

Abstract
The significance of RET in thyroid cancer comes from solid evidence that, when inherited, an RET activating mutation primes C-cells to transform into medullary carcinomas. Moreover, environmental exposure to radiation also induces rearranged transforming RET “isoforms” that are found in papillary thyroid cancer. The RET gene codes for a tyrosine kinase receptor that targets a diverse set of intracellular signaling pathways. The nature of RET point mutations predicts differences in the mechanisms by which the receptor becomes activated and correlates with different forms of clinical presentation, age of onset, and biological aggressiveness. A number of RET-targeting Tyrosine Kinase Inhibitors (TKIs) are currently undergoing clinical trials to evaluate their effectiveness in the treatment of thyroid cancer, and it is conceivable that the RET genotype may also influence response to these compounds. The question that now emerges is whether, in the future, the rational for treatment of refractory thyroid cancer will be based on the management of an abnormal RET signal. In this paper we address the RET-targeting TKIs and review studies about the signaling properties of distinct RET mutants as a means to predict response and design combinatorial therapies for the soon to be available TKIs.
1. The RET Tyrosine Kinase Receptor Targets a Diverse Spectrum of Intracellular Signaling Pathways
RET (Rearranged during Transfection) encodes a membrane receptor tyrosine kinase (RTK) composed of four extracellular cadherin-like motifs and a cysteine-rich region, a transmembrane portion, and an intracellular domain with tyrosine kinase activity [1]. The RET signaling pathways are outlined in (Figure 1). RET signals through a ligand/coreceptor/RET multiprotein complex instead of the usual receptor/ligand binding. To date, several ligands of the glial-derived neurotrophic factor (GDNF) family, which include GDNF, artemin, neurturin, and persephin and a family of GPI-linked RET coreceptors (GFR1-4), have been identified [2]. The formation of ligand/coreceptor and RET complexes results in RET dimerization and triggers autophosphorylation at intracellular tyrosine residues. Phosphorylated tyrosine 687 (Y687), serine 696 (S696), Y752, Y791, Y806, Y809, Y826, Y864, Y900, Y905, Y928, Y952, Y981, Y1015, Y1029, Y1062, Y1090, and Y1096 constitute docking sites for numerous intracellular adaptor proteins such as RAC1-guanine exchange factor (GEF) [3], growth factor receptor-bound (GRB) docking proteins GRB7/10 [4], chicken Rous sarcoma virus oncogene (c-Src), focal adhesion kinase (FAK) [5], phospholipase C-γ (PLC-γ) and Src homologue collagen (Shc), insulin receptor substrate 1/2 (IRS1/2), fibroblast growth factor substrate 2 (FRS2), or downstream of kinase 1/4/5 (DOK1/4/5) (reviewed by De Groot et al. [6]).
Figure 1.
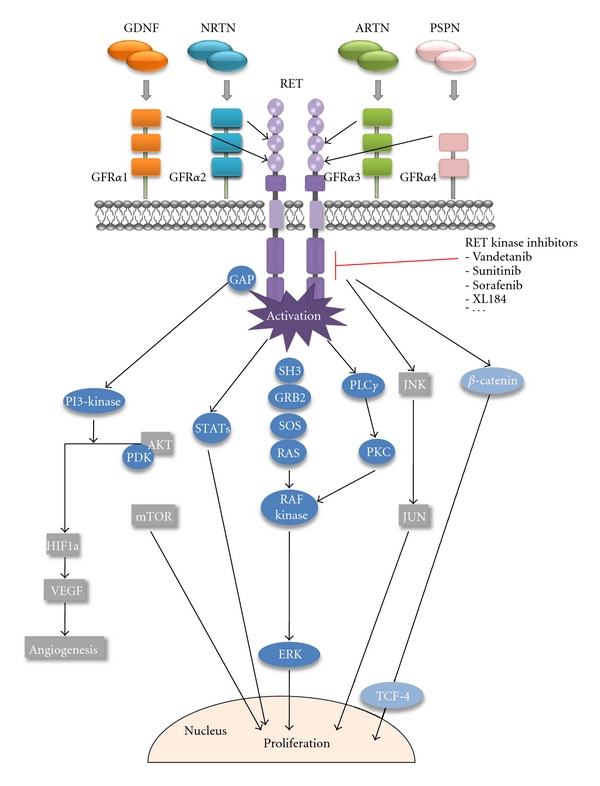
Outline of RET signalling pathways.
