acid (TCA) cycle are also necessary for normal regulation of HIF-1α. In addition to O2, PHDs require the substrates 2-OG and ascorbic acid as a cofactor, to catalyze hydroxylation of HIF-1α. This reaction produces succinate and CO2 as byproducts. 2-OG is a metabolite of the TCA cycle. As 2-OG is required for the functionality of PHDs, activity of the TCA cycle regulates PHD activity and thus HIF-1α stability. Furthermore, inactivating mutations in enzymes of the TCA cycle leads to direct stabilisation of HIF-1α protein [18, 19]. Mutations in fumarate hydratase (FH) and succinate dehydrogenase (SDH) lead to accumulation of fumarate and succinate. Both fumarate and succinate inhibit PHDs by competing with 2-OG for binding to the active site [20, 21]. Inactivating mutations in SDH have been found in a range of cancers associated with enhanced HIF-1 and VEGF signalling. These include renal cell carcinomas, gastrointestinal stromal (GIST) tumours, and Carney's syndrome [22, 23].
3.3. Growth Factor Signalling Pathways
For thyroid cancer the most important mutational changes occur in the MAPK/Ras—extracellular signal-regulated kinase (ERK) and the PI3K/AKT signalling cascades [24]. These pathways are dominant regulators of many cellular processes including growth, metabolism, cell survival, and angiogenesis and are important in the coordinated interaction of cells with the microenvironment [25, 26]. Both the MAPK and PI3K signalling pathways increase HIF-1 signalling in many cancer types including thyroid [27–30].
3.4. HIF-1 and the MAPK Pathway
A number of MAPK (p38 and p38γ) and ERK1/2 (p42 and p44) isoforms have been found to regulate HIF-1α activity in a cell-specific manner. MAPK signalling enhances the transactivation of HIF-1α by phosphorylation of HIF-1α co-activators and by direct phosphorylation of HIF-1α itself. MAPK stimulation leads to the phosphorylation of p300. This phosphorylation facilitates binding of p300/CBP to the C-TAD and promotes the transactivation activity of both p300/CBP and HIF-1α. Studies suggest that MAPK does not phosphorylate FIH, and therefore, this signalling cascade is not involved in the oxygen-dependent regulation of HIF-1α [31]. Thus, even though MAPK signalling does not depend on sensing oxygen tension, activation of this signalling cascade augments the HIF response.
Studies have shown that MAPK can directly phosphorylate the TADs of HIF-1α. Sang et al. found that MAPK signalling leads to indirect phosphorylation of C-TAD constructs by MAPK and direct phosphorylation of N-TAD constructs, when expressed in Escherichia coli. However, this direct phosphorylation of the TADs was not necessary for binding of p300 and activation of HIF-1 [31]. Other studies suggest that direct phosphorylation of HIF-1α by MAPKs has functional consequences on HIF-1 activity. In HeLa and CCL39 cells, p42/44 MAPK (but not p38 MAPK and JNK) increased the transcriptional activity of HIF-1 by phosphorylation of HIF-1α and not by increasing the level of HIF-1α protein. This was blocked by the MEK inhibitor PD098059 [32]. Conversely, in Hep3B and HEK293 cells, activation of the small GTPase Rac1 by hypoxia leads to Rac1-dependent p38 MAPK signalling resulting in both enhanced phosphorylation of the N-TAD and transcriptional activity of HIF-1α [33, 34]. Another study, however, has shown that Rac1 reduces HIF-1 activity by activating NAPDH oxidases resulting in enhanced generation of ROS leading to reduced HIF-1 in both normoxia and hypoxia [35]. The effect of Rac1 on MAPK signalling was not shown in this study. Furthermore, the latter group used 8% O2 for hypoxia, whereas the first study used 5 and 1% O2 for hypoxia. These functional differences observed by Rac1 may, therefore, be dependent upon O2 concentration. Furthermore, different cell lines were used in these studies suggesting that these differences in Rac1 signalling may be dependent on the activating mutation status of the MAPK pathway in different cell lines. Studies in HeLa cells have also shown that direct phosphorylation of HIF-1α by p42/44 MAPKs not only leads to enhanced HIF-1 transcriptional activity, but also promotes nuclear accumulation of HIF-1α [36]. Thus, depending on the cell type, the MAPK signalling cascade can enhance HIF-1 activity via activation of selective kinases which either directly phosphorylate HIF-1α or co-activators of HIF-1α (Figure 2).
Figure 2.

Regulation of HIF-1α by the MAPK pathway. Stimulation of receptor tyrosine kinases (RTKs) by growth factors, or activating mutations in the RAS family of GTPases, such as KRAS, results in activation of a range of MAPK-signalling proteins. Activation of ERK1/2 by MEK1/2 increases HIF-1 signalling (a) by phosphorylating and promoting the interaction of the coactivator p300 with HIF-1α and (b) by phosphorylating and activating the translational regulatory protein p70S6K and by phosphorylating and disrupting the repressive interaction of 4E-BP1 with eIF-4E. Activation of these translational machinery proteins leads to increased protein synthesis of HIF-1α. Activation of MAPKs (p38/p38γ) leads to the direct phosphorylation and activation of HIF-1α. Additionally, under hypoxia, activation of the small GTPase Rac1 leads to a Rac1 dependent increase in HIF-1α activity via Rac-1 induced activation of p38 MAPK. Genetic mutations in members of the MAPK signalling pathway such as those in BRAF (BRAFV600E) lead to hyperactive MEK/ERK signalling and enhanced HIF-1α activity.
In addition to MAPK signalling leading to enhanced activity of HIF-1, studies have also shown that the MAPK pathway may increase HIF-1α protein synthesis in certain cancer types. Fukuda et al. showed that MAPK signalling can increase HIF-1α protein synthesis in HCT116 colon carcinoma cells. Inhibitors of MAPK blocked HIF-1α protein synthesis, and the overexpression of a constitutively active MAPK kinase (MEK2) induced HIF-1α protein [37] (Figure 2).
Genetic mutations in the v-raf murine sarcoma viral oncogene homolog B1 (BRAF) gene, which encodes a serine-threonine kinase, result in hyperactive MAPK signalling. BRAFV600E is the most common genetic mutation within papillary thyroid carcinomas (PTCs). On average, approximately 50% of all PTCs harbour the BRAFV600E mutation [38]. This BRAF mutant is constitutively active and phosphorylates MEK1/2 leading to hyperactive MAPK signalling (Figure 2). It is thought that BRAF and, thus, MAPK signalling are important in the early stages of thyroid tumour development and predispose cells to become dedifferentiated tumours [39–41]. This has recently been substantiated in a mouse model of a thyroid-specific knock-in of oncogenic BRAF (LSL-BRAFV600E/TPO-Cre), which leads to a high frequency of invasive PTCs. These mice had high levels of thyroid stimulating hormone that acted cooperatively with oncogenic BRAF to drive tumour initiation [42].
The GTPase V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) is one of three members of the RAS family of GTPases. KRAS interacts with and activates a number of effector proteins including BRAF and PI3K. Genetic mutations in RAS leading to constitutive activity results in hyperactivation of the PI3K/MAPK pathways, promoting tumour progression in many cancer types including thyroid [43]. Recently, it has been shown that mutations in KRAS or BRAF differentially regulate HIF-1α and HIF-2α in colon carcinoma cells. Mutant KRAS enhanced expression of HIF-1α only, whereas mutant BRAF enhanced expression of both HIF-1α and -2α. KRAS-induced HIF-1α was dependent on PI3K activation, whereas only HIF-2α was inhibited by the MEK inhibitor PD089059 in BRAF mutant cells [44]. These data highlight the complexity of HIF induction by oncogenic signalling cascades and may contribute to the phenotypic differences observed not only in colon cancer but other cancers harbouring these mutations such as thyroid.
The interplay and activation of MAPK by additional signalling pathways promotes HIF-1 activity. In colon cancer cells, loss of the tumour suppressor SMAD4 (mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein SMA-4) led to enhanced HIF-1α activity through the activation of MEK-ERK and p38 MAPK by Transforming growth factor-β (TGF-β) [45].
Stimulation of the MAPK cascade enhances HIF-1 signalling mainly by increasing the transcriptional activity of HIF-1α. This will lead to increased expression of HIF-1 downstream targets under normoxic conditions and in the presence of hypoxia will lead to a cooperative enhancement of expression of HIF-1 target genes.
3.5. HIF-1 and the PI3K Pathway
Aberrant activation of the PI3K pathway occurs by (a) hyperactive stimulation of receptor tyrosine kinases (RTKs) including the insulin-like growth factor-1 receptor (IGF-IR), the human epidermal growth factor receptor 2 (HER2neuR), and the epidermal growth factor receptor (EGFR) and (b) by mutational events that occur in negative regulators of PI3K signalling such as those in Phosphatase and tensin homolog (PTEN) and p53. Activation of the PI3K pathway is pivotal in the development and progression of thyroid cancer aggressiveness and appears to be predominantly involved in the metastatic behaviour of these tumours [41]. Hyperactive PI3K signalling leads to stabilisation of HIF-1α in normoxia. This depends on the activation of AKT and subsequently mammalian target of rapamycin (mTOR), which increases the translation and activity of HIF-1α [37, 41, 46, 47].
mTOR is a serine/threonine kinase consisting of multiple protein-binding motifs that enable interaction and phosphorylation of many proteins and is considered a central signalling molecule for convergence and crosstalk between multiple pathways. mTOR phosphorylates and activates the translational machinery proteins: eukaryotic translation initiation factor 4E- (eIF-4E-) binding protein (4E-BP1) and p70 S6 kinase. Upon phosphorylation, 4E-BP1 no longer interacts with and represses eIF-4E, and p70S6 phosphorylates and activates the 40 S ribosomal protein. Activation of both eIF-4E and the 40 S ribosomal protein initiates translation of HIF-1α mRNA (Figure 3) [48, 49].
