

Introduction
Asthma is a syndrome characterized by intermittent attacks of breathlessness, wheezing and cough, often nocturnal. The disease most commonly onsets in childhood, where it can usually be controlled with inhaled medications, and runs a variable natural history; as many as one in four may continue to have symptoms that persist or recur in adulthood. The childhood disease is often associated with other allergic disorders, such as atopic dermatitis (eczema) and seasonal rhinitis (hayfever). The latter manifestations can precede the recognition of full-blown asthma, a sequence denoted ‘atopic march’. Patients who have persistent disease into adulthood, or who have onset in adulthood, are more likely to include subgroups with more frequent and severe episodes that require intensification of therapy. In general, however, the severity of the disease is usually established at or near the time of onset.
A breakthrough in the understanding of asthma pathogenesis was made with the recognition that chronic inflammation of the conducting airways characterizes the disease, even during asymptomatic periods. Inflammation included T helper-2 CD4 T (Th2) cells and Th2-associated cytokines, as well as eosinophils, a finding that greatly informed studies in experimental animals seeking to understand the initiation and maintenance of allergic immunity (Robinson et al., 1992). More recent findings have suggested that IL-17-associated neutrophilic airway inflammation and even ‘pauci-granulocytic’ subtypes may also occur (Wenzel, 2006). Whether alternative forms of inflammation represent distinct subtypes of the disease or heterogeneity in the response to currently available therapeutics, or both, is unknown. Persistent inflammation, possibly fueled by immune responses to repeated inhalations of environmental allergens, such as pollens, grasses, animal danders, molds and excreta from urban-dwelling insects, particularly dust mites and cockroaches, results in compensatory structural airway alterations, including epithelial mucus metaplasia, smooth muscle hypertrophy and enhanced deposition of subepithelial matrix glycoproteins. These changes, collectively termed ‘airway remodeling’, are thought to lead to the persistent, poorly reversible airflow limitation and airway hyperresponsiveness found in some patients with chronic asthma. Airway hyperresponsiveness can be revealed using aerosolized agonist drugs, such as methacholine, that induce smooth muscle contraction, or by respiratory irritants, including pollutants (e.g., sulfur dioxide, diesel fuel particles) and recurrent respiratory virus infections, such as by rhinoviruses or respiratory syncytial viruses. Despite many advances, it is clear that no animal models completely simulate human disease, and more access to human tissues will be required to gain deeper understanding of the natural history, breadth and pathogenesis of asthma.
Scope of the problem
Asthma and allergic inflammation, including food allergies, have increased over the past 50 years to become the most prevalent chronic illnesses of childhood in developed countries. The estimated asthma worldwide prevalence is 300 million persons. In the United States, childhood asthma prevalence more than doubled between 1980 and the mid-1990’s, with 2007 estimates projecting 16.2 million adults and 6.7 million children with the disease – over 8% of the population (Moorman et al., 2007). Although mortality is infrequent, asthma patients drive extensive use of the health care system, accounting for approximately 10 million office visits, 400,000 hospitalizations and 200,000 emergency room visits; in some communities, one in four emergency room patients are seen for asthma. Annual economic costs approach $20 billion. Over half of patients with asthma have additional allergic diseases, including food allergies. The latter has reached awareness levels such that peanut butter has been essentially banned from grade schools in many metropolitan areas of the United States.
The increase in allergic diseases over relatively short time periods implicates environmental factors as overwhelming determinants of disease risk. Genetic predisposition to asthma is clear, however, with family and twin studies suggesting hereditary contributions approaching 60%. Genome-wide studies of asthma in carefully phenotyped populations remain few, and, similar to other chronic complex human diseases, suggest relatively small contributions by many loci, none of which have been rigorously quantified (Rogers et al., 2009). Posited genome-environment interactions, as proposed for most complex human diseases, are only vaguely defined. Genetic disparities that drive ethnic differences in asthma risk, severity or drug responsiveness also obscure analysis (Burchard et al., 2003). Replication against larger cohorts of well-defined populations will be needed. Intriguing links with genes involved in the pathway for keratinocyte terminal differentiation to barrier cells secreting the protein-lipid matrix that regulates skin permeability have suggested a connection with epithelial barrier dysfunction. Common loss-of-function mutations in filaggrin, present in up to 10% of Western Europeans and their descendents, have been associated with severe atopic dermatitis, and, in some studies, with risk for asthma (Muller et al., 2009). Loss of barrier function may establish breaches for allergen entry and priming (Fallon et al., 2009) and/or lead to changes in the commensal microbial flora that alter the homeostatic regulation of immunity in ways that predispose to allergy, perhaps mediated by elaboration of the epithelial cell cytokine, thymic stromal lymphopoietin (TSLP; Demehri et al., 2009). Epidemiologic studies have repeatedly identified more severe viral upper respiratory infections, most commonly rhinoviruses, early in life as a major risk, particularly in families with a history of atopy. An urbanized life-style in early life also increases risk, suggesting that post-natal exposure to indoor antigens or urban pollutants affects the quality of subsequent mucosal immunity. Alternatively, loss of microbial exposures associated with rural environments may shape the infant immune system.