Phosphorylation of intracellular target proteins activates several downstream pathways which include mitogen-activated protein kinase cascade: rat sarcoma oncogene/rapidly accelerated fibrosarcoma/extracellular regulated kinase 1/2 (RAS/RAF/ERK1/2), the phosphatidylinositol 3-kinase/protein kinase B pathway (PI3K/AKT) [7, 8], the c-Jun N-terminal kinase pathway (JNK) [9], p38, enigma extracellular regulated kinase 5 (ERK5), the cAMP-responsive element-binding protein, and the signal transducer and activator of transcription 3 (STAT3) (for a review see Arighi et al. [10] and De Groot et al. [6]. More recently, Gujral et al. [11] have shown that RET mediates direct tyrosine phosphorylation of beta-catenin, which associated with an induction of the WNT pathway, that accounts for a part of RET tumorigenic ability in vivo [11].
Many of the above-mentioned intracellular signalling pathways are otherwise known to be general signal transducing pathways targeted not only by RET, but by other RTKs as well. Yet, RET is the main RTK targeted for genetic lesions in thyroid cancer. The transforming ability of activated RET, which was actually on the basis of its isolation as an oncogene [12], could be attributable to the diversity of its signalling which covers several hallmarks of cancer [13].
Increased growth signals and proliferation result from the activation of the RAS/RAF/ERK1/2 cascade and phosphorylation of STAT3 [14, 15].
Cell migration is dependent on RET-mediated activation of RAC1 and JNK [3, 16], and FAK [5] is also reported to play a role in cell migration and to be required for invasion and metastatic behaviour [5, 17].
Inflammation (regarded as the 7th hallmark of cancer [18]) has also been shown to operate as a major component downstream of oncogenic RET mutations. In freshly isolated human thyrocytes, the activation of RET generates a transcriptional program that is similar to that which occurs during inflammation [19] inducing the expression of various inflammatory factors [19–21]. Furthermore, key protein components of the RET-activated “inflammatory” program were found in tumor specimens taken by biopsy, and larger amounts of these inflammatory molecules were found in the primary tumors of patients with lymph-node metastasis than in primary tumors in the absence of lymph-node metastasis (reviewed in [22]). These and other results ([23, 24]; [25]) connect the activation of RET to inflammation.
2. Hereditary MTC-Associated Activating Mutations Cluster at Specific Functional Domains of the RET Receptor Kinase
Overall, as stated before, varied signalling properties, covering multiple hallmarks of cancer, might afford explanation for the ability of RET to transform certain cell types. Nonetheless, the most solid grounds for the significance of RET as a cancer gene come from the fact that, when inherited, an RET germline point mutation alone primes a specific spectrum of tissues to develop endocrine tumors [26, 27]. Carriers of RET germline mutations develop hereditary medullary thyroid carcinoma (hMTC) as the first and most common clinical presentation. Along with hMTC, patients present with pheochromocytoma (tumor of the adrenal medulla) and parathyroid adenomas. This syndromic condition is referred to as Multiple Endocrine Neoplasia type 2 (MEN2) [28]. Penetrance for hMTC is near complete, which highlights the critical role of RET activation in the development of MTC and can be further taken to pinpoint RET as a relevant therapeutic target for MTC.
In hMTC, RET mutations occur in a specific spectrum of codons and result in gain of function, increased kinase activity, and receptor activation [29]. Mutational hotspots are located at the cysteine-rich region of the extracellular domain and in the intracellular tyrosine kinase domain [28]. The clustering of mutations in hotspots might be explained by the fact that proto-oncogene activation requires changes at residues that specifically interact in specific ways with receptor function, and thus mutations cannot occur in a widespread manner. A comprehensive description of all known germline RET variations can be found at the MEN2 RET database (http://www.arup.utah.edu/database/MEN2/MEN2_Welcome). The most common RET germline mutations are missense substitutions of extracellular cysteine residues, occurring at cysteine codon 634 in 80% of cases. Cysteine codons 609, 611, 618, 620, and 630 are less frequently affected. Other noncysteine extracellular domain mutations, located at exons 5 and 8, have been detected [30]. Tyrosine kinase domain mutations affect a more varied spectrum of amino acids, and most frequently recurring mutations replace Met918, Val804, Leu790, Tyr791, and Ala883. Less frequently, residues 768, 876, 891, 886, and 912 are affected. Rare mutations found in isolated families have been reported, comprising homozygous mutations [31], duplications [32], and double mutations [33].
Besides the point mutations found in MTC, an alternative somatic genetic event that causes RET activation is found in the papillary type of thyroid carcinoma (PTC). This involves chromosomal translocations between RET and a number of other loci, referred in general as RET/PTC rearrangements, which interestingly occur as alternative events to the V600E somatic BRAF mutation [34].