Figure 3.
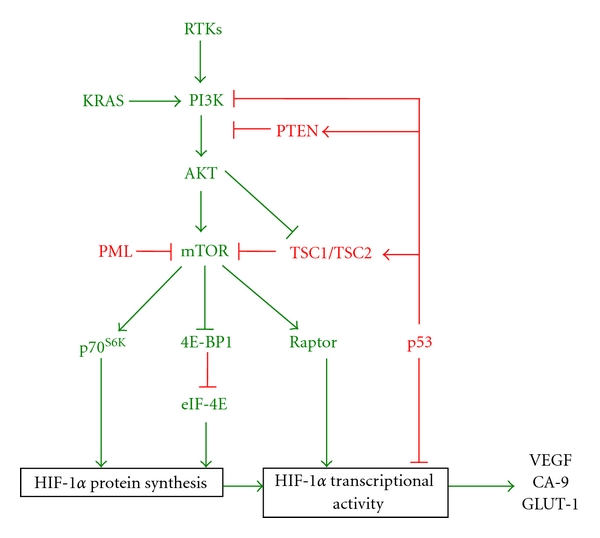
Regulation of HIF-1α by the PI3K pathway. Stimulation of receptor tyrosine kinases (RTKs) by growth factors or activating mutations in the RAS family of GTPases, such as KRAS, results in activation of PI3K. Activated PI3K enables the phosphorylation and activation of AKT. AKT in turn activates mammalian mTOR by disrupting the inhibitory interaction of the TSC1/TSC2 complex with mTOR. mTOR enhances HIF-1 signalling in a multimechanistic way (a) By activating the translational machinery proteins p70S6K and EIF-4E leading to enhanced protein synthesis of HIF-1α and (b) By promoting the interaction of HIF-1α with the coactivator p300, possibly by direct phosphorylation of HIF-1α via interaction with the scaffold protein raptor. Additionally, deletion or loss of function of negative regulators of the PI3K pathway leads to enhanced signalling and activation of HIF-1. These include PTEN, p53, PML, and TSC1/2. The phosphatase PTEN dephosphorylates the products of PI3K, inhibiting activation of AKT and subsequent downstream targets. p53 inhibits PI3K signalling at multiple levels; p53 inhibits transcription of the catalytic subunit of PI3K (p110α), enhances transcription of PTEN and TSC2, interacts with HIF-1α, and recruits MDM2, targeting HIF-1α for proteosomal degradation. PML inhibits mTOR under hypoxia and TSC1/2 interacts with and prevents activation of mTOR in the absence of PI3K activation.
Stimulation of PI3K signalling by activation of different RTKs in a variety of cancer types induces protein synthesis of HIF-1α. This has been shown in colon and breast cancer cells via activation of IGF-1R and HER2nueR by IGF-1 and heregulin, respectively, [37, 50]. Stimulation of PI3K/AKT/mTOR induces the activation of the translational machinery proteins and protein synthesis of HIF-1α. HER2neu is an indicator for poor prognosis in breast cancer and tumours overexpressing HER2 have increased angiogenesis due to increased expression of the HIF-1 target gene VEGF. Similarly, overexpression of HIF-1α and VEGF has been found in prostate cancer cells that have hyperactive PI3K/AKT/mTOR activity as a result of enhanced EGFR signalling [51].
Depending on the type of RTK that is activated and on the type of cancer, increased HIF-1α protein may be PI3K dependent even in the presence of activated MAPK signalling or may be dependent on both signalling pathways. Oestrogen-dependent tumours usually display hyperactive, PI3K, HIF-1α, and VEGF signalling. Studies on the effect of oestrogen induced HIF-1α and VEGF in rat uterine tissue have shown that although oestrogen receptor signalling stimulates both the PI3K and MAPK signalling cascades, induction of HIF-1α and VEGF was only dependent upon PI3K signalling [52]. However, in HCT116 colon cancer cells, IGF-1 induced HIF-1α protein synthesis involved both the PI3K and MAPK signaling pathways [37]. Enhanced activity of HIF-1 by growth factor receptor signalling pathways is both receptor and cell specific and highlights the complexity of HIF-1 regulation by these signalling cascades.
Loss of negative regulators of the PI3K pathway including PTEN, p53 and the tumour-suppressor proteins tuberous sclerosis proteins TSC1 (hamartin), TSC2 (tuberin), and promyelocytic leukemia (PML) leads to hyperactive PI3K-signalling. PTEN is a phosphatase that dephosphorylates the PI3K products, and thus inhibits PI3K downstream signalling. Loss of PTEN or loss of function of PTEN has been found in a range of cancers including prostate, glioma, and thyroid [53–55]. Loss of PTEN has been associated with enhanced angiogenesis and cancer progression possibly through PI3K-induced HIF-1α and downstream targets including VEGF. Loss of function or deletion of the tumour suppressor p53 is common in many cancer types including thyroid. p53 negatively regulates the PI3K pathway by inhibiting transcription of the PIK3CA gene that encodes the catalytic subunit of PI3K; p110α and by activating transcription of PTEN and TSC2 [56, 57]. Furthermore, p53 can directly interact with HIF-1α and recruit MDM2, which targets HIF-1α for ubiquitination and degradation [28, 58]. mTOR activity is negatively regulated by the heterodimer TSC1 (hamartin) and TSC2 (tuberin). This complex is disrupted and functionally inactivated by AKT leading to enhanced activity of mTOR and upregulation of HIF-1α. This has recently been confirmed in irradiated tumours where HIF-1α is activated through this pathway [59]. HIF-1α is not only stimulated through PI3K/AKT activation of mTOR via loss of the TSC1/2 complex, but also via mutational changes leading to loss of TCS2 function. Subsequently, HIF-1α downstream targets including VEGF and Glucose transporter-1(GLUT-1) are increased [60]. Hypoxia inhibits mTOR activity in both a HIF-dependent and independent manner [61]. Under hypoxia, PML negatively regulates mTOR by directly interacting with and preventing activation of mTOR by the small GTPase Rheb1. Loss of PML is observed in a number of sporadic tumours and correlates with increased VEGF and HIF-1α expression via attenuation of hypoxic mTOR inhibition [62]. Characteristically, patients with mTOR-associated hamartoma and tumours harbouring mutations in TSC2 and PML are highly vascularised, a common phenotype of tumours arising from mutations in VHL, which have hyperactive HIF-1 and VEGF signalling [60, 62]. Collectively, these tumour types highlight the importance of HIF-1 in tumour angiogenesis and progression.
Studies in human embryonic kidney cells suggest that mTOR may directly phosphorylate HIF-1α, promoting binding of p300/CBP and enhancing HIF-1α activity. mTOR interacts with the scaffold protein raptor (regulatory-associated protein of mTOR). Raptor directly interacts with HIF-1α under conditions favouring stabilisation of HIF-1α, that is, hypoxia, resulting in enhanced activity, which was not due to mTOR induced stabilisation of HIF-1α protein [63].
The pathophysiology behind increased HIF-1α in many solid tumours is not only restricted to hypoxia, growth factor signalling cascades, or mutations in components of the oxygen-dependent regulatory mechanism of HIF-1, but also may be mediated by other signalling pathways. A hyper-active Wnt/β-catenin signalling cascade may similarly result in increased activity of HIF-1 due to the interaction of β-catenin with HIF-1α [64, 65]. Collectively, these data highlight that many pathways are involved in the fine tuning of HIF-1α regulation by oncogenic signalling cascades such as the ERK-MAPK and PI3K/AKT pathways.
4. The Role of HIF in Cancer
The majority of solid tumours encounter hypoxic stress as a result of (a) limited oxygen diffusion due to the rapid proliferation of tumour cells which outgrow the existing vascular network and (b) by perfusion deficits mediated by abnormal blood vessel structure and function within the tumour [3, 66, 67]. Exposure to low oxygen tensions results in enhanced expression and activity of HIF-1α leading to increased tumour cell survival and growth under stressful environments. Over one hundred HIF target genes have been identified. HIF promotes tumour cell survival and progression by regulating multiple genes including those involved in angiogenesis (VEGF), anaerobic metabolism (GLUT-1), control of intracellular pH (CA-9), regulation of cell cycle, DNA damage response, proliferation and apoptosis (p21 and p27) and extracellular matrix remodelling and cell migration (lysyl oxidase (LOX), and matrix-metalloprotease-1 and -9 (MMP-1 and -9)) [68–71].
Clinically, hypoxia and HIF-1 has been associated with poor prognosis in a range of cancers including uterine, breast, and nonsmall cell lung cancer as well as poor treatment response in cancers such as nasopharyngeal cancer [1–3, 72]. Additionally, coexpression of HIF-1α with other pro-oncogenic proteins has been shown to be a significant prognostic predictor of survival. In patients with nonsmall cell lung cancer, coexpression of HIF-1α with Snail or TWIST1 has been shown to reduce overall survival and recurrence-free survival [73]. Expression of HIF-2α and coexpression with EGFR and insulin-like growth factor-binding protein-2 (IGFBP-2) has also been linked to reduced survival in patients with higher grade astrocytomas [74].
The HIFs act as the most important sensors of oxygen homeostasis. Constant HIF activation is a common feature in many cancers and is becoming increasingly recognized as a target for therapeutic intervention.