Whatever underlies the rising prevalence of asthma, it is recognized that some patients, although the exact proportion is unknown, progress to irreversible lung changes characterized by the increased subepithelial fibrosis and structural hyperplasia of ‘airway remodeling’. When severe, these patients can overlap the pathologic spectrum of patients with chronic bronchitis. The impact of primary and secondary smoking exposure has complicated analysis, although cases can occur in nonsmokers. Animal models have been very informative in increasing understanding of asthma, but these are imperfect, and lack of access to patient tissues has hampered the ability to delineate fully the natural history and full spectrum of human disease.
Pathogenesis: current status
Asthma and food allergies are initiated at mucosal surfaces where environmental allergens contact epithelia. The physical properties that endow environmentally widespread entities with allergenic properties are of much interest, but in asthma, at least minimally involve the capacity to be sustained in aerosolized forms that reach conducting airways and evade mucociliary clearance. Allergens may share properties such as stability, protease activity or molecular mimicry, which endows them with the capacity to penetrate mucus and epithelial barriers and induce cytokine release by engaging innate pattern recognition receptors on epithelia and resident lung myeloid cells, including macrophages, dendritic cells and mast cells. Natural allergens are typically complex biologic mixtures of constituents that share many individual attributes, a feature infrequently addressed in experimental models (see below). Airway inflammation is sustained from these initial encounters through the interactions between epithelia of the airways and sentinel antigen processing cells, likely resident dendritic cells, although recent studies suggest that basophils may subserve this function in allergic inflammation in some situations (see below). In this way, adaptive immunity is engaged through the development of antigen-specific memory T cells and antibody-secreting plasma cells, thus establishing the potential for chronicity upon repeated exposures to allergens.
The subsequent pathways are based on amalgamation of mouse studies, often in the setting of over-expression or deletion of various genes, and human studies based largely on in situ analysis and studies of isolated cells and cell lines. Differences in cells, receptors and anatomy of the airways between the species suggest caveats in creating a unifying hypothesis of current understanding. Of necessity over-simplified, the reader is referred to recent reviews (Barrett and Austen, 2009; Lambrecht and Hammad, 2009; Saenz et al.; 2008). Briefly, current models envision the release of epithelial cytokines, particularly IL-25, IL-33 and TSLP, and CC family chemokines, as proximal events important in initiating allergic inflammation (Fig. 1). Although few studies are available, IL-25, IL-33 and TSLP can be elicited from epithelia when activated by allergenic stimuli or by helminths, and can induce IL-13-dependent allergic inflammation when administered individually to mice or when over-expressed as transgenes. In the lung, TSLP may be released from other sources as well, as epithelial production at this site is lower than in skin and bowel. These cytokines also target resident hematopoietic cells to induce the influx of inflammatory cells from the circulation and the activation and mobilization of dendritic cells. Although all of these cytokines can target multiple cell types, IL-33 potently activates mast cells to secrete vasoactive amines, lipid mediators, chemokines and cytokines, and as such may contribute to anaphylaxis (Pushparaj et al., 2009). Basophils, initially fewer in number in parenchymal tissues, are also activated by IL-33 to secrete cytokines, particularly IL-4. IL-25 and IL-33 potently activate incompletely characterized IL-25R+ lymphoid cells in tissues (Hurst et al., 2002; Moro et al., 2009) that respond with production of IL-5 and IL-13, key cytokines involved in mediating, respectively, the survival of eosinophils, and the induction of chemokines, the differentiation of mucus-secreting goblet cells, a pro-fibrogenic stromal environment, alternative macrophage activation and smooth muscle alterations that contribute to enhanced airway hyperresponsiveness; IL-4, which shares use of the type II IL-4 receptor with IL-13, contributes to these latter functions, making this shared receptor an attractive pharmaceutical target. TSLP targets dendritic cells and mediates mobilization, activation and induction of the TNF superfamily member, OX40L. In both mouse and human studies, OX40L-positive dendritic cells activate naïve CD4 T cells to an IL-4-competent state, consistent with an important priming step in Th2 cell differentiation.
Figure 1.
