

Abstract
TP53, encoding p53, is one of the most famous tumor suppressor genes. The majority of human cancers demonstrate the inactivation of the p53 pathway. Mutant p53 not only, no longer, functions as a tumor suppressor but can also exert tumor-promoting effects. The basic function of p53 is to respond to cellular stress. We herein review the recent advances in p53 research and focus on apoptosis, cell cycle arrest, and senescence in response to stress. We also review the clinical applications of p53-based therapy for human cancer.
1. Introduction
The TP53 gene, which encodes p53, is one of the most frequently mutated genes in human cancers. It is reported that approximately half of all cancers have inactivated p53 [1]. The p53 protein has broad range of biological functions, including regulation of the cell cycle, apoptosis, senescence, DNA metabolism, angiogenesis, cellular differentiation, and the immune response. Numerous publications have reported various functions of p53 including transcriptional, posttranscriptional, and posttranslational roles.
In this paper, we will focus on issues concerning p53 and application of p53-based cancer therapies. As reviewed by Vousden and Prives [2], the major functions of p53 are the regulation of growth arrest and apoptosis.
2. Normal Functions of p53
Numerous studies have shown p53 to be a transcription factor that targets many genes and microRNAs in response to cellar stress. The key role of p53 as a tumor suppressor is to block cell cycle progression and/or to induce apoptosis, in response to cellular stresses such as DNA damage. Impaired p53 activity promotes the accumulation of DNA damage in cells, which leads to a cancer phenotype. As a transcription factor, p53 forms a diverse and complex gene regulatory network. There has been extensive investigation to clarify the target sequences that p53 recognizes, the p53 response element (RE), as recently reviewed by Riley et al. [3] p53 has a very wide range of biological activities, so this review will focus on the role of p53 as a tumor suppressor and its implications for cancer therapy.
2.1. Human Cancers and p53 Mutations
More than 26,000 somatic mutation data of p53 appear in the international agency for research on cancer (IARC) TP53 database version R14 (http://www-p53.iarc.fr/) [4].
The frequency of TP53 mutation varies from ~10% (hematopoietic malignancies) to 50–70% (ovarian, colorectal, and head and neck malignancies) [5]. Germline mutation of TP53 causes Li-Fraumeni syndrome, which is a familial cancer syndrome including breast cancer, soft tissue sarcoma, and various other types of cancer [6]. Most TP53 mutations in human cancers result in mutations within the DNA-binding domain, thus preventing p53 from transcribing its target genes. However, mutant p53 has not only led to a loss of normal function of the wild-type protein but also led to new abilities to promote cancer [5]. The first report of this gain of function by mutant p53 was the observation that transfection of mutant p53 into p53-null cells enhances tumor formation in mice [7]. Numerous subsequent studies have confirmed this finding [8].
2.2. p53 as a Tumor Suppressor
2.2.1. p53 as a Sensor of DNA Damage
Genetic instability is one of the most prominent features of malignant tumors. There are very sophisticated systems for detecting DNA damage and repairing the genome. p53 plays an important role in such “caretaker” systems. When p53 responds to DNA damage, it elicits either cell cycle arrest or apoptosis [9].
It was shown in 1991 that induction of wild-type p53 can induce apoptosis in leukemia cells [10]. Mice that have a specific p53 mutant lack the ability to induce cell cycle arrest, but retain the ability to induce apoptosis, allowing them to efficiently suppress oncogene-induced tumors [11], thus suggesting that the proapoptotic function of p53 may play a more important role in its antitumor effects than in its induction of cell cycle arrest.
2.2.2. p53 and Apoptosis
Numerous reports have described the mechanism by which p53 induces apoptosis. As p53 functions mainly as a transcription factor, it is important to explore the genes regulated by p53 that contribute to the regulation of apoptosis. Early studies showed that wild-type p53 can bind the bax gene promoter region and regulate bax gene transcription [12, 13]. bax is a member of the Bcl-2 family, which forms heterodimers with Bcl-2, inhibiting its activity [14]. The Bcl-2 protein family plays an important role in apoptosis and cancer [15, 16]. For example, Bcl-2 controls the release of cytochrome c from the mitochondria, which activates the apoptotic pathway by activating caspase 9. Caspase 9 then activates executioner caspase 3. Both caspases play key roles in the apoptotic pathway.