5. HIF and Chemo/Radiotherapy
5.1. HIF and Chemoresistance
HIF-1α has been found to upregulate expression of the ATP-binding cassette (ABC) transporter family of proteins and multidrug resistance related proteins (MRPs) in certain tumours and cancer cell lines. The ABC-transporter proteins can efflux chemotherapeutic agents out of the cell, namely, taxanes or anthracyclines resulting in reduced cytotoxicity and cell death [75–77].
The HIF-1 downstream target VEGF has also been implicated in chemoresistance. Blockade of VEGF signalling can promote normalisation of the tumour vasculature resulting in enhanced delivery of chemotherapeutics into the tumour. This has been observed in anaplastic thyroid carcinoma xenografts, where bevacizumab (inhibitory monoclonal antibody against VEGF) lowered tumour interstitial pressure and reduced vascular permeability [78]. Due to enhanced VEGF signalling, the tumour vasculature is erratic, dysfunctional, and leaky, resulting in poorly perfused regions and high tumour interstitial pressure. This can hinder diffusion of chemotherapeutics into the tumours [79]. Qayum et al. recently showed that blockade of the EGFR, RAS, and PI3K pathways leads to long-term morphological changes that promote increased perfusion and allow prolonged and enhanced drug delivery to the tumour. These changes may also improve tumour response to radiotherapy [80]. In addition to VEGF being a HIF-1 downstream target, VEGF itself can induce HIF-1α and promote HIF-1 activity leading to enhanced chemo-resistance.
Other HIF-1 target genes implicated in chemo-resistance include CA-9. CA-9 plays a role in acidification of the tumour microenvironment by catalysing the hydration of CO2 to bicarbonate and protons. Due to this, anticancer drugs that are weakly basic are unable to be taken up effectively into tumour cells as a result of raised intracellular pH and low extracellular pH, leading to reduced cytotoxicity [81–83]. Additionally, our group has shown that inhibition of HIF-1α either genetically or pharmacologically can enhance chemo-sensitivity when used in combination with drugs that lack efficacy in hypoxic cells such as etoposide [84].
Collectively, these data highlight the importance of HIF-1 in chemo-resistance and suggest that combination treatments with HIF inhibitors may be useful in improving therapeutic response to existing drugs and especially those associated with hypoxic chemo-resistance.
5.2. HIF and Radioresistance
Tumour hypoxia is associated with radioresistance due to the lack of oxygen leading to a reduction in the level of radiation-induced free radicals. These free radicals induce single- and double-stranded DNA breaks, leading to cell death by necrosis, apoptosis, or mitotic catastrophe [85, 86]. Within a tumour, the level of hypoxia has been shown to inversely correlate with radiosensitivity [66]. HIF-1 signalling promotes cell survival following exposure to ionising radiation. Clinically, high HIF-1 activity after radiation is associated with poor prognosis [2, 87]. HIF-1 activity has been found to increase with increasing doses of radiation in tumour xenograft models. No effect of radiation on HIF-1 activity was observed in tumour cells in vitro, suggesting that increased activity is dependent on radiation-induced physical changes within the intact tumour [88]. Conversely, other studies suggest that radiation itself can increase HIF-1α expression in addition to hypoxia in vitro. HIF-1 reporter activity was found to increase in hypoxic cells following exposure to radiation in range of carcinoma cell lines [89]. As a result of radiation-induced HIF-1 signalling, expression of HIF-1 downstream targets such as VEGF, plasminogen activator inhibitor-1, and CA-9 are increased and promote radio-resistance [90].
Within a tumour, mechanisms thought to contribute to radiation induced increases in HIF-1 include tumour reoxygenation. Radiation increases the level of O2 within a tumour. This is due to cell death of well-oxygenated cells that frees up O2 molecules. Additionally, as there are fewer tumour cells due to cell death, the vasculature is able to expand and grow, leading to enhanced oxygenation. This reoxygenation causes the generation of free radical such as reactive nitrogen species that lead to inhibition of the PHDs, thus reducing proteosomal degradation of HIF-1α [91].
Hyperactivity of growth factor signalling pathways such as EGFR, RAS, and PI3K/AKT cascades have been linked to radio-resistance and tumour cell survival following radiotherapy [92, 93]. As HIF-1 is a downstream target of PI3K, PI3K-induced HIF-1 activity may contribute to radio-resistance. Therefore, it is feasible to assume that tumour cells associated with hyperactive PI3K signaling, and thus, HIF-1 activity may respond better to radiotherapy when used in combination with a PI3K inhibitor. Currently, inhibition at the level of the EGFR in combination with radiotherapy has been used clinically and with success in certain tumour types [92, 94]. However, as there are many different points of activation within the PI3K signalling pathway, inhibition of PI3K itself may be useful in a whole range of tumours that have hyperactive PI3K signalling which is not dependent on EGFR activation but are dependent on other RTKs. Enhanced radiosensitivity has been observed in colon carcinoma cells treated with the selective PI3K inhibitor PI-103 [95] and in glioma xenografts treated with selective small molecule inhibitors of p110α [96]. Thus, these data provide promising evidence that in addition to inhibiting the downstream effects of PI3K that promote cell survival after radiation, indirect targeting of HIF-1 by PI3K inhibitors may significantly contribute to enhanced radiosensitivity.
6. HIF and Thyroid Hormones
Thyroid hormones play a key role in growth, metabolism and development. The effects of these hormones are brought about, namely, by the thyroid hormone ligand 3,3,5-triiodothyronine (T3) binding to the nuclear thyroid hormone receptors (TR) β1, β2, or α1. The ligand-nuclear receptor complex recruits additional proteins and acts as transcription factors that regulate gene expression by binding to the thyroid hormone response elements (TREs) in the promoter region of target genes. These genomic actions of thyroid hormones usually involve the binding of T3 to monomers, homodimers, or as heterodimers of the TRs with another member of the nuclear hormone receptor family such as the retinoid X receptor. This results in shedding of corepressors and recruitment of co-activators leading to alterations in transcription of thyroid hormone responsive genes [97, 98]. This contrasts to the rapid, nongenomic actions of thyroid hormones on the activity of ion pumps, cytosolic signalling, mitochondria, and the intracellular protein trafficking from cytosol to nucleus [98].
Thyroid hormones can activate both the PI3K and MAPK-signalling cascades [98–101]. Furthermore, thyroid hormones have been found to directly regulate expression of HIF-1α via activation of these signal transduction pathways. Microarray analysis of T3 regulated genes in human skin fibroblasts revealed an upregulation of HIF-1α and HIF-1 target genes GLUT1, platelet-type phosphofructokinase (PFK) and monocarboxylate transporter-4 (MCT-4) via T3/TRβ signalling. The specificity of this upregulation by T3 was confirmed in fibroblasts from patients with an inactivating mutation in TRβ. These fibroblasts did not show any such changes in gene expression of HIF-1α and target genes [99]. The direct upregulation of HIF-1α by T3 resulted in a T3 indirect increase of GLUT-1, PFK, and MCT-4 expression via enhanced HIF-1α activity. This direct increase in HIF-1α activity was due to T3/TRβ activation of PI3K (Figure 4). Other studies have also shown that a TRβ mutant (TRβPV/PV) interacts with the PI3K-regulatory subunit p85α leading to enhanced PI3K signalling, promoting thyroid carcinogenesis in a mouse model of spontaneous follicular thyroid cancer (thyroid hormone receptorPV/PV mice) [100]. This may lead to enhanced HIF-1α signalling in vivo.
Figure 4.
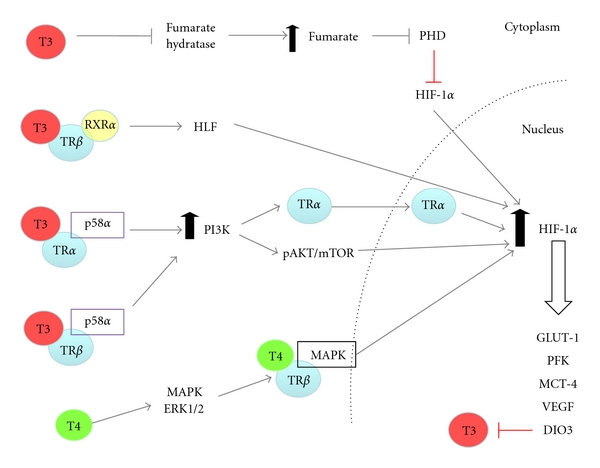
Thyroid hormone-dependent activation of HIF-1α: T3 and T4 induce HIF-1α activity by both genomic and nongenomic mechanisms. Genomically, T3 indirectly increases HIF-1α mRNA by increasing expression of the transcription factor hepatic leukemia factor (HLF), which initiates transcription of HIF-1α. Non-genomically, T3 stimulates PI3K signalling by promoting the interaction of both TRα and β with the PI3K regulatory subunit p85 leading to enhanced PI3K/AKT/mTOR activity and translation of HIF-1α mRNA. T3-induced PI3K promotes nuclear shuttling of TRα leading to increased HIF-1α expression. T3-induced PI3K signalling by either TRα or β is cell specific. T3 inhibits the enzyme fumarate dehydrogenase resulting in the accumulation of fumarate. Fumarate inhibits PHD2 leading to reduced hydroxylation of HIF-1α and increased protein stabilisation. T4 increases HIF-1α by stimulating MAPK signalling, leading to enhanced T4/TRβ activity and expression of HIF-1α. Activated HIF-1α by T3/T4 results in the upregulation of target genes, known to promote tumour cell survival and progression. These include GLUT-1, PFK, MCT-4, and VEGF. Additionally, HIF-1 upregulates DIO3, which inhibits T3 by catalysing the conversion of T3 to the metabolically inactive T2.