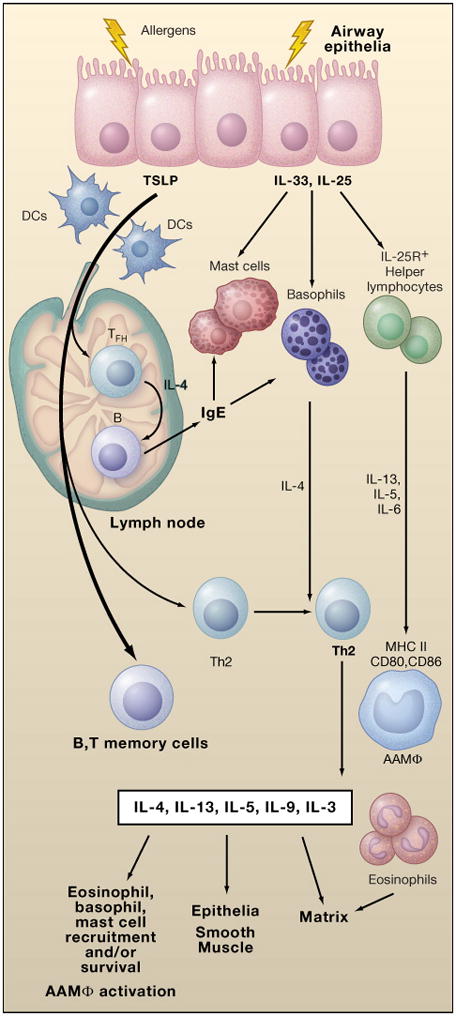
Initiation of allergic immunity is mediated through interactions of allergens with epithelia that result in release of the cytokines TSLP, IL-33, and IL-25. TSLP mediates DC migration and maturation that primes IL-4 competency (green) in helper T cells. Competent cells moving into follicular areas mature into IL-4-secreting TFH cells, and mediate B cell isotype-switching to IgE (and, in the mouse, to IgG1, in the germinal center reaction), which, in turn, binds to mast cells and basophils, thus extending their survival and facilitating allergen-specific activation of these cells. IL-33 promotes IL-4 release from basophils, and IL-33 and IL-25 promote IL-13 and IL-5 release from tissue IL25R+ cells (these cells also release IL-6 which supports B cell maturation and impairs T regulatory cell function). IL-4, IL-5 and IL-13 from these innate cells promote terminal differentiation and/or recruitment of Th2 cells to tissues, as well as alternative macrophage activation (AAMφ) and eosinophil recruitment. These effects are greatly augmented by activation of tissue Th2 cells, which contribute a diverse set of cytokines that feedback to facilitate the survival and activation of innate cells and the effects of Th2-associate cytokines on epithelia, smooth muscle and the stromal matrix. Memory T and B cells are generated that can facilitate more rapid responses to repeated stimulation, particularly if the regulatory T cell response is impaired (not shown).
Once activated, IL-4 competent T cells in lymph nodes migrate to B cell zones and differentiate to T follicular helper (TFH) cells, or exit into the draining lymph and thence the circulation to complete maturation as Th2 cells (Fig. 1). IL-4-secreting TFH cells in parafollicular B cell areas mediate IgE switching and conjugate with B cells to drive IgG1 (and possibly IgG4 in human) switching and affinity maturation in the germinal center reaction (Reinhardt et al., 2009). The understanding of TFH cells has been confusing due to the inability to distinguish these cells by surface phenotype, which is shared among most activated T cells in the lymph node. Most lymph node IL-4-secreting cells during allergic immunity are actually TFH cells that mediate B cell help, and not Th2 cells, which exit the lymph node and go to peripheral sites of inflammation. In tissues, effector Th2 cells augment the survival of eosinophils and basophils by secretion of IL-5 and IL-3, respectively, facilitate mast cell survival via IL-9, and contribute antigen-specific elaboration of IL-4 and IL-13 required for epithelial and smooth muscle manifestations of the disease. Eosinophils, when sustained, mediate tissue remodeling and fibrosis. Macrophages assume an alternatively activated state in the setting of IL-4 and IL-13 stimulation, and secrete proteases and enzymes involved in matrix remodeling; these cells also participate in restraining tissue inflammation. Many manifestations of the response resemble wound healing and likely target resolution of the mucosal irritation. Over time, allergen-specific IgE (and, in the mouse, IgG1) antibodies occupy the high-affinity IgE Fc immunoglobulin receptor, FcεRI, expressed on mast cells and basophils (and, in the mouse, the respective IgG1-binding FcγRIII on these cells as well) to provide alternative mechanisms for cell activation by antibody crosslinking. With repeated allergen exposure, FcεRI activation leads to the rapid production of vasoactive amines and lipid mediators that underlie the acute fall in respiratory function after allergen challenge, while also driving the elaboration of cytokines, chemokines and lipid mediators that regulate the delayed phase of the inflammatory process amplified by recruited cells. A common emergent theme is the role of adaptive immunity in providing cytokines and growth factors that sustain the recruitment, retention and extended lifespan of a select group of myeloid cells, including eosinophils, basophils and mast cells, in involved tissues. Occupancy of FcεRI by IgE can itself enhance the survival of mast cells and basophils, and serum IgE levels in humans correlate with FcεRI expression levels on DCs, where, unlike in the mouse, components of the FcεRI are constitutively present on additional types of hematopoietic cells (Kraft and Kinet, 2007). Even relatively rare cell types, such as basophils, are increased in tissues from cases of fatal asthma (Kepley et al., 2001).
Control of inflammation by regulatory T cells, Treg, may also be compromised in asthma, although the precise pathways remain unclear. TGF-β and IL-10 have been implicated in the resolution of allergic inflammation by Treg but dysregulated expression of these cytokines has been connected with progressive fibrosis and with a diminished capacity to restrain respiratory viruses, respectively. Further study of the natural history of Treg and their specificity in allergic lung inflammation is needed (Lloyd and Hawrylowicz, 2009). With repeated allergen exposure, eosinophil and basophil infiltration become sustained, the mast cell pool enlarges, the epithelia undergo enhanced goblet cell differentiation with copious mucus production, thickening of the basal lamina reticularis is apparent, and increases in smooth muscle mass occur. The result is hyperresponsiveness to common airway irritants. Although much insight has been gained in understanding the pathogenesis of asthma associated with Th2 cells and allergic pathology, very little is understood regarding the mechanisms that sustain airway inflammation among patients with neutrophilic or ‘pauci-granulocytic’ presentations that make up a substantial number of adult-onset cases of asthma.