Several human cancers, including colon and stomach cancer, have altered expression of Bcl-2 [17–19]. However, the importance of the expression level of Bcl-2 on the prognosis of cancer patients is still being investigated. The contribution of bax expression to the prognosis of cancer patients and the response to therapy is also unclear. In breast cancer, a study showed that a low level of expression of bax is associated with a poor prognosis [20], whereas other reports have shown no correlation between the bax expression level and prognosis [21]. Further research will therefore be necessary.
CD95 (also called Fas and Apo-1) is a “death receptor” indicating its major role in apoptosis. The first report of CD95 showed that an anti-CD95 antibody reduced the growth of human B-cell xenograft tumors [22]. Since then, numerous reports have been published about the CD95-induced signaling pathway in apoptosis. Cytotoxic agents, such as chemotherapeutic drugs, can induce apoptosis in drug-sensitive cells. It is therefore important to clarify which signaling pathway(s) contributes most to apoptosis. Elucidating this information would be helpful for drug discovery.
Several reports have indicated the CD95 pathway to play an important role in apoptosis induced by cytotoxic agents, and that this system involves the activation of wild-type p53 [23, 24]. Therefore, the p53 status may influence chemosensitivity via CD95 signaling. However, a recent report indicated that CD95 could promote tumor growth [25]. Programmed cell death is very complicated and depends on a variety of factors.
A Bcl-2 subfamily exists which contains only the BH3 domain. Several BH3-only proteins have been identified, and p53 acts as a transcription factor for PUMA [26, 27] and NOXA [28], which both belong to this class. PUMA is also a key mediator of the apoptotic pathway induced by p53.
When PUMA is disrupted in colon cancer cells, p53-induced apoptosis is prevented [27]. PUMA may play a pivotal role in determining cell fate (programmed cell death versus cell cycle arrest) in response to p53 activation. A report about PUMA knockout mice [29] showed that knockout of PUMA recapitulates the apoptotic deficiency observed in p53 knockout mice. PUMA is an essential mediator for p53-dependent and -independent apoptosis in vivo [30]. Because evading apoptosis is one of the hallmarks of cancer [9], PUMA may also play an important role during carcinogenesis.
Recent reports [31, 32] have shown, however, that in certain situations, apoptosis can promote carcinogenesis. Michalak et al. and Labi et al. showed that loss of PUMA ablated gamma-radiation-induced thymic lymphomagenesis. PUMA-deficient hematopoietic stem cells are protected from gamma-irradiation-induced cell death, which reduces compensatory proliferation and replication. On the other hand, wild-type mice experience massive cell death when they received gamma irradiation, which subsequently led to repopulation of the region by stem/progenitor cells. These reports indicate that the homeostasis stem/progenitor structure of tissue may suppress tumor formation. This phenomenon will need to be confirmed, but should be kept in mind when treating patients with strategies such as chemotherapy and/or radiation.
2.2.3. p53 and Cell Cycle Arrest
The p53 protein suppresses tumor formation not only by inducing apoptosis but also by causing cell cycle arrest. Depending on the type of cellular stress, p53 can induce G1 arrest through activation of transcription of the cyclin-dependent kinase inhibitor p21. This process is well known and has been extensively studied [33]. p53 also regulates the G2/M transition. For example, p53 can block cell entry into mitosis by inhibition of Cdc2. Cdc2 needs to bind to cyclin B1 in order to function. Repression of cyclin B1 by p53 also arrests of cells in G2 [34]. However, transient cell cycle may not lead to tumor eradication, because a cell with oncogenic potential that cannot be repaired may resume proliferation [2].