3. Distinct RET Mutations Determine Different Clinical Presentations of MEN2 and Predict Age of Onset of hMTC
In MEN2 there are consistent genotype/phenotype correlations that underlie aspects such as clinical manifestation, RET activation mechanisms, and disease penetrance, allowing for a mutation-specific classification of MEN2 [28]. In clinical terms, three disease phenotypes can be recognized: MEN2A, MEN2B, and a familial form of medullary thyroid carcinoma (FMTC). MEN2A was found to be associated with substitutions at one of six specific cysteine residues in exons 10 (609, 611, 618, 620) and 11 (630 and 634). MEN2A cysteine mutations give rise to MTC at young age (onset at 5 to 25 years), along with variable expression of pheochromocytoma (50%) and hyperparathyroidism (15–30%) [28]. MEN2B, on the other hand, is mainly caused by a specific missense mutation located at the RET tyrosine kinase domain (Met918Thr), which accounts for 95% of cases [35]. A second tyrosine kinase domain substitution (Ala883Phe) has been detected in a small proportion of MEN2B patients [36]. Additionally, double mutations affecting codons 804 and 805 and 804 and 806 were described in individual MEN2B cases [33, 37]. MEN2B kinase domain mutations give rise to a more complex clinical phenotype characterized by an early onset (sometimes <1 year old) and very aggressive form of MTC, concomitant with pheochromocytoma in 50% of cases and accompanied by other nonneoplastic features, such as mucosal neuromas of the tongue, lips, and eyelids, ganglioneuromatosis of the gastrointestinal tract, thickening of corneal nerves, and Marfanoid habitus [38]. In FMTC the only disease manifestation is MTC, which usually occurs in adult age, with no additional endocrinopathies. RET mutations with low clinical expression, involving codons 321, 533, 768, 790, 791, 804, and 891, may be found in these families [28]. Occasionally, patients with these mutations may also develop the MEN2A phenotype, showing that FMTC and MEN2A represent a continuum of clinical expression in a common genetically related disorder [39–42]. Age-dependent penetrance for MTC in MEN2 is also codon specific, and classification of the risk of developing MTC can be done based on the genotype (reviewed by Raue and Frank-Raue in [43]). This is of clinical relevance because the ideal timing of prophylactic thyroidectomy should take into consideration the balance between the adverse effects of thyroidectomy at early ages and the individual risk of developing MTC. Comprehensive guidelines have been issued by the American Thyroid Association concerning this aspect [44]. In general, RET mutations with a very high risk of producing MTC (risk level D), comprising all the MEN2B mutations, require surgery before 1 year of age. RET mutations at codon Cys634 constitute risk level C and are managed by thyroidectomy before 5 years old. Level B mutations encompass the changes in the remaining extracellular cysteine codons 609, 611, 618, 620, and 630. In these cases, surgery is advised before 5 years old; however it can be postponed until calcitonin level rise. Risk level A accounts for the FMTC mutations, for which surgery before 5 years old is not required and can be delayed until calcitonin levels rise.
4. The Nature of Somatic RET Mutations Influences the Prognosis of Sporadic MTC
Aside from germline mutations, a somewhat similar spectrum of somatic mutations is observed in about 50 to 60% of the cases with sporadic MTC. A catalogue of somatic mutations can be found at the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/cosmic/). The most frequent somatic lesion is the prototypic MEN2B Met918Thr mutation at exon 16, which comprises up to 60% of the mutation positive cases. Moreover, patients in which tumors harbor MEN2B mutations have a higher prevalence and number of lymph node metastases, present more often with multifocal tumors and with persistent disease at advanced stage, indicating that among the sporadic MTCs, cases with somatic MEN2B mutations are associated with the worst prognosis [45, 46]. Interestingly, cases with RET mutations at the cysteine cluster have the most indolent course, and those with no RET mutations have an intermediate risk [46].
5. Mutations Activate RET by Distinct Mechanisms and Confer Somewhat Different Oncogenic Signaling Properties
The functional basis for the differences in clinical expression of distinct RET genotypes might be explained by the recognition of mutation-specific mechanisms of activating the RET proto-oncogene. Mutations in the extracellular cysteine-rich region result in the replacement of a cysteine residue by another amino acid, subsequently leading to loss of an intramolecular disulfide bond. As a consequence, one cysteine residue becomes available for the formation of an intermolecular disulfide bond, which results in covalently bound receptors that are constitutively active because of ligand-independent receptor dimerization [29]. These mutations commonly associate with MEN2A and FMTC. In contrast, the intracellular MEN2B-specific mutations and other tyrosine kinase domain mutations affect receptor activation in a totally different way. By altering the conformation of the catalytic core of the tyrosine kinase domain they increase catalytic activity and alter the spectrum of intracellular substrates, resulting in remarkable changes of the signalling properties of the receptor [29].