New directions and needs
Common environmental allergens are complex mixtures of bioactive entities. House dust mite allergens that elicit IgE reactivity include a papain-like cysteine protease; a lipid-binding MD-2 molecular mimetic capable of augmenting TLR4 signaling (Trompette et al., 2009); trypsin-like, chymotrypsin-like and serine proteases; fatty acid- and lipid-binding proteins; chitinases; and at least ten other proteins (Thomas et al., 2002). Chitin and a variety of polymeric glycans are also present in mite and cockroach fecal pellets (see below). Cockroach allergens are similarly diverse and include active proteases and lipid-binding proteins involved in food digestion that are secreted in the fecal pellets. Fungi, a highly diverse spectrum of organisms, can contribute up to 10% of the particulate mass in air of sufficient size to be inhaled into the terminal airways and alveoli (Frohlich-Nowoisky et al., 2009). Fungi contain cell wall chitin and β-glucans that potently elicit eosinophil and neutrophil tissue infiltration in experimental models, and also secrete proteases implicated in allergic lung immunity (Porter et al., 2009). Chitinases and β-glucanases are present in plants, and are induced in edible fruits and berries as part of evolutionarily ancient components of defense against invasive chitin-bearing fungi and insects. Like chitinases in insect excreta, these plant-derived proteins, together with a number of lipid-binding proteins, are common targets of IgE reactivity in atopic individuals. Cross-reactivity of IgE may occur among these shared group responses. Such cross-reactivity with chitin- and β-glucan-binding domain-like structures in natural latex rubber has been proposed to contribute to the prevalence of ‘latex-fruit syndrome’; similar cross-reactivity may underlie the emergence of ‘pollen-food allergy syndrome’. Immunologists are only beginning to explore these complex antigen mixtures in experimental models of allergic lung disease, which, in humans, is sustained by ‘cocktails’ of diverse natural substances that intersect multiple innate immune pathways and contribute to allergenic priming and inflammation.
Cell wall β-glucans in fungi are potent stimuli of dectin C-type lectin receptors on dendritic cells, macrophages and neutrophils, and initiate IL-17-driven neutrophilic infiltration that may be relevant to the understanding of inflammatory subsets of asthma (Goodridge et al., 2009). Chitin, an insoluble polymer of N-acetyl-β-D-glucosamine, is a structural element shared by fungi, crustacea, helminths and insects, and constitutes, after cellulose, the most widespread natural biopolymer. When administered to the airways of mice, chitin induces a leukotriene-mediated infiltrate of eosinophils and basophils and drives alternative macrophage activation in both resident and recruited macrophages (Reese et al., 2007). A conserved epithelial and macrophage chitinase, AMCase, is rapidly induced by a Stat6-dependent pathway and degrades chitin, thus solubilizing the particulate structure. Chitin is a component of the fungal cell wall and a constituent of the insect peritrophic matrix (PM). The PM is a complex network of chitin, chitin-binding proteins, glycoproteins and glycans that encloses the food bolus with a mix of intestinal digestive enzymes (Hegedus et al., 2009). Spatially compartmentalizing the digestive process in the insect, the PM is ultimately excreted as fecal particles enveloping the digestive enzymes, including the allergenic proteases and lipid-binding proteins. Further work is necessary to establish whether environmental chitin from molds and insect excreta contributes to their sensitizing capacity, and whether such associations contribute to links between asthma risk and prevalent human variants of AMCase (Siebold et al., 2009). Intriguingly, variants of the chitin-binding chi-lectin, YKL-40, have also been associated with asthma (Ober et al., 2008).
Several recent reports have used approaches to isolate MHC class II expression to basophils in experimental mouse systems involving proteases, helminths, IgE-mediated immunity and airways challenge to suggest a requisite role for basophils as antigen processing and presenting cells in allergic immunity (Niu et al., 2009; Perrigoue et al., 2009; Sokol et al., 2009; Yoshimoto et al., 2009). Together, the findings in multiple systems using creative reagents demand further scrutiny of the possibility, although caution will be required before divesting dendritic cells from a role in Th2-associated lung immunity. Mechanisms for purifying and identifying basophils, and for maintaining their viability, remain imperfect, and reliance on FcεRI antibodies for basophil deletion and detection may be problematic if mouse DCs in allergic inflammatory states can be induced to express this receptor (Grayson et al., 2007). Novel reagents that direct MHC class II to CD11c-expressing cells were used to demonstrate the lack of need for classical DCs, but do not recapitulate wild-type expression of MHC class II on DCs, particularly on migratory DCs required for the efficient induction of effector immunity (Allenspach et al., 2008). Further, CD11c is expressed on other cells during allergic inflammation, including eosinophils (Voehringer et al., 2004). Indeed, all myeloid cells, including neutrophils, basophils, eosinophils, mast cells and monocyte/macrophages can be induced to express MHC class II and some have been linked with antigen presentation (Kambayashi et al., 2009; Wang et al., 2007). In each case, expression of MHC class II is low as compared to mature dendritic cells, and is more compatible with a role for these cells in augmenting inflammation through interactions with effector Th2 cells in peripheral tissues rather than in priming naïve T cells in lymph nodes, where activation thresholds are greater. Indeed, activation of naïve T cells to gain IL-4 competence is IL-4 independent during priming in lymph nodes (van Panhuys et al., 2008), but entry into and/or maturation of Th2 cells in tissues is highly Stat6-dependent, suggesting the need for exogenous IL-4 in mediating localization, maturation and/or activation of Th2 cells in affected tissues rather than in lymph node priming. Here, imaging studies (see below) would allow more compelling evidence that basophils, as compared to DCs, can mediate efficient activation of naïve T cells in lymph nodes. Accurate lineage marking of basophils has not been accomplished and remains a pressing need. Regardless, the confluence of the recent studies requires further investigation, including extension to humans, in order not to overlook findings of fundamental importance. Contributions of other IL-13-producing cells, such as incompletely characterized IL-25R+ lymphoid cells in lung and intestinal tissues, need to be examined, particularly in humans, as targeting IL-25, a potent activator of these cells, has substantial benefits in animal models of allergic lung inflammation (Ballantyne et al., 2007).