Therefore, the other mechanism, cellular senescence, may play an important role in p53-mediated tumor suppression. Cellular senescence is permanent cell cycle arrest. There are many reports regarding the correlation between tumor development, p53, and senescence [35, 36]. We will discuss a few of these factors from the standpoint of cancer therapy. Oncogenic ras expressed in human and rodent primary cells results in cellular senescence. This process is due to the accumulation of p53 and p16. Inactivation of p53 or p16 prevents ras-induced senescence [37]. This report implies that cellar senescence has an important role in suppressing tumor development. The inactivation of p53, as is present in most human cancers, allows cells to evade cellar senescence, thus resulting in macrolevel tumor development.
p53 also seems to prevent premalignant lesions from developing into malignant tumors by activating senescence programs [2]. Cellular senescence induced by p53 is important not only for cancer prevention but also for the anticancer effect induced by any wild-type p53 introduced in established tumors.
Xue et al. constructed a mouse model of liver cancer with conditionally regulated endogeneous p53. Reactivation of endogeneous p53 in p53-deficient tumors results in complete tumor regressions. Interestingly, the primary response to p53 was not apoptosis, but the induction of a cellar senescence program. This program triggered an innate immune response that eliminated the tumor from the host [38].
We have reviewed two important roles of p53 as a tumor suppressor. Among the various functions of p53, apoptosis and senescence are the main mechanisms responsible for its tumor suppression. However, precisely how p53 determines whether or not the activation of the senescence program or the apoptosis program occurs still remains to be elucidated. This question is especially important for the development of p53-based cancer therapy, including approaches in combination with conventional chemotherapy. Most conventional chemotherapeutic agents achieve elimination of cancer cells by killing them. Therefore, if p53 induces senescence rather than apoptosis, a conflict will emerge. Indeed, it has been reported that some types of breast cancer are protected from cytotoxic chemotherapeutic agents by endogeneous p53 [39].
Therefore, multiple factors, such as the quality and quantity of cellular stress, the tissue type, and the cellar microenvironment, determine the fate of the cell. Vousden and Prives proposed a model wherein the decision between life and death can be determined by the extent of damage or the duration of stress [2]. In their model, a low level of stress which can be repaired elicits a DNA repair/survival response, while a high level of stress that cannot be repaired induces an apoptotic or senescence response. This dual nature of p53, killer and protector, indicates the possibility that p53 may also act as tumor promoter. The antiapoptotic function of p53 may lead to the survival of damaged cells, which may increase the possibility for malignant transformation (Figure 1).
Figure 1.
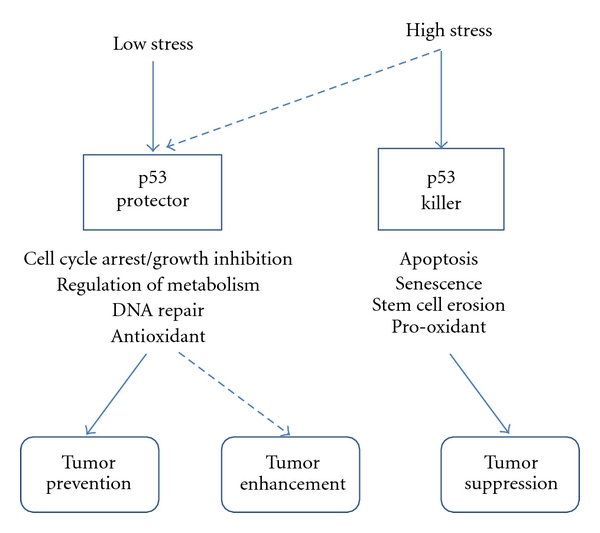
Vousden and Prives [2] proposed a model of the dual mechanism of p53 function in tumors. The result of p53 activation depends on multiple variables. In this model, the p53 response to low stress results in cell cycle arrest, growth inhibition, DNA repair, and so on. This response enables cells to restore the damage induced by the cellular stress. But when the cells receive high stress that cannot be restored, p53 acts as killer that can induce apoptosis or senescence, preventing proliferation of defective cells. If p53 mistakenly responds as a protector when cells receive a high stress that cannot be repaired, the cells keep the genetic damage, which can lead to or contribute to cancer progression (dotted line).