T3 can also increase HIF-1α expression through interaction with the TRα members. In endothelial cells, T3 promotes the interaction of TRα1 with the PI3K subunit p85α, leading to increased phosphorylation and activation of AKT and endothelial nitric oxide synthase (eNOS). This is thought to contribute to the vasodilatory and cardiovascular protective effects of thyroid hormones [101]. It is likely that this T3-induced PI3K activity in endothelial cells would enhance HIF-1α activity, which may contribute to the vascular effects of thyroid hormone, as eNOS is a HIF-1 regulated gene [102]. In glioma cells, stimulation of PI3K/AKT by T3 induces shuttling of TRα from the cytoplasm to the nucleus, and promotes transcription of HIF-1α mRNA (Figure 4). This T3-dependent process was inhibited by treatment with the PI3K inhibitor LY294002 but not by the ERK inhibitor PD098095 [98]. T3-induced expression of HIF1-α and the downstream target VEGF has been verified in a gastric cancer cell line and in vivo mouse model. Accumulation of HIF-1α was due to T3 dependent activation of the PI3K signalling cascade. Interestingly, in addition to PI3K-signalling, HIF-1α overexpression by T3 may be regulated by fumarate accumulation; this accumulation was enhanced by T3-mediated inactivation of fumarate hydratase and reduced in the presence of 2-oxoglutarate [103] (Figure 4). These data link thyroid hormones with metabolic proteins and the HIF-1 signalling pathway in carcinogenesis and provide a means for understanding the adaptive mechanisms of tumour cells to metabolic stress.
Interestingly, T4 but not T3 has been found to impact on HIF-1α expression via interaction with the TRβ1 in glioblastoma (U-87) cells. This process was dependent on MAPK (ERK1/2) signalling as PD098095 inhibited T4-induced HIF-1α expression [98]. In CV-1 cells transfected with TRβ1, T4-activated MAPK signalling led to the interaction of MAPK with TRβ1 and enhanced phosphorylation and TRβ1 signalling [104]. Other studies have shown that both T3 and T4 can promote HIF-1α activity by activating the PI3K pathway. In hepatoma cells, T3 and to a lesser extent T4 increased expression of the HIF-1 target genes EPO and adrenomedullin (ADM), by stimulating the PI3K pathway, thus leading to increased translation of HIF-1α mRNA. No effects on HIF-1α protein stability/proteosomal degradation were observed [105].
In addition to the rapid nongenomic induction of HIF-1α by thyroid hormones via activation of growth factor signalling cascades, thyroid hormones have been shown to induce HIF-1α via the classic genomic actions of TRs. T3 can indirectly increase HIF-1α mRNA in a range of carcinoma cell lines that express hepatic leukemia factor (HLF). This regulation appears to be independent of a major protein kinase pathway, as the inhibition of members of both the MAPK and PI3K kinase pathways, including mTOR, had no effect on T3-mediated HIF-1α expression. T3 induction of HIF-1α was dependent on HLF-mediated gene expression of HIF-1α. This stimulation of HLF was in turn mediated by T3-activation of the TRβ-retinoid X receptor α heterodimer. Increased HIF-1α was, therefore, solely at the mRNA level and independent of translation [106] (Figure 4).
A close link between thyroid hormones and HIF is further substantiated by data on the activity of deiodinase type 3 (DIO3). Hypoxia induces DIO3, the physiological inactivator of T3, which catalyses the conversion of T3 to the metabolically inactive T2 (Figure 4). As this effect was also mimicked by hypoxia mimetics such as cobalt chloride, a direct action of HIF-1 was suspected. HIF-1α directly interacts with the DIO3 promoter, and thus stimulates DIO3 activity. This, in turn, induces a reduction of T3-dependent activation of metabolic pathways and represents a protective mechanism to reduce metabolic rate in tissues subjected to hypoxia [107].
The effect of T3 and T4 on HIF-1α is both receptor and cell specific. Furthermore, interaction of these hormones with additional signalling cascades provides insight into the importance of thyroid hormones in driving carcinogenesis by upregulating prosurvival pathways and those involved in the adaptation to the stressful conditions of the tumour microenvironment. An understanding of how these hormones and their receptors differentially regulate HIF-1α in both normal tissue and cancer may help identify the most appropriate drug treatment to improve therapeutic response.
7. HIF and Thyroid Cancer
7.1. Clinical Significance of HIF-1 and Thyroid Carcinomas
Recently we expanded on the potential pathophysiological importance of HIF-1α in thyroid carcinomas by showing that HIF-1α protein was variably expressed in primary thyroid carcinomas associated with advancing tumour grade. Tumour samples were representative of papillary (PTC), follicular (FTC), and anaplastic (ATC) thyroid carcinomas [26]. HIF-1α expression was absent from normal thyroid tissue. Interestingly and consistent with previous findings in a range of cancers, HIF-1α expression was highest in the most aggressive dedifferentiated anaplastic thyroid carcinomas (ATCs). However, analysis of vessel number and distribution via Von Willebrand factor (vWF) immunostaining were unable to link HIF-1α expression and vessel distribution. This may have been expected due to the short half-life of HIF-1α and transient regions of hypoxia that occur as a result of the erratic and dysfunctional vasculature, HIF-1α expression may not always be detected. Similarly to HIF-1α, highest expression of GLUT-1 was observed in ATCs and cell lines derived from ATCs, with lower expression observed in cell lines derived from differentiated tumours. These increases, however, were only slight and were not significant. In contrast to GLUT-1, the HIF-1 downstream target CA-9 closely mimicked the pattern of HIF-1α expression with significantly higher levels observed in the ATCs. This suggests that CA-9 as in other cancer types may serve as a potential new biomarker of aggressive disease in thyroid carcinomas. This observation has been supported by previous work by Jubb et al. who reported high expression levels of HIF-1α in all thyroid carcinoma types particularly in FTCs [29, 108]. Additionally, data from a preliminary study in our group looking at HIF-1α and CA-9 in PTC, FTC, and ATC xenografts showed a similar correlation to that seen in clinical samples; both HIF-1α and CA-9 expression was highest in the tumours derived from ATC (8505c) cells. Furthermore, tumours with high expression of HIF-1α and CA-9 had a greater number of spontaneous metastatic colonies to the lungs (Figure 5). Collectively, these data from primary tumour tissue and xenografts suggest that HIF-1α expression is involved in the adaption of thyroid carcinomas to hypoxia and supports a pathophysiological role for thyroid tumour progression, aggressiveness and metastasis.
Figure 5.

HIF-1α and CA-9 protein expression and metastatic potential are highest in tumours derived from follicular (WRO) and anaplastic (8505c) cell lines. Briefly, BcPAP, FTC133, WRO and 8505c cells were implanted sub-cutaneously in female CBA nu/nu nude mice. Once tumours reached 450 mm3, tumour tissue and fresh lung tissue were excised. Tumour sections were immuno-stained for HIF-1α. Freshly excised lung tissue was enzymatically digested, plated out in serial dilution and left until visible colonies formed. Colonies were stained, counted and number of metastatic colonies per mg lung tissue calculated. (For full methods, see supplementary materials and methods which is located at doi: 10.406/2011/762905). (a) Immunohistochemical staining for HIF-1α (brown staining). One example of HIF-1α staining for each tumour type: BcPAP, FTC133, WRO and 8505c are displayed. (b) Semiquantitative analysis of HIF-1α and CA-9 immunostaining quantified according to Allred et al. [109]. The total score is representative of intensity and proportion of staining. HIF-1α was highly expressed in all tumour types with highest expression observed in the 8505c tumours. CA-9 was also expressed in all tumour types with significantly higher expression observed in 8505c tumours (P < .05, one way Anova, with Tukey post test). (c) The number of spontaneous metastatic colonies cultured from enzymatically digested lung tissue that was excised from mice bearing WRO, 8505c, FTC133, and BcPAP tumours. The highest number of colonies counted were from the lungs of mice bearing FTC (WRO) and ATC (8505c) tumours. Data are representative of the mean ± S.D. of 5 independent mice for each tumour type.
7.2. Hypoxia and HIF-1 in Thyroid Carcinomas
A number of functional studies in thyroid carcinoma cell lines and in immortalized cells derived from normal thyroid tissue substantiate the importance of hypoxia-induced HIF-1 in promoting the expression of proteins that drive thyroid tumour progression. HIF-1α was predominantly localized in the cytoplasm under normoxia but stabilised and translocated to the nucleus in hypoxia [29]. Enhanced nuclear localisation of HIF-1α has also been observed in the BcPAP cell line harbouring the BRAFV600E mutation versus a cell line with WT BRAF under normoxia [30]. The functional response of both normal and carcinoma thyroid cell lines to hypoxia was further confirmed as graded hypoxia-induced a marked increase in expression of HIF-1α and downstream targets CA-9, VEGF and GLUT1. Hypoxia induced HIF-1α was further supported by HIF-1α reporter activation in cells exposed to lowering oxygen tensions. Furthermore, the degree of hypoxia induced HIF-1α activity related to the level of thyroid carcinoma aggressiveness; lowest activity was observed in PTC cell lines, with highest seen in ATC cells. We have also confirmed that hypoxia induces expression of HIF-2α in follicular and anaplastic thyroid carcinoma cell lines (unpublished, data not shown). This is of interest, as the HIFs are known to regulate genes differentially in many different cancer types. Therefore, expression in thyroid carcinoma may account for some of the phenotypic differences seen between the different classifications of thyroid carcinoma.