The absence of efficient imaging modalities has stymied efforts to more accurately assess components of allergic inflammation in the lung and draining lymph nodes. The lung presents a complex highly branched tissue optimized for gas exchange but offering a large mucosal surface for antigen surveillance. A heterogeneous group of lung DCs and macrophages patrol epithelia from the large conducting airways to the alveolae, and the balance of toleragenic and stimulatory responses is only beginning to be unraveled (Lambrecht and Hammad, 2009). The precise flow of soluble allergens such as proteases or particulates like chitin from conducting airways to the mediastinal lymph nodes is not well studied, although anatomic studies in rodents suggest precise partitioning of materials ferried from the lung to draining lymph nodes. Although not without limitations, thick sections of agarose-inflated lung can be maintained in oxygenated media for up to five days, and offer an approach yet to be fully explored for live imaging (Delmotte and Sanderson, 2006). Fluorescent molecular tomography (FMT) uses protease-sensitive probes to detect inflammation by transillumination in living animals. The depth of penetration and scale of resolution allow reconstructive analysis of three-dimensional tissues, including the lung. The pathogenic role of proteases in asthma and allergy suggests that FMT can be utilized with cell lineage markers to more accurately study the organization of the inflammatory milieu in the lung (Cortez-Retamozo et al., 2008). Computerized tomographic imaging is being paired with functional studies using inhaled hyperpolarized 3He magnetic resonance imaging to investigate more completely the mechanisms driving airway collapse and airspace trapping that underlies the ventilation heterogeneity in asthma. Although thick-walled airways and luminal narrowing are evident, more surprising is the extended time required for recovery of ventilatory abnormalities after allergen challenge in asthmatic as compared to non-asthmatic individuals, and the finding that ventilation defects in a given subject tend to be fixed in the same areas with repeated studies over time, suggesting an irreversible structural component to the process (de Lange et al., 2009; Tzeng et al., 2009). Asthma has complex effects on the lung due to the interconnections between intrinsic tissue and conducting airway mechanics, and the functional consequences of airspace collapse on both. Further efforts linking the physical and immunological realms are needed.
The big picture
Despite the presence of homologs and paralogs in flies and other insects that reveal vestiges of immunity against bacteria, fungi and viruses mediated by macrophage-like phagocytic cells, similar evolutionary evidence for the origins of allergic immunity is not obvious. The increasingly complex nutrient needs of larger organisms with prolonged parturition has been linked with expanded mucosal surfaces required for tissue oxygenation and caloric uptake. The latter has driven a symbiotic developmental relationship with a highly diverse microbial commensal flora intimately involved in optimizing nutrient extraction and mucosal immune homeostasis that has no exact counterpart in invertebrates (Backhed et al., 2005; Round and Mazmanian, 2009). The constituents of the bacterial flora and their contributions to human health are being increasingly defined, but it is likely that additional intestinal resident organisms – namely, parasitic heminths – also drove mutualism in the mucosa. In natural environments worldwide the great majority of vertebrates are infected with intestinal helminths, including humans in rural environments where exposure to contaminated soils and water is high. Oetzi the iceman, a mummified individual over 50 centuries old found in the Alps of Europe, was infected with whipworm (Aspock et al., 1996). Parasitic helminths include nematodes (roundworms, such as hookworms and whipworms) and platyhelminths (flatworms, including flukes and tapeworms) and afflict billions of people in developing countries. The diversity in natural environments is astounding (Parkinson et al., 2004). Nematodes alone have been estimated to make up 80% of all animals on the earth and comprise possibly a million species. Among the five nematode clades, all include parasitic species of plants and animals.