2.3. Applications of p53-Based Cancer Therapy
Because most, if not all, human cancers harbor altered p53, the concept of restoration of p53 for cancer therapy is very attractive. An animal model showed the reactivation of wild-type p53 to result in efficient tumor regression, including regression of lymphoma [40, 41] and liver carcinoma [38].
2.3.1. Reactivating Mutant p53
There is class of small molecules that reactivate the wild-type functions of mutant p53. PhiKan083 is a carbazole derivative found from in silico screening of the crystal structure of p53. By binding mutated p53, PhiKan083 raises the melting temperature of mutated p53, which results in the reactivation of its function [42]. PRIMA-1 is another small molecule identified by cell-based screening which restored sequence-specific DNA binding and the active conformation of p53 [43]. CP-31398 is also a small molecule that can restore the protein folding of mutated p53 to a more natural conformation that permits a wild-type function [44].
2.3.2. p53 Stabilization
MDM2 is an E3 ubiquitin ligase which controls p53 degradation. Many tumors overexpress MDM2 [45], even tumors without p53 mutations [46]. Targeting MDM2 for p53 stabilization seems to be promising, so many reports on targeting MDM2 or the MDM2-p53 have been published.
For example, the nutlins are cis-imidazoline compounds that act as antagonists of the MDM2-p53 interaction. Analysis of the crystal structure showed that nutlin binds in the pocket of MDM2 to prevent the p53-MDM2 interaction. Nutlin can activate the p53 pathway, thereby inducing cancer cells and xenograft tumors in mice to undergo cell cycle arrest, apoptosis, and growth inhibition [47].
MI-219 is another small molecule that inhibits the MDM2-p53 interaction. MI-219 also activates the p53 pathway in cells with wild-type p53. Apoptosis and cell cycle arrest were observed in xenograft tumors which resulted in tumor regression [48].
However, MDM2 inhibition and p53 activation in normal tissue may be harmful. Ringshausen et al. showed that p53 is spontaneously activated in many tissues in MDM2-deficient mice. Moreover, p53 triggered fatal pathologies that included the ablation of classically radiosensitive tissues [49].
2.3.3. Other Classes of Drugs for p53 Stabilization
Tenovin was found by a cell-based drug screen to activate p53. Tenovin acts as an inhibitor of the protein-deacetylating activities of SirT1 and SirT2. The intraperitoneal administration of tenovin-6 has been demonstrated to induce a regression of xenograft tumors in a mouse mode [50].
Issaeva et al. screened a chemical library and found the small molecule RITA (reactivation of p53 and induction of tumor cell apoptosis), which binds to p53 and inhibits the p53-MDM2 interaction both in vitro and in vivo. RITA induced apoptosis in various cancer cells that retained wild type p53 [51]. They also found that the p53 released from MDM2 by RITA promotes p21 and hnRNP K (a p53 cofactor), thus implying that p21 plays a major role in regulating the cancer cell fate after p53 reactivation [52] (Figure 2).
Figure 2.
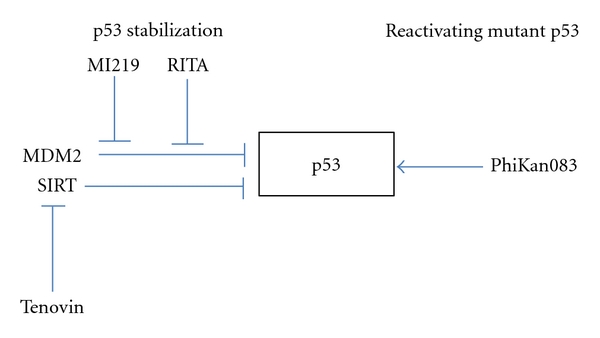
Strategies for p53 stabilization and reactivating mutant p53.
2.4. p53 Inhibition for Cancer Therapy
The inhibition of p53 can protect normal cells during genotoxic chemotherapy or radiation therapy. The side effects of genotoxic therapy for cancer are largely caused by p53-mediated apoptosis. The small molecule pifithrin-alpha can block p53-dependent transcriptional activity and protect mice from the lethal side effects associated with anticancer treatment [53]. If we can avoid dose-limiting genotoxic stress to normal cells during chemotherapy or radiotherapy for cancer, it will thus allow a higher dose to be used for patients who are not sufficiently responsive to conventional chemotherapy.