7.3. Growth-Factor Signalling Pathways and HIF-1 in Thyroid Carcinomas
We found that in addition to hypoxia, growth factor-signalling pathways also induced HIF-1α expression and activity in thyroid carcinoma cell lines. Induction of HIF-1α by pathways such as the MAPK and PI3K signalling cascades have been described in a range of cancer types (see oxygen-independent regulation of HIF) [110]. The majority of thyroid carcinomas have mutations in growth factor signalling pathways. Mutations in the BRAF gene (BRAFV600E) leading to hyperactive MAPK/ERK signalling have been found in approximately 50% of all PTCs [38]. Recently, this mutation has been reported to increase the transcription and expression of HIF-1α protein [30].
KRAS has been found to differentially regulate HIFs in certain cancer types [44]. Activating mutations in the RAS family of GTPases, such as those found in KRAS, have been found in approximately 20% of PTCs and more specifically the follicular variant of PTC suggesting the mutation promotes aggressive phenotype [43]. As KRAS activates both BRAF and PI3K, it would be interesting to determine the role of KRAS in the context of HIF activity and aggressive phenotype in thyroid tumours and thyroid carcinoma cell lines harbouring this mutation.
Studies have shown that highest levels of HIF-1α expression and activity were detected in thyroid tumours and tumour cell lines harbouring classic mutations in BRAF and PI3K [29, 30]. Thus, the current data supports the importance of oncogenic activation of HIF-1 in thyroid cancer and aggressive disease [46]. Direct intervention with pharmacological inhibitors of these pathways (LY294002 for PI3K, PD098059 for ERK/MAPK, and sorafenib for inhibition of RAF-1 kinase/BRAF) or by genetic modulation; (re-expression of PTEN in PTEN null cell lines and silencing of mutant BRAF), further support the close dependency of HIF expression on oncogenic signalling. Interestingly, inhibition of HIF-1α reporter activity was much more substantial as a consequence of LY294002 treatment under varying O2 tensions compared to PD98059 even in cell lines with no known mutation in the PI3K pathway. These findings are particularly interesting with regard to the frequent mutations in the PI3K/AKT pathway found in up to 15% of PTC and approximately 50% of FTC [111, 112], where PI3K-pathway mutations have been directly linked to tumour aggressiveness [41]. HIF-1α silencing reduced expression of HIF-1 target genes like CA-9. Thus, blocking HIF-1α, either directly or through inhibition of the PI3K pathway in thyroid carcinomas, may decrease aggressiveness and thus open new therapeutic options [46].
7.4. HIF and VEGF in Thyroid Carcinomas
VEGF serum levels are elevated in thyroid carcinomas, and this is associated with poor tumour prognosis [113–116]. As one of the most important downstream targets of HIF-1 signalling, VEGF has not yet been linked clinically to HIF activity in thyroid carcinomas. A number of studies using antagonists of VEGF signalling for the treatment of advanced thyroid carcinomas have been reported with some positive results ([117–120]. However, no data are currently available on the effect on VEGF levels by direct targeting of HIF-1. We have reported that VEGF levels are significantly increased by anoxia in thyroid carcinoma cells lines derived from PTC, FTC and ATC. Additionally, highest basal levels of VEGF were observed in the PTEN—null FTC133 follicular thyroid carcinoma cell lines [29]. This suggests that the two different signalling pathways may act synergistically to increase VEGF. The anoxia-induced increases in VEGF expression did not, however, match the pattern of HIF-1α reporter activity in varying thyroid carcinoma cell lines. This suggests that the level of dependency on either the HIF-1 or growth factor-signalling pathways to induce VEGF is specific to the type of thyroid carcinoma cell line. Current unpublished data from our group supports a prominent role and an important link between the PI3K and HIF-1 pathways in the regulation of VEGF. Furthermore, we have found that inhibition of these pathways has profound effects on VEGF expression in thyroid carcinoma cell lines. These data provide a new angle for therapeutic approaches of thyroid carcinoma as drugs which target HIF-1 signalling may be more efficacious in downregulating VEGF dependent signalling in these tumours.
7.5. HIF-1, Migration, and Metastasis in Thyroid Carcinomas
Hypoxia and HIF-1 have been associated with increased migration and metastasis in a range of cancer types [121–123]. We have found that hypoxia significantly increased migration in cell lines derived from PTCs and FTCs. Additionally, we have exciting data that supports a prominent role of PI3K and HIF-1 signalling in metastatic characteristics of FTC and ATC (unpublished).
Studies have shown a link between hypoxia/HIF-1α and MNNG HOS transforming gene (MET) upregulation in clinical PTCs. MET is a receptor tyrosine kinase that is stimulated by hepatocyte growth factor (HGF). Stimulation results in the activation of signal transduction pathways such as PI3K, which promote migration, invasion, and the release of cytokines and proangiogenic factors such as VEGF [124, 125]. Additionally, MET activation promotes migration and invasion by stimulating the translocation of β-catenin from the cytoplasm to the nucleus [126]. MET is highly expressed in PTC, and this high expression of MET is thought to be due to HIF-1 induced upregulation in hypoxic regions of PTCs. Furthermore, higher expression of MET and HIF-1α mRNA was found at the periphery of tumours (in cells located at the invading front) compared to the centre in 44% of PTCs [127]. These were characterised to have a more aggressive phenotype. Collectively, these data suggest that HIF-1 may play a role in promoting migration and aggressiveness in PTC.
Taken together, these data highlight the importance of hypoxia, HIF-1α and PI3K signalling as a means of promoting metastatic characteristics and driving aggressive disease in thyroid carcinomas (Figure 6).
Figure 6.
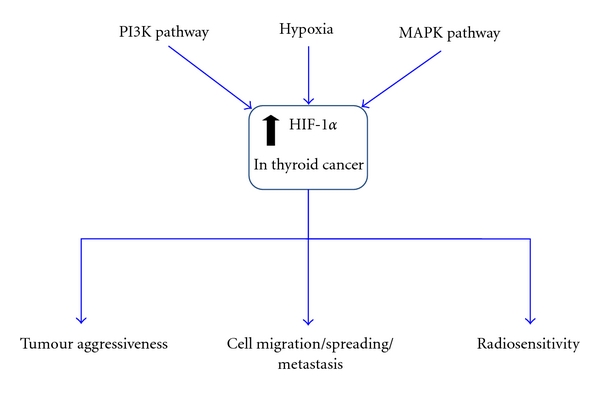
Mechanisms of induction of HIF-1α and downstream effects of HIF-1 signalling in thyroid carcinomas. HIF-1 activity is induced not only by hypoxia but by the oncogenic MAPK and PI3K signalling pathways. Clinically, expression of HIF-1α and the downstream target CA-9 have been linked with thyroid carcinoma aggressiveness. It is well known that HIF-1 plays an important role in tumour development, metastasis, and aggressiveness and is ultimately correlated with poor prognosis. Enhanced HIF-1 signalling by hypoxia and the PI3K/MAPK pathways may play an important role in the metastatic characteristics of thyroid carcinoma. HIF-1-induced MET upregulation has been implicated in promoting invasiveness of PTC. HIF-1 signalling may also play a prominent role in the therapeutic response to radiotherapy in thyroid carcinoma.
8. HIF-1 and Sensitivity to Radiotherapy in Thyroid Carcinomas
There is increasing evidence for the role of HIF in tumour cell sensitivity to chemo- and importantly radiotherapy [77, 84, 128]. As discussed above, HIF-1 stabilisation and action depend on growth factor-signalling pathways. Thus, many inhibitors of growth factor signalling affect HIF-1 expression and downstream targets. Anticancer drugs that target these pathways possess antiangiogenic properties and include the BCR-ABL kinase inhibitor imatinib (Gleevec), EGFR inhibitors such as gefitinib, erlotinib, and cetuximab, the HER2neu inhibitor trastuzumab, and the mTOR inhibitors rapamycin, temsirolimus, and everolimus. Collectively, these drugs all target the translation of HIF-1α mRNA into protein [129]. Modulation of HIF-1α expression can increase tumour sensitivity, in particular to radiotherapy in certain cancer types (see HIF and radio-resistance). Thus, HIF-1 signalling may be a particularly important feature for the sensitivity of dedifferentiated thyroid carcinomas to external beam radiation. This has yet to be investigated.
It has been shown that tumour cell apoptosis is increased when targeted radioiodide therapy is combined with angiogenic inhibitors, which represents an indirect approach to circumvent HIF-1—dependent radio-resistance [130]. Data discussed here support the view that targeting of HIF-1 may serve as a more direct approach to overcome HIF dependent adaptation of thyroid carcinoma cells to radiotherapy. Preliminary data from our group in FTC and ATC cell lines supports this notion of HIF-1 signalling playing a role in radio-resistance (unpublished). As described earlier, PI3K has been found to play an important role in HIF-1α induced expression and activity in thyroid carcinoma cell lines under both normoxia and anoxia. Therefore, targeting HIF-1 either directly or by use of a PI3K-inhibitor may improve the therapeutic response of thyroid carcinoma to radiotherapy.
Currently, there is no data available on whether inhibition of HIF-1 may reflect on the sensitivity of more differentiated tumours to standard radiotherapy. It is conceivable that in patients receiving external beam radiation treatment and particularly in patients with a poor response but positive uptake of radioiodine, any augmentation of the sensitivity towards irradiation-induced apoptosis would be favourable (Figure 6). Studies in our group on animal models of metastatic follicular and anaplastic thyroid carcinomas will hopefully allow us to answers some of these questions in the future.