In experimental studies, the response to helminths shares many attributes with asthma and allergic inflammation, including requirements for IL-4 and IL-13, the presence of eosinophil-rich inflammatory infiltrates, elevated IgE and alterations in smooth muscle contractility, goblet cell hyperplasia and mucus production, and increased epithelial cell turnover, all of which are felt to underlie the ‘weep and sweep’ response to intestinal infestation. Helminths secrete an array of proteases and lipid-binding proteins, and express chitin in eggs and the cuticular pharynx. Despite the inclination to associate allergic inflammation with host immunity to helminths, it is not clear that helminths have not co-evolved with the immune response to fashion a niche in which to establish a symbiotic relationship in the nutrient-rich intestines. Examples have been noted in which helminths exploit eosinophilia for infection, induce alternative macrophage activation and elicit cytokines, such as TSLP and IL-25, that mitigate mucosal injury, and moderate Treg development in involved tissues (Maizels et al., 2009). In healthy Ugandans, eosinophilia is the single most common laboratory ‘abnormality’ as based on Western hematologic reference values and contributes to the difficulty in conducting vaccine trials against HIV and other pathogens due to exclusion criteria (Eller et al., 2008). It is more likely, however, that eosinophilia and increased numbers of tissue basophils and mast cells were ‘normal’ hematologic responses during human evolution, and represent homeostatic responses induced to maintain healthy tissues in the face of universal intestinal parasitism. Inflammatory diseases such as asthma, but also obesity, metabolic syndrome and atherosclerosis, have increased in areas where intestinal parasitism has decreased. Further investigations are needed to assess whether the symbiosis of early humans with intestinal helminths as compared to the widespread absence in urbanized humans today has led to significant modifications in the microbial commensal flora, changes in the availability of micronutrients such as vitamins A and D involved in mucosal immune homeostasis, or even alterations in the kinetics of the development of the mucosal lymphoid system. Early alterations of airway microbial flora have been noted in infants at later risk for asthma (Bisgaard et al., 2007). Such speculation has been advanced previously as a component of the ‘hygiene hypothesis’ (Yazdanbakhsh et al., 2002). Further study of the impact of intestinal parasitism on mucosal bacterial commensals and immune homeostasis in humans might prove fruitful in attempting to understand the increasing prevalence of allergic diseases among humans in a modern world (Fig. 2). Obesity, itself a contributor to asthma severity, may also result from alterations in pathways impacting the human microbiota (Blaser and Falkow, 2009). Until better insights regarding the nature and function of allergic immunity are known, tempered caution is offered that we not eliminate all facets of these responses in order that basic homeostatic pathways are not inadvertently compromised.
Figure 2.

The loss of universal helminth infection as occurred in earlier human evolution may alter the numbers or types of bacterial and fungal commensals and thus affect normal mucosal tissue homeostasis. In susceptible or highly exposed individuals, such alterations might alter the balance between immunotolerance, immunosurveillance and nutrient extraction. This imbalance may contribute to the appearance of inflammatory systemic dysregulation at mucosal surfaces, resulting in increases in asthma and allergic diseases, particularly in the setting of environmental changes that have increased exposure to indoor allergens and pollutants, and even to increases in obesity, which can be a risk factor for severe asthma.
Acknowledgments
I gratefully acknowledge helpful discussions and input from faculty and members of my laboratory at UCSF and support from the Howard Hughes Medical Institute, the National Institutes of Health and the Strategic Asthma Basic Research Center at UCSF. I apologize for being unable to cite the extensive primary literature due to space limitations.
References
- Allenspach EJ, Lemos MP, Porrett PM, Turka LA, Laufer TM. Migratory and lymphoid-resident dendritic cells cooperate to efficiently prime naïve CD4 T cells. Immunity. 2008;29:1–12. doi: 10.1016/j.immuni.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspock H, Auer H, Picher O. Trichuria trichiura eggs in the Neolithic glacier mummy from the alps. Parasitol Today. 1996;12:255–256. [Google Scholar]
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, Sturton G, Wong SH, McKenzie AN. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2007;120:1324–1331. doi: 10.1016/j.jaci.2007.07.051. [DOI] [PubMed] [Google Scholar]
- Barrett NA, Austen F. Innate cells and T helper 2 cell immunity in airway inflammation. Immunity. 2009;31:425–437. doi: 10.1016/j.immuni.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bonnelykke K, Brasholt M, Heltberg A, Vissing NH, Thorsen SV, Stage M, Pipper CB. Childhood asthma after bacterial colonization of the airway in neonates. New Engl J Med. 2007;357:1487–1495. doi: 10.1056/NEJMoa052632. [DOI] [PubMed] [Google Scholar]
- Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchard EG, Ziv E, Coyle N, Gomez SL, Tang H, Karter AJ, Mountain JL, Perez-Stable EJ, Sheppard D, Risch N. The importance of race and ethnic background in biomedical research and clinical practice. New Engl J Med. 2003;348:1170–1175. doi: 10.1056/NEJMsb025007. [DOI] [PubMed] [Google Scholar]
- Cortez-Retamozo V, Swirski FK, Waterman P, Yuan H, Figueiredo JL, Newton AP, Upadhyay R, Vinegoni C, Kohler R, Blois J, Smith A, Nahrendorf M, Josephson L, Weissleder R, Pittet MJ. Real-time assessment of inflammation and treatment response in a mouse model of allergic airway inflammation. J Clin Invest. 2008;118:4058–4066. doi: 10.1172/JCI36335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lange EE, Altes TA, Patrie JT, Battiston JJ, Juersivich AP, Mugler JP, Platts-Mills TA. Changes in regional airflow obstruction over time in the lungs of patients with asthma: evaluation with 3He MR imaging. Radiology. 2009;250:567–575. doi: 10.1148/radiol.2502080188. [DOI] [PubMed] [Google Scholar]
- Delmotte P, Sanderson MJ. Ciliary beat frequency is maintained at a maximal rate in the small airways of mouse lung slices. Am J Respir Cell Mol Biol. 2006;35:110–117. doi: 10.1165/rcmb.2005-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demehri S, Morimoto M, Holtzman MJ, Kopan R. Skin-derived TSLP triggers progression from epidermal barrier defects to asthma. PLOS Biol. 2009;7:e1000067. doi: 10.1371/journal.pbio.1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, Callanan JJ, Kawasaki H, Shiohama A, Kubo A, Sundberg JP, Presland RB, Fleckman P, Shimizu N, Kudoh J, Irvine AD, Amagai M, McLean WH. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41:602–608. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eller LA, Eller MA, Ouma B, Kataaha P, Kyabaggu D, Tumusiime R, Wandege J, Sanya R, Sateren WB, Wabwire-Mangen F, Kibuuka H, Robb ML, Michael NL, de Souza MS. Reference intervals in healthy adult Ugandan blood donors and their impact on conducting international vaccine trials. PLoS ONE. 2008;3:e3919. doi: 10.1371/journal.pone.0003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich-Nowoisky J, Pickersgill DA, Despres VR, Poschl U. High diversity of fungi in air particulate matter. Proc Natl Acad Sci USA. 2009;106:12814–12819. doi: 10.1073/pnas.0811003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge HS, Wolf AJ, Underhill DM. Beta-glucan recognition by the innate immune system. Immunol Rev. 2009;230:38–50. doi: 10.1111/j.1600-065X.2009.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson MH, Cheung D, Rohlfing MM, Kitchens R, Spiegel DE, Tucker J, Battaile JT, Alevy Y, Yan L, Agapov E, Kim EY, Holtzman MJ. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med. 2007;204:2759–2769. doi: 10.1084/jem.20070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus D, Erlandson M, Gillott C, Toprak U. New insights into peritrophic matrix synthesis, architecture, and function. Annu Rev Entomol. 2009;54:285–302. doi: 10.1146/annurev.ento.54.110807.090559. [DOI] [PubMed] [Google Scholar]
- Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, Menon S, Seymour B, Jackson C, Kung TT, Brieland JK, Zurawski SM, Chapman RW, Zurawski G, Coffman RL. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol. 2002;169:443–453. doi: 10.4049/jimmunol.169.1.443. [DOI] [PubMed] [Google Scholar]
- Kambayashi T, Allenspach EJ, Chang JT, Zou T, Shoag JE, Reiner SL, Caton AJ, Koretzky GA. Inducible MHC class II expression by mast cells supports effector and regulatory T cell activation. J Immunol. 2009;182:4686–4695. doi: 10.4049/jimmunol.0803180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepley CL, McFeeley PJ, Oliver JM, Lipscomb MF. Immunohistochemical detection of human basophils in postmortem cases of fatal asthma. Am J Respir Crit Care Med. 2001;164:1053–1058. doi: 10.1164/ajrccm.164.6.2102025. [DOI] [PubMed] [Google Scholar]
- Kraft S, Kinet JP. New developments in FcεRI regulation, function and inhibition. Nat Rev Immunol. 2007;7:365–378. doi: 10.1038/nri2072. [DOI] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–424. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Lloyd CM, Hawrylowicz Regulatory T cells in asthma. Immunity. 2009;31:438–449. doi: 10.1016/j.immuni.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizels RM, Pearce EJ, Artis D, Yazdanbakhsh M, Wynn TA. Regulation of pathogenesis and immunity in helminth infections. J Exp Med. 2009;206:2059–2066. doi: 10.1084/jem.20091903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman JE, Rudd RA, Johnson CA, King M, Minor P, Bailey C, Scalia MR, Akinbami LJ. Surveillance for asthma – United States, 1980–2004. Morbid Mortal Weekly Rep. 2007;56:1–14. [PubMed] [Google Scholar]
- Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S. Innate production of Th2 cytokine by adipose tissue-associated c-kit+Sca-1+ lymphoid cells. Nature. 2009 doi: 10.1038/nature08636. (in press) [DOI] [PubMed] [Google Scholar]