2.5. p53 Gene Therapy
The first p53-based gene therapy was reported in 1996. A retroviral vector containing the wild-type p53 gene under the control of an actin promoter was injected directly into tumors of nonsmall cell lung cancer patients [54]. After development of a replication-defective recombinant p53 virus (Ad5CMV-p53) [55], many clinical trials have been performed, including one in esophageal cancer patients [56]. A few trials reached phase III, but final approval from the FDA has not yet been granted [57]. Recently, p53-based gene therapy has been developing in China [58].
2.6. p53-Based Immunotherapy
Tumor-associated antigen-specific cytotoxic T lymphocyte can mediate immune response of host against cancer in vivo [59]. P53 protein, especially targeting missense mutation of p53, can be candidate of tumor antigen [60]. Some cancer patients have antibodies against p53 [61], the frequency and clinical significance are still under debate [62].
Speetjens et al. reported clinical trials of a p53-specific synthetic long peptide (p53-SLP) vaccine for metastatic colorectal cancer patients [63]. Ten patients were vaccinated with p53-SLP in a Phase I and Phase II trial. Toxicity was tolerable, and p53-specific immune response was detected in 9 of 10 patients. P53-specific T-cell reactivity persisted more than 6 month in 6 of 9 patients. Although the trial was Phase I/II, the clinical benefit may be hard to obtain because most patients had T-helper cells that lacked key cytokines [64]. Preclinical phase I/ II trial of INGN-225 (Introgen), a p53-modified adenovirus-induced dendritic cell vaccine for small cell lung cancer (SCLC) patients, has been reported [65]. INGN-225 was well tolerated and induced p53-specific immune response in 18/43 (41.8%) patients and sensitized SCLC to subsequent chemotherapy.
3. Conclusion
In this paper, we focused on the functions of p53 and clinical applications targeting p53 for cancer therapy. However, despite recent advances in the research on p53's cytoplasmic function, it appears that various roles remain to be elucidated other than its function as a nuclear factor [66]. p53 has been reported to induce apoptosis independent of its transcription of genes as early as 1994 [67]. Surprisingly, activated p53 can induce apoptosis in the cytoplasm by a bax-dependent mechanism [68].
These reports indicate that cytoplasmic p53 can activate a transcription-independent apoptotic program. The next generation of p53-based cancer therapeutic approaches should therefore be developed to take advantage of this cytosolic function. This may be safer than regulating the transcription modulation of wild-type p53, which can induce both prosurvival and proapoptotic effects in tumor cells, as discussed above.
Recent reports have showed that p53 regulates the process of self-renewal of neural stem cells [69] and hematopoietic stem cells [70]. The cancer stem model insists that tumors are maintained by a small population of cancer stem cells that can divide both symmetrically and asymmetrically. Loss of p53 promotes acute myeloid leukemia by aberrant self-renewal [71].
Mammary stem cells with the targeted mutation of p53 have been reported to show the same properties as cancer stem cells. The reactivation of p53 restored the asymmetric cell division of cancer stem cells and induced tumor growth inhibition [72]. As a result, further investigation of the link between the p53 function and cancer stem cells may therefore be one of the most important research fields for uncovering new paradigms in cancer therapy.