9. Conclusions
HIF-1 is potently induced by both hypoxia and oncogenic signalling pathways in thyroid carcinoma, and its expression and activity have been correlated with aggressiveness. Current literature suggests that PI3K and MAPK pathways promote aggressive and metastatic disease in part via the upregulation of HIF-1 activity. With the known effects of hypoxia, PI3K/MAPK pathways, and HIF-1 on desensitisation to radiotherapy, HIF-1 may be a new and important therapeutic target in reducing local tumour growth, metastatic burden, and radio-resistance in thyroid carcinoma.
Financial Support
Cancer Research UK (K. J. Williams C7820/A8696; supporting N. Burrows) EU FP7 Metoxia Grant agreement no. 222741 (K. J. Williams supporting M. Babur), and Wellcome Trust project grant, UK 082794 (G. Brabant, supporting J. Resch).
Acknowledgment
K. J. Williams and G. Brabant contributed equally to the work.
References
- 1.Birner P, Schindl M, Obermair A, Breitenecker G, Oberhuber G. Expression of hypoxia-inducible factor 1alpha in epithelial ovarian tumors: its impact on prognosis and on response to chemotherapy. Clinical Cancer Research. 2001;7(6):1661–1668. [PubMed] [Google Scholar]
- 2.Aebersold DM, Burri P, Beer KT, et al. Expression of hypoxia-inducible factor-1alpha: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Research. 2001;61(7):2911–2916. [PubMed] [Google Scholar]
- 3.Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. Journal of the National Cancer Institute. 2001;93(4):266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 4.Brahimi-Horn MC, Pouysségur J. Harnessing the hypoxia-inducible factor in cancer and ischemic disease. Biochemical Pharmacology. 2007;73(3):450–457. doi: 10.1016/j.bcp.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Hirota K, Semenza GL. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochemical and Biophysical Research Communications. 2005;338(1):610–616. doi: 10.1016/j.bbrc.2005.08.193. [DOI] [PubMed] [Google Scholar]
- 6.Pouysségur J, Mechta-Grigoriou F. Redox regulation of the hypoxia-inducible factor. Biological Chemistry. 2006;387(10-11):1337–1346. doi: 10.1515/BC.2006.167. [DOI] [PubMed] [Google Scholar]
- 7.Patel SA, Simon MC. Biology of hypoxia-inducible factor-2α in development and disease. Cell Death & Differentiation. 2008;15(4):628–634. doi: 10.1038/cdd.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loboda A, Jozkowicz A, Dulak J. HIF-1 and HIF-2 transcription factors—similar but not identical. Molecules & Cells. 2010;29(5):435–442. doi: 10.1007/s10059-010-0067-2. [DOI] [PubMed] [Google Scholar]
- 9.Aprelikova O, Wood M, Tackett S, Chandramouli GVR, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Research. 2006;66(11):5641–5647. doi: 10.1158/0008-5472.CAN-05-3345. [DOI] [PubMed] [Google Scholar]
- 10.Hu CJ, Wang LIY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Molecular and Cellular Biology. 2003;23(24):9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warnecke C, Zaborowska Z, Kurreck J, et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. The FASEB Journal. 2004;18(12):1462–1464. doi: 10.1096/fj.04-1640fje. [DOI] [PubMed] [Google Scholar]
- 12.Holmquist-Mengelbier L, Fredlund E, Löfstedt T, et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell. 2006;10(5):413–423. doi: 10.1016/j.ccr.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka T, Wiesener M, Bernhardt W, Eckardt KU, Warnecke C. The human HIF (hypoxia-inducible factor)-3alpha gene is a HIF-1 target gene and may modulate hypoxic gene induction. Biochemical Journal. 2009;424(1):143–151. doi: 10.1042/BJ20090120. [DOI] [PubMed] [Google Scholar]
- 14.Maxwell PH, Wlesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 15.Gnarra JR, Ward JM, Porter FD, et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(17):9102–9107. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clifford SC, Cockman ME, Smallwood AC, et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis invon Hippel-Lindau disease. Human Molecular Genetics. 2001;10(10):1029–1038. doi: 10.1093/hmg/10.10.1029. [DOI] [PubMed] [Google Scholar]
- 17.Kapitsinou PP, Haase VH. The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death & Differentiation. 2008;15(4):650–659. doi: 10.1038/sj.cdd.4402313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schofield CJ, Ratcliffe PJ. Signalling hypoxia by HIF hydroxylases. Biochemical and Biophysical Research Communications. 2005;338(1):617–626. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 19.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature Reviews Urology. 2010;7(5):277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollard PJ, Wortham NC, Tomlinson IP. The TCA cycle and tumorigenesis: the examples of fumarate hydratase and succinate dehydrogenase. Annals of Medicine. 2003;35(8):632–639. doi: 10.1080/07853890310018458. [DOI] [PubMed] [Google Scholar]
- 21.Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha; prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 22.Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nature Reviews Cancer. 2005;5(11):857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- 23.Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. Journal of Internal Medicine. 2009;266(1):19–42. doi: 10.1111/j.1365-2796.2009.02111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fagin JA, Mitsiades N. Molecular pathology of thyroid cancer: diagnostic and clinical implications. Best Practice & Research Clinical Endocrinology & Metabolism. 2008;22(6):955–969. doi: 10.1016/j.beem.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roymans D, Slegers H. Phosphatidylinositol 3-kinases in tumor progression. European Journal of Biochemistry. 2001;268(3):487–498. doi: 10.1046/j.1432-1327.2001.01936.x. [DOI] [PubMed] [Google Scholar]
- 26.Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT—A major therapeutic target. Biochimica et Biophysica Acta. 2004;1697(1-2):3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Berra E, Milanini J, Richard DE, et al. Signaling angiogenesis via p42/p44 MAP kinase and hypoxia. Biochemical Pharmacology. 2000;60(8):1171–1178. doi: 10.1016/s0006-2952(00)00423-8. [DOI] [PubMed] [Google Scholar]
- 28.Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends in Molecular Medicine. 2002;8(4, supplement):S62–S67. doi: 10.1016/s1471-4914(02)02317-1. [DOI] [PubMed] [Google Scholar]
- 29.Burrows N, Resch J, Cowen RL, et al. Expression of hypoxia-inducible factor 1alpha in thyroid carcinomas. Endocrine-Related Cancer. 2010;17(1):61–72. doi: 10.1677/ERC-08-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zerilli M, Zito G, Martorana A, et al. BRAF mutation influences hypoxia-inducible factor-1alpha expression levels in papillary thyroid cancer. Modern Pathology. 2010;28(3):1052–1060. doi: 10.1038/modpathol.2010.86. [DOI] [PubMed] [Google Scholar]
- 31.Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. The Journal of Biological Chemistry. 2003;278(16):14013–14019. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richard DE, Berra E, Gothié E, Roux D, Pouysségur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-reducible factor (HIF-1alpha) and enhance the transcriptional activity of HIF-1. The Journal of Biological Chemistry. 1999;274(46):32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- 33.Hirota K, Semenza GL. Rac1 Activity is Required for the Activation of Hypoxia-inducible Factor 1. The Journal of Biological Chemistry. 2001;276(24):21166–21172. doi: 10.1074/jbc.M100677200. [DOI] [PubMed] [Google Scholar]
- 34.Semenza GL. Signal transduction to hypoxia-inducible factor 1. Biochemical Pharmacology. 2002;64(5-6):993–998. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- 35.Görlach A, Berchner-Pfannschmidt U, Wotzlaw C, et al. Reactive oxygen species modulate HIF-I mediated PAI-I expression: involvement of the GTPase RacI. Thrombosis & Haemostasis. 2003;89(5):926–935. [PubMed] [Google Scholar]
- 36.Mylonis I, Chachami G, Samiotaki M, et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. The Journal of Biological Chemistry. 2006;281(44):33095–33106. doi: 10.1074/jbc.M605058200. [DOI] [PubMed] [Google Scholar]
- 37.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. The Journal of Biological Chemistry. 2002;277(41):38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 38.Tang KT, Lee CH. BRAF mutation in papillary thyroid carcinoma: pathogenic role and clinical implications. Journal of the Chinese Medical Association. 2010;73(3):113–128. doi: 10.1016/S1726-4901(10)70025-3. [DOI] [PubMed] [Google Scholar]
- 39.Ciampi R, Nikiforov YE. Minireview: RET/PTC rearrangements and braf mutations in thyroid tumorigenesis. Endocrinology. 2007;148(3):936–941. doi: 10.1210/en.2006-0921. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Nakamura M, Kakudo K. Targeting of the BRAF gene in papillary thyroid carcinoma (review) Oncology Reports. 2009;22(4):671–681. doi: 10.3892/or_00000487. [DOI] [PubMed] [Google Scholar]
- 41.Paes JE, Ringel MD. Dysregulation of the phosphatidylinositol 3-kinase pathway in thyroid neoplasia. Endocrinology and Metabolism Clinics of North America. 2008;37(2):375–387. doi: 10.1016/j.ecl.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franco AT, Malaguarnera R, Refetoff S, et al. Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(4):1615–1620. doi: 10.1073/pnas.1015557108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greco A, Borrello MG, Miranda C, Degl’Innocenti D, Pieroiti MA. Molecular pathology of differentiated thyroid cancer. Quarterly Journal of Nuclear Medicine and Molecular Imaging. 2009;53(5):440–454. [PubMed] [Google Scholar]