- Muller S, Marenholz I, Lee YA, Sengler C, Zitnik SE, Griffioen RW, Meglio P, Wahn U, Nickel R. Association of filaggrin loss-of-function mutations with atopic dermatitis and asthma in the early treatment of the atopic child (ETAC) population. Pediatr Allergy Immunol. 2009;20:358–61. doi: 10.1111/j.1399-3038.2008.00808.x. [DOI] [PubMed] [Google Scholar]
- Niu N, Laufer T, Homer RJ, Cohn L. Cutting edge: limiting MHC class II expression to dendritic cells alters the ability to develop Th2-dependent allergic airway inflammation. J Immunol. 2009;183:1523–1527. doi: 10.4049/jimmunol.0901349. [DOI] [PubMed] [Google Scholar]
- Ober C, Tan Z, Sun Y, Possick JD, Pan L, Nicolae R, Radford S, Parry RR, Heinzmann A, Deichmann KA, Lester LA, Gern JE, Lemanske RF, Nicolae DL, Elias JA, Chupp GL. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. New Engl J Med. 2008;358:1652–1691. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J, Mitreva M, Whitton C, Thomson M, Daub J, Martin J, Schmid R, Hall N, Barrell B, Waterston RH, McCarter JP, Blaxter ML. A transcriptome analysis of the phylum Nematoda. Nat Genet. 2004;36:1259–1267. doi: 10.1038/ng1472. [DOI] [PubMed] [Google Scholar]
- Perrigoue JG, Saenz SA, Siracusa MC, Allenspach EJ, Taylor BC, Giacomin PR, Nair TM, Artis D. MHC class II-dependent basophil-CD4 T cells interactions promote Th2 cytokine-dependent immunity. Nat Immunol. 2009;10:697–705. doi: 10.1038/ni.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter P, Susarla SC, Polikepahad S, Qian Y, Hampton J, Kiss A, Vaidya S, Sur S, Ongeri V, Yang T, Delclos GL, Abramson S, Kheradmand F, Corry DB. Link between allergic asthma and airway mucosal infection suggested by proteinase-secreting household fungi. Mucosal Immunol. 2009;2:504–517. doi: 10.1038/mi.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushparaj PN, Tay HK, H’ng SC, Pitman N, Xu D, McKenzie A, Liew FY, Melendez AJ. The cytokine interleukin-33 mediates anaphylactic shock. Proc Natl Acad Sci USA. 2009;106:9773–9778. doi: 10.1073/pnas.0901206106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt RL, Liang H-E, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol. 2009;10:385–393. doi: 10.1038/ni.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant Th2-like bronchoalveolar T-lymphocyte population in atopic asthma. New Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- Rogers AJ, Raby BA, Lasky-Su JA, Murphy A, Lazarus R, Klanderman BJ, Sylvia JS, Ziniti JP, Lange C, Celedon JC, Silverman EK, Weiss ST. Assessing the reproducibility of asthma candidate gene associations, using genome-wide data. Am J Respir Crit Care Med. 2009;179:1084–1090. doi: 10.1164/rccm.200812-1860OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold MA, Reese TA, Choudhry S, Salam MT, Beckman K, Eng C, Atakilit A, Meade K, Lenoir M, Watson HG, Thyne S, Kumar R, Weiss KB, Grammer LC, Avila P, Schleimer RP, Fahy JV, Rodriguez-Santana J, Rodriguez-Clintron W, Boot RG, Sheppard D, Gilliland FD, Locksley RM, Burchard EG. Differential enzymatic activity of common haplotypic versions of human acidic mammalian chitinase protein. J Biol Chem. 2009;284:19650–19658. doi: 10.1074/jbc.M109.012443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive responses at mucosal sites. Immunol Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol CL, Chu N-Q, Yu S, Nish SA, Laufer TM, Medzhitov R. Basophils function as antigen-presenting cells for an allergen-induced T helper type 2 response. Nat Immunol. 2009;10:713–720. doi: 10.1038/ni.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WR, Smith W-A, Hales BJ, Mills KL, O’Brien RM. Characterization and immunobiology of house dust mite allergens. Int Arch Allergy Immunol. 2002;129:1–18. doi: 10.1159/000065179. [DOI] [PubMed] [Google Scholar]
- Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, Thorne PS, Wills-Karp M, Gioannini TL, Weiss JP, Karp CL. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng YS, Lutchen K, Albert M. The difference in ventilation heterogeneity between asthmatic and healthy subjects quantified using hyperpolarized 3He MRI. J Appl Physiol. 2009;106:813–22. doi: 10.1152/japplphysiol.01133.2007. 2008. [DOI] [PubMed] [Google Scholar]
- Van Panhuys N, Tang SC, Prout M, Camberis M, Scarlett D, Roberts J, Hu-Li J, Paul WE, Le Gros G. In vivo studies fail to reveal a role for IL-4 or STAT6 signaling in Th2 lymphocyte differentiation. Proc Natl Acad Sci USA. 2008;105:12423–12428. doi: 10.1073/pnas.0806372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004;20:267–277. doi: 10.1016/s1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]
- Wang H-B, Ghiran I, Matthaei K, Weller PF. Airway eosinophils: allergic inflammation recruited professional antigen-presenting cells. J Immunol. 2007;179:7585–7592. doi: 10.4049/jimmunol.179.11.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;368:804–13. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- Yazdanbakhsh M, Kremsner PG, van Ree R. Allergy, parasites, and the hygiene hypothesis. Science. 2002;296:490–494. doi: 10.1126/science.296.5567.490. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Yasuda K, Tanaka H, Nakahira M, Imai Y, Fujimori Y, Nakanishi K. Basophils contribute to Th2-IgE responses in vivo via IL-4 production and presentation of peptide-MHC class II complexes to CD4 T cells. Nat Immunol. 2009;10:706–712. doi: 10.1038/ni.1737. [DOI] [PubMed] [Google Scholar]