References
- 1.Soussi T, Béroud C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nature Reviews Cancer. 2001;1(3):233–240. doi: 10.1038/35106009. [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 3.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nature Reviews Molecular Cell Biology. 2008;9(5):402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 4.Petitjean A, Achatz MIW, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26(15):2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 5.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nature Reviews Cancer. 2009;9(10):701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 6.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 7.Wolf D, Harris N, Rotter V. Reconstitution of p53 expression in a nonproducer Ab-MuLV-transformed cell line by transfection of a functional p53 gene. Cell. 1984;38(1):119–126. doi: 10.1016/0092-8674(84)90532-4. [DOI] [PubMed] [Google Scholar]
- 8.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harbor Perspectives in Biology. 2010;2(2) doi: 10.1101/cshperspect.a001107. Article ID a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 10.Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352(6333):345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- 11.Toledo F, Krummel KA, Lee CJ, et al. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell. 2006;9(4):273–285. doi: 10.1016/j.ccr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Miyashita T, Krajewski S, Krajewska M, et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9(6):1799–1805. [PubMed] [Google Scholar]
- 13.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80(2):293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 14.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, bax, that accelerates programmed cell death. Cell. 1993;74(4):609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 15.Cotter TG. Apoptosis and cancer: the genesis of a research field. Nature Reviews Cancer. 2009;9(7):501–507. doi: 10.1038/nrc2663. [DOI] [PubMed] [Google Scholar]
- 16.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27(50):6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 17.Ayhan A, Yasui W, Yokozaki H, Seto M, Ueda R, Tahara E. Loss of heterozygosity at the bcl-2 gene locus and expression of bcl-2 in human gastric and colorectal carcinomas. Japanese Journal of Cancer Research. 1994;85(6):584–591. doi: 10.1111/j.1349-7006.1994.tb02400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hague A, Moorghen M, Hicks D, Chapman M, Paraskeva C. BCL-2 expression in human colorectal adenomas and carcinomas. Oncogene. 1994;9(11):3367–3370. [PubMed] [Google Scholar]
- 19.Krajewska M, Moss SF, Krajewski S, Song KI, Holt PR, Reed JC. Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas. Cancer Research. 1996;56(10):2422–2427. [PubMed] [Google Scholar]
- 20.Krajewski S, Blomqvist C, Franssila K, et al. Reduced expression of proapoptotic gene bax is associated with poor response rates to combination chemotherapy and shorter survival in women with metastatic breast adenocarcinoma. Cancer Research. 1995;55(19):4471–4478. [PubMed] [Google Scholar]
- 21.Sjöström J, Blomqvist C, Von Boguslawski K, et al. The predictive value of bcl-2, bax, bcl-xL, bag-1, fas, and fasL for chemotherapy response in advanced breast cancer. Clinical Cancer Research. 2002;8(3):811–816. [PubMed] [Google Scholar]
- 22.Trauth BC, Klas C, Peters AM, et al. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245(4915):301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- 23.Muller M, Strand S, Hug H, et al. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. The Journal of Clinical Investigation. 1997;99(3):403–413. doi: 10.1172/JCI119174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller M, Wilder S, Bannasch D, et al. P53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. Journal of Experimental Medicine. 1998;188(11):2033–2045. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Park SM, Tumanov AV, et al. CD95 promotes tumour growth. Nature. 2010;465(7297):492–496. doi: 10.1038/nature09075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Molecular Cell. 2001;7(3):683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 27.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Molecular Cell. 2001;7(3):673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 28.Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288(5468):1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 29.Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4(4):321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 30.Yu J, Zhang L. No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell. 2003;4(4):248–249. doi: 10.1016/s1535-6108(03)00249-6. [DOI] [PubMed] [Google Scholar]
- 31.Labi V, Erlacher M, Krumschnabel G, et al. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes & Development. 2010;24(15):1602–1607. doi: 10.1101/gad.1940210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michalak EM, Vandenberg CJ, Delbridge AR, et al. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes & Development. 2010;24(15):1608–1613. doi: 10.1101/gad.1940110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. Journal of Cellular Physiology. 2006;209(1):13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 34.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20(15):1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 35.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nature Reviews Molecular Cell Biology. 2007;8(9):729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 37.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 38.Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bertheau P, Espié M, Turpin E, et al. TP53 status and response to chemotherapy in breast cancer. Pathobiology. 2008;75(2):132–139. doi: 10.1159/000123851. [DOI] [PubMed] [Google Scholar]