- 44.Kikuchi H, Pino MS, Min Z, Shirasawa S, Chung DC. Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1alpha and -2alpha in colon cancer. Cancer Research. 2009;69(21):8499–8506. doi: 10.1158/0008-5472.CAN-09-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papageorgis P, Cheng K, Ozturk S, et al. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Research. 2011;71(3):998–1008. doi: 10.1158/0008-5472.CAN-09-3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bärdos JI, Ashcroft M. Hypoxia-inducible factor-1 and oncogenic signalling. BioEssays. 2004;26(3):262–269. doi: 10.1002/bies.20002. [DOI] [PubMed] [Google Scholar]
- 47.Saji M, Ringel MD. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Molecular & Cellular Endocrinology. 2010;321(1):20–28. doi: 10.1016/j.mce.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes & Development. 2001;15(7):807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 49.Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin-associated protein. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(8):4438–4442. doi: 10.1073/pnas.96.8.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Molecular & Cellular Biology. 2001;21(12):3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Research. 2000;60(6):1541–1545. [PubMed] [Google Scholar]
- 52.Kazi AA, Koos RD. Estrogen-induced activation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor expression, and edema in the uterus are mediated by the phosphatidylinositol 3-kinase/Akt pathway. Endocrinology. 2007;148(5):2363–2374. doi: 10.1210/en.2006-1394. [DOI] [PubMed] [Google Scholar]
- 53.Deocampo ND, Huang H, Tindall DJ. The role of PTEN in the progression and survival of prostate cancer. Minerva Endocrinologica. 2003;28(2):145–153. [PubMed] [Google Scholar]
- 54.Zundel W, Schindler C, Haas-Kogan D, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes and Development. 2000;14(4):391–396. [PMC free article] [PubMed] [Google Scholar]
- 55.Eng C. Role of PTEN, a lipid phosphatase upstream effector of protein kinase B, in epithelial thyroid carcinogenesis. Annals of the New York Academy of Sciences. 2002;968:213–221. doi: 10.1111/j.1749-6632.2002.tb04337.x. [DOI] [PubMed] [Google Scholar]
- 56.Astanehe A, Arenillas D, Wasserman WW, et al. Mechanisms underlying p53 regulation of PIK3CA transcription in ovarian surface epithelium and in ovarian cancer. Journal of Cell Science. 2008;121(5):664–674. doi: 10.1242/jcs.013029. [DOI] [PubMed] [Google Scholar]
- 57.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nature Reviews Cancer. 2006;6(3):184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 58.Ravi R, Mookerjee B, Bhujwalla ZM, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia- inducible factor 1alpha. Genes & Development. 2000;14(1):34–44. [PMC free article] [PubMed] [Google Scholar]
- 59.Harada H, Itasaka S, Kizaka-Kondoh S, et al. The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. The Journal of Biological Chemistry. 2009;284(8):5332–5342. doi: 10.1074/jbc.M806653200. [DOI] [PubMed] [Google Scholar]
- 60.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4(2):147–158. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 61.Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cellular Signalling. 2009;21(6):827–835. doi: 10.1016/j.cellsig.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 62.Bernardi R, Guernah I, Jin D, et al. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442(7104):779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- 63.Land SC, Tee AR. Hypoxia-inducible factor 1aplha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. The Journal of Biological Chemistry. 2007;282(28):20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 64.Kaidi A, Williams AC, Paraskeva C. Interaction between alpha-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nature Cell Biology. 2007;9(2):210–217. doi: 10.1038/ncb1534. [DOI] [PubMed] [Google Scholar]
- 65.Baldewijns MM, Van Vlodrop IJH, Vermeulen PB, Soetekouw PMMB, Van Engeland M, De Bruïne AP. VHL and HIF signalling in renal cell carcinogenesis. The Journal of Pathology. 2010;221(2):125–138. doi: 10.1002/path.2689. [DOI] [PubMed] [Google Scholar]
- 66.Höckel M, Vaupel P. Biological consequences of tumor hypoxia. Seminars in Oncology. 2001;28(2, supplement 8):36–41. [PubMed] [Google Scholar]
- 67.Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist. 2004;9(supplement 5):4–9. doi: 10.1634/theoncologist.9-90005-4. [DOI] [PubMed] [Google Scholar]
- 68.Chavez A, Miranda LF, Pichiule P, Chavez JC. Mitochondria and hypoxia-induced gene expression mediated by hypoxia-inducible factors. Annals of the New York Academy of Sciences. 2008;1147:312–320. doi: 10.1196/annals.1427.021. [DOI] [PubMed] [Google Scholar]
- 69.Jiang Y, Zhang W, Kondo K, et al. Gene expression profiling in a renal cell carcinoma cell line: dissecting VHL and hypoxia-dependent pathways. Molecular Cancer Research. 2003;1(6):453–462. [PubMed] [Google Scholar]
- 70.Kaelin WG., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nature Reviews Cancer. 2008;8(11):865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 71.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nature Reviews Cancer. 2008;8(12):967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Milani M, Harris AL. Targeting tumour hypoxia in breast cancer. European Journal of Cancer. 2008;44(18):2766–2773. doi: 10.1016/j.ejca.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 73.Hung JJ, Yang MH, Hsu HS, Hsu WH, Liu JS, Wu KJ. Prognostic significance of hypoxia-inducible factor-1alpha, TWIST1 and Snail expression in resectable non-small cell lung cancer. Thorax. 2009;64(12):1082–1089. doi: 10.1136/thx.2009.115691. [DOI] [PubMed] [Google Scholar]
- 74.Scrideli CA, Carlotti CG, Mata JF, et al. Prognostic significance of co-overexpression of the EGFR/IGFBP-2/HIF-2A genes in astrocytomas. Journal of Neuro-Oncology. 2007;83(3):233–239. doi: 10.1007/s11060-007-9328-0. [DOI] [PubMed] [Google Scholar]
- 75.Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Research. 2002;62(12):3387–3394. [PubMed] [Google Scholar]
- 76.Park S, Shimizu C, Shimoyama T, et al. Gene expression profiling of ATP-binding cassette (ABC) transporters as a predictor of the pathologic response to neoadjuvant chemotherapy in breast cancer patients. Breast Cancer Research and Treatment. 2006;99(1):9–17. doi: 10.1007/s10549-006-9175-2. [DOI] [PubMed] [Google Scholar]
- 77.Yasuda H. Solid tumor physiology and hypoxia-induced chemo/radio-resistance: novel strategy for cancer therapy: nitric oxide donor as a therapeutic enhancer. Nitric Oxide—Biology and Chemistry. 2008;19(2):205–216. doi: 10.1016/j.niox.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 78.Salnikov AV, Heldin NE, Stuhr LB, et al. Inhibition of carcinoma cell-derived VEGF reduces inflammatory characteristics in xenograft carcinoma. International Journal of Cancer. 2006;119(12):2795–2802. doi: 10.1002/ijc.22217. [DOI] [PubMed] [Google Scholar]
- 79.Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Research. 1990;50(15):4478–4484. [PubMed] [Google Scholar]
- 80.Qayum N, Muschel RJ, Jae HI, et al. Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Research. 2009;69(15):6347–6354. doi: 10.1158/0008-5472.CAN-09-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pastorekova S, Kopacek J, Pastorek J. Carbonic anhydrase inhibitors and the management of cancer. Current Topics in Medicinal Chemistry. 2007;7(9):865–878. doi: 10.2174/156802607780636708. [DOI] [PubMed] [Google Scholar]
- 82.Rademakers SE, Span PN, Kaanders JHAM, Sweep FCGJ, Van der Kogel AJ, Bussink J. Molecular aspects of tumour hypoxia. Molecular Oncology. 2008;2(1):41–53. doi: 10.1016/j.molonc.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raghunand N, Gillies RJ. pH and chemotherapy. Novartis Foundation Symposium. 2001;240:199–211. doi: 10.1002/0470868716.ch14. [DOI] [PubMed] [Google Scholar]
- 84.Brown LM, Cowen RL, Debray C, et al. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Molecular Pharmacology. 2006;69(2):411–418. doi: 10.1124/mol.105.015743. [DOI] [PubMed] [Google Scholar]
- 85.Abend M. Reasons to reconsider the significance of apoptosis for cancer therapy. International Journal of Radiation Biology. 2003;79(12):927–941. doi: 10.1080/09553000310001632958. [DOI] [PubMed] [Google Scholar]
- 86.Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumor Biology. 2010;31(4):363–372. doi: 10.1007/s13277-010-0042-8. [DOI] [PubMed] [Google Scholar]
- 87.Koukourakis MI, Giatromanolaki A, Sivridis E, et al. Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. International Journal of Radiation Oncology Biology Physics. 2002;53(5):1192–1202. doi: 10.1016/s0360-3016(02)02848-1. [DOI] [PubMed] [Google Scholar]
- 88.Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5(5):429–441. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 89.Chadderton N, Cowen RL, Sheppard FCD, et al. Dual responsive promoters to target therapeutic gene expression to radiation-resistant hypoxic tumor cells. International Journal of Radiation Oncology Biology Physics. 2005;62(1):213–222. doi: 10.1016/j.ijrobp.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 90.Koukourakis MI, Bentzen SM, Giatromanolaki A, et al. Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2 alpha and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. Journal of Clinical Oncology. 2006;24(5):727–735. doi: 10.1200/JCO.2005.02.7474. [DOI] [PubMed] [Google Scholar]
- 91.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brüne B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Molecular Biology of the Cell. 2003;14(8):3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Milas L, Mason K, Hunter N, et al. In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clinical Cancer Research. 2000;6(2):701–708. [PubMed] [Google Scholar]
- 93.Kim TJ, Lee JW, Song SY, et al. Increased expression of pAKT is associated with radiation resistance in cervical cancer. British Journal of Cancer. 2006;94(11):1678–1682. doi: 10.1038/sj.bjc.6603180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. New England Journal of Medicine. 2006;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 95.Prevo R, Deutsch E, Sampson O, et al. Class I PI3 kinase inhibition by the pyridinylfuranopyrimidine inhibitor PI-103 enhances tumor radiosensitivity. Cancer Research. 2008;68(14):5915–5923. doi: 10.1158/0008-5472.CAN-08-0757. [DOI] [PubMed] [Google Scholar]
- 96.Chen JS, Zhou LJ, Entin-Meer M, et al. Characterization of structurally distinct, isoform-selective phosphoinositide 3’-kinase inhibitors in combination with radiation in the treatment of glioblastoma. Molecular Cancer Therapeutics. 2008;7(4):841–850. doi: 10.1158/1535-7163.MCT-07-0393. [DOI] [PubMed] [Google Scholar]
- 97.Li X, Kimbrel EA, Kenan DJ, McDonnell DP. Direct interactions between corepressors and coactivators permit the integration of nuclear receptor-mediated repression and activation. Molecular Endocrinology. 2002;16(7):1482–1491. doi: 10.1210/mend.16.7.0860. [DOI] [PubMed] [Google Scholar]
- 98.Lin HY, Sun M, Tang HY, et al. L-thyroxine vs. 3,5,3’-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. American Journal of Physiology. 2009;296(5):C980–C991. doi: 10.1152/ajpcell.00305.2008. [DOI] [PubMed] [Google Scholar]
- 99.Moeller LC, Cao X, Dumitrescu AM, Seo H, Refetoff S. Thyroid hormone mediated changes in gene expression can be initiated by cytosolic action of the thyroid hormone receptor beta through the phosphatidylinositol 3-kinase pathway. Nuclear Receptor Signaling Atlas. 2006;4:p. e020. doi: 10.1621/nrs.04020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Furuya F, Hanover JA, Cheng SY. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone β receptor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1780–1785. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hiroi Y, Kim HH, Ying H, et al. Rapid nongenomic actions of thyroid hormone. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(38):14104–14109. doi: 10.1073/pnas.0601600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Palmer LA, Semenza GL, Stoler MH, Johns RA. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. American Journal of Physiology. 1998;274(2):L212–L219. doi: 10.1152/ajplung.1998.274.2.L212. [DOI] [PubMed] [Google Scholar]
- 103.Liu R, Li Z, Bai S, et al. Mechanism of cancer cell adaptation to metabolic stress: proteomics identification of a novel thyroid hormone-mediated gastric carcinogenic signaling pathway. Molecular & Cellular Proteomics. 2009;8(1):70–85. doi: 10.1074/mcp.M800195-MCP200. [DOI] [PubMed] [Google Scholar]
- 104.Lin HY, Zhang S, West BL, et al. Identification of the putative MAP kinase docking site in the thyroid hormone receptor-alpha1 DNA-binding domain: functional consequences of mutations at the docking site. Biochemistry. 2003;42(24):7571–7579. doi: 10.1021/bi0273967. [DOI] [PubMed] [Google Scholar]
- 105.Ma Y, Freitag P, Zhou J, Brüne B, Frede S, Fandrey J. Thyroid hormone induces erythropoietin gene expression through augmented accumulation of hypoxia-inducible factor-1. American Journal of Physiology. 2004;287(3):R600–R607. doi: 10.1152/ajpregu.00115.2004. [DOI] [PubMed] [Google Scholar]
- 106.Otto T, Fandrey J. Thyroid hormone induces hypoxia-inducible factor 1alpha gene expression through thyroid hormone receptor beta/retinoid x receptor alpha-dependent activation of hepatic leukemia factor. Endocrinology. 2008;149(5):2241–2250. doi: 10.1210/en.2007-1238. [DOI] [PubMed] [Google Scholar]
- 107.Simonides WS, Mulcahey MA, Redout EM, et al. Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. The Journal of Clinical Investigation. 2008;118(3):975–983. doi: 10.1172/JCI32824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jubb AM, Pham TQ, Hanby AM, et al. Expression of vascular endothelial growth factor, hypoxia inducible factor 1alpha, and carbonic anhydrase IX in human tumours. Journal of Clinical Pathology. 2004;57(5):504–512. doi: 10.1136/jcp.2003.012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Allred DC, Clark GM, Elledge R, et al. Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. Journal of the National Cancer Institute. 1993;85(3):200–206. doi: 10.1093/jnci/85.3.200. [DOI] [PubMed] [Google Scholar]
- 110.Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441(7092):437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 111.Hou P, Liu D, Shan Y, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clinical Cancer Research. 2007;13(4):1161–1170. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- 112.Wang Y, Hou P, Yu H, et al. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/Akt pathway in thyroid tumors. Journal of Clinical Endocrinology and Metabolism. 2007;92(6):2387–2390. doi: 10.1210/jc.2006-2019. [DOI] [PubMed] [Google Scholar]
- 113.Bunone G, Vigneri P, Mariani L, et al. Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical pathological features. American Journal of Pathology. 1999;155(6):1967–1976. doi: 10.1016/S0002-9440(10)65515-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kilicarslan AB, Ogus M, Arici C, Pestereli HE, Cakir M, Karpuzoglu G. Clinical importance of vascular endothelial growth factor (VEGF) for papillary thyroid carcinomas. APMIS. 2003;111(3):439–443. doi: 10.1034/j.1600-0463.2003.t01-1-1110209.x. [DOI] [PubMed] [Google Scholar]
- 115.Lennard CM, Patel A, Wilson J, et al. Intensity of vascular endothelial growth factor expression is associated with increased risk of recurrence and decreased disease-free survival in papillary thyroid cancer. Surgery. 2001;129(5):552–558. doi: 10.1067/msy.2001.112592. [DOI] [PubMed] [Google Scholar]
- 116.Vieira JM, Santos SC, Espadinha C, et al. Expression of vascular endothelial growth factor (VEGF) and its receptors in thyroid carcinomas of follicular origin: a potential autocrine loop. European Journal of Endocrinology. 2005;129(5):552–558. doi: 10.1530/eje.1.02009. [DOI] [PubMed] [Google Scholar]
- 117.Kloos RT, Shah MH, Ringel MD, et al. Phase II trial of sorafenib in metastatic thyroid cancer. Journal of Clinical Oncology. 2009;27(10):1675–1684. doi: 10.1200/JCO.2008.18.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wells SA, Jr., Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. American Journal of Clinical Oncology. 2010;28(5):767–772. doi: 10.1200/JCO.2009.23.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gupta-Abramson V, Troxel AB, Nellore A, et al. Phase II trial of sorafenib in advanced thyroid cancer. Journal of Clinical Oncology. 2008;26(29):4714–4719. doi: 10.1200/JCO.2008.16.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bass MB, Sherman SI, Schlumberger MJ, et al. Biomarkers as predictors of response to treatment with motesanib in patients with progressive advanced thyroid cancer. The Journal of Clinical Endocrinology & Metabolism. 2010;95(11):5018–5027. doi: 10.1210/jc.2010-0947. [DOI] [PubMed] [Google Scholar]
- 121.Erler JT, Bennewith KL, Nicolau M, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440(7088):1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 122.Zhao XY, Chen TT, Xia L. Hypoxia inducible factor-1 mediates expression of galectin-1: the potential role in migration/invasion of colorectal cancer cells. Carcinogenesis. 2010;31(8):1367–1375. doi: 10.1093/carcin/bgq116. [DOI] [PubMed] [Google Scholar]
- 123.Chen J, Imanaka N, Chen J, Griffin JD. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. British Journal of Cancer. 2010;102(2):351–360. doi: 10.1038/sj.bjc.6605486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Scarpino S, Stoppacciaro A, Ballerini F, et al. Papillary carcinoma of the thyroid: hepatocyte growth factor (HGF) stimulates tumor cells to release chemokines active in recruiting dendritic cells. American Journal of Pathology. 2000;156(3):831–837. doi: 10.1016/S0002-9440(10)64951-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Scarpino S, D’Alena FC, Di Napoli A, Ballarini F, Prat M, Ruco LP. Papillary carcinoma of the thyroid: evidence for a role for hepatocyte growth factor (HGF) in promoting tumour angiogenesis. The Journal of Pathology. 2003;199(2):243–250. doi: 10.1002/path.1278. [DOI] [PubMed] [Google Scholar]
- 126.Brabletz T, Jung A, Reu S, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(18):10356–10361. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Scarpino S, d’Alena FC, Di Napoli A, Pasquini A, Marzullo A, Ruco LP. Increased expression of Met protein is associated with up-regulation of hypoxia inducible factor-I (HIF-I) in tumour cells in papillary carcinoma of the thyroid. The Journal of Pathology. 2004;202(3):352–358. doi: 10.1002/path.1522. [DOI] [PubMed] [Google Scholar]
- 128.Moeller BJ, Dewhirst MW. HIF-1 and tumour radiosensitivity. British Journal of Cancer. 2006;95(1):1–5. doi: 10.1038/sj.bjc.6603201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Magnon C, Opolon P, Ricard M, et al. Radiation and inhibition of angiogenesis by canstatin synergize to induce HIF-1α-mediated tumor apoptotic switch. The Journal of Clinical Investigation. 2007;117(7):1844–1855. doi: 10.1172/JCI30269. [DOI] [PMC free article] [PubMed] [Google Scholar]