- 40.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127(7):1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 42.Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(30):10360–10365. doi: 10.1073/pnas.0805326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nature Medicine. 2002;8(3):282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 44.Rippin TM, Bykov VJ, Freund SMV, Selivanova G, Wiman KG, Fersht AR. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene. 2002;21(14):2119–2129. doi: 10.1038/sj.onc.1205362. [DOI] [PubMed] [Google Scholar]
- 45.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358(6381):80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 46.Reifenberger G, Liu L, Ichimura K, Schmidt EE, Collins VP. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Research. 1993;53(12):2736–2739. [PubMed] [Google Scholar]
- 47.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 48.Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(10):3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ringshausen I, O’Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell. 2006;10(6):501–514. doi: 10.1016/j.ccr.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 50.Lain S, Hollick JJ, Campbell J, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13(5):454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nature Medicine. 2004;10(12):1321–1328. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 52.Enge M, Bao W, Hedström E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell. 2009;15(3):171–183. doi: 10.1016/j.ccr.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 53.Komarov PG, Komarova EA, Kondratov RV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285(5434):1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 54.Roth JA, Nguyen D, Lawrence DD, et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nature Medicine. 1996;2(9):985–991. doi: 10.1038/nm0996-985. [DOI] [PubMed] [Google Scholar]
- 55.Zhang WW, Fang X, Mazur W, French BA, Georges RN, Roth JA. High-efficiency gene transfer and high-level expression of wild-type p53 in human lung cancer cells mediated by recombinant adenovirus. Cancer Gene Therapy. 1994;1(1):5–13. [PubMed] [Google Scholar]
- 56.Shimada H, Matsubara H, Shiratori T, et al. Phase I/II adenoviral p53 gene therapy for chemoradiation resistant advanced esophageal squamous cell carcinoma. Cancer Science. 2006;97(6):554–561. doi: 10.1111/j.1349-7006.2006.00206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the P53 pathway. Nature Reviews Cancer. 2009;9(12):862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 58.Guo J, Xin H. Chinese gene therapy. Splicing out the West? Science. 2006;314(5803):1232–1235. doi: 10.1126/science.314.5803.1232. [DOI] [PubMed] [Google Scholar]
- 59.Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10(3):281–287. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- 60.DeLeo AB. P53-based immunotherapy of cancer. Critical Reviews in Immunology. 1998;18(1-2):29–35. doi: 10.1615/critrevimmunol.v18.i1-2.40. [DOI] [PubMed] [Google Scholar]
- 61.Sangrajrang S, Sornprom A, Chernrungroj G, Soussi T. Serum p53 antibodies in patients with lung cancer: correlation with clinicopathologic features and smoking. Lung Cancer. 2003;39(3):297–301. doi: 10.1016/s0169-5002(02)00509-3. [DOI] [PubMed] [Google Scholar]
- 62.Vojtesek B, Kovarik J, Dolezalova H, et al. Absence of p53 autoantibodies in a significant proportion of breast cancer patients. British Journal of Cancer. 1995;71(6):1253–1256. doi: 10.1038/bjc.1995.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Speetjens FM, Kuppen PJK, Welters MJP, et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clinical Cancer Research. 2009;15(3):1086–1095. doi: 10.1158/1078-0432.CCR-08-2227. [DOI] [PubMed] [Google Scholar]
- 64.Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nature Reviews Clinical Oncology. 2011;8(1):25–37. doi: 10.1038/nrclinonc.2010.174. [DOI] [PubMed] [Google Scholar]
- 65.Chiappori AA, Soliman H, Janssen WE, Antonia SJ, Gabrilovich DI. INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opinion on Biological Therapy. 2010;10(6):983–991. doi: 10.1517/14712598.2010.484801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458(7242):1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caelles C, Helmberg A, Karin M. P53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature. 1994;370(6486):220–223. doi: 10.1038/370220a0. [DOI] [PubMed] [Google Scholar]
- 68.Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4(5):371–381. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 69.Meletis K, Wirta V, Hede SM, Nistér M, Lundeberg J, Frisén J. P53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133(2):363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- 70.Liu Y, Elf SE, Miyata Y, et al. P53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4(1):37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao Z, Zuber J, Diaz-Flores E, et al. P53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes & Development. 2010;24(13):1389–1402. doi: 10.1101/gad.1940710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cicalese A, Bonizzi G, Pasi CE, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138(6):1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]