These observations highlight that distinct clinical presentations can arise from differences in the RET activation mechanism and the corresponding output in terms of oncogenic signalling. However, not much is known about the specific differences in signalling properties of the different RET mutants. Studies have shown that wild-type and mutated RET display differences in the autophosphorylation levels of docking sites, which are likely to lead to differential activation of downstream cascades [47]. Support for this paradigm comes from evidence that there are marked differences between MEN2A and MEN2B mutations in terms of their capacity for downstream PI3K/AKT activation. This pathway seems to be more active in MEN2B than in MEN2A [7], and this difference might be attributed to an enhanced autophosphorylation of Y1062 caused by the MEN2B mutation [48].
Another example concerns RET-induced activation of STAT3. The MEN2A mutation Cys634Arg activates STAT3 independently of Janus tyrosine kinases (JAKs) [15]. However, the FMTC mutants, Tyr791Phe and Ser891Ala, seem to do so through a different route and need the involvement of Src and JAKs in order to constitutively activate STAT3 [49].
Thus, on the basis of the above-mentioned evidence that distinct signalling properties are displayed by RET mutants, it is conceivable that different sensitivity to the action of tyrosine kinase inhibitors can occur due to the potentially different conformations of the receptor in each of the RET mutants.
6. RET-Targeting Tyrosine Kinase Inhibitors
The small molecule tyrosine kinase inhibitors (TKIs) mechanism of action is based on the principle that sterically blocking the ATP-binding pocket results in impaired phosphorylation activity, inhibits signal transduction, and prevents activation of intracellular signalling pathways relevant to tumor growth and angiogenesis.
The finding of various compounds (Table 1) capable of inhibiting oncogenic RET (mutated or rearranged), such as PP1 and PP2 [50], ZD6474 (Vandetanib) [51], RPI-1 [52], CEP-701, CEP-751 [66], Imatinib [67], Sunitinib (SU5416, SU11248) [53], Gefitinib [55], Sorafenib (BAY 43-9006) [57], Motesanib (AMG706) [59], Axitinib (AG013736) [61] and XL 184, has brought further clinical relevance to the classification of the pharmacological sensitivity of RET mutants, as metastatic MTC is the most common cause of death in patients with MEN2 [68]. In addition, these compounds could find application in radioactive iodine-refractory PTC with RET/PTC rearrangements.
Table 1.
Molecules used in preclinical and clinical trials as RET tyrosine kinase inhibitors.
| Compound | Trade name | Structure | Targets | Clinical trials | Refs |
|---|---|---|---|---|---|
| PP1 | Zaleplon | Pyrazolopyrimidine | RET | — | [50] |
| PP2 | |||||
| ZD6474 | Vandetanib | Anilinoquinazoline | RET; VEGFR; EGFR | Phase II | [51] |
| RPI-1 | — | Indolinone | RET; MET | — | [52] |
| SU5416 | Sunitinib | Butanedioic acid | VEGFR-2; PDGFR; c-KIT; RET; CSF-1R | Phase II | [53, 54] |
| SU11248 | |||||
| ZD1839 | Gefitinib | Anilinoquinazoline | EGFR | Phase II | [55, 56] |
| BAY43-9006 | Sorafenib | Bis-aryl urea | RAF-1; BRAF; VEGFR-2/-3; PDGFR-B; Flt-3; c-KIT; RET | Phase II | [57, 58] |
| AMG706 | Motesanib diphosphate | Diphosphate salt | VEGFR; PDGFR; KIT; RET | Phase II | [59, 60] |
| AG-013736 | Axitinib | Benzamide | RET; VEGFR; PDGFR; c-KIT | Phase II | [61] |
| XL184/XL880 | VEGFR2; RET and MET | Phase III | [***] | ||
The pyrazolopyrimidines PP1 and PP2 and the 4-anilinoquinazoline Vandetanib inhibit RET-rearrangement-derived oncoproteins with a half maximal inhibitor concentration (IC50) below 100 nM. These molecules were shown to inhibit RET enzymatic activity and phosphorylation of downstream targets, such as ERK1/2. Vandetanib has also been found to inhibit RET signalling in two human PTC cell lines and to reduce tumorigenicity of RET/PTC-transformed fibroblasts injected into nude mice [50]. Vandetanib blocks in vivo phosphorylation and signalling mediated by RET/PTC3 oncoprotein and of an epidermal growth factor- (EGF-) activated EGF-receptor/RET chimeric receptor. Finally, it blocks anchorage-independent growth of RET/PTC3-transformed NIH3T3 fibroblasts and the formation of tumors after injection of NIH-RET/PTC3 cells into nude mice [51].
Sorafenib (BAY 43-9006) was designed originally as a RAF inhibitor [69]. Nonetheless, preclinical studies have shown that Sorafenib can inhibit the kinase activity and signalling of wild-type and oncogenic RET. Sorafenib inhibited oncogenic RET kinase activity at an IC50 of 50 nM or less in NIH3T3 cells. It arrested the growth of NIH3T3 and RAT1 fibroblasts transformed by oncogenic RET and of thyroid carcinoma cells that harbour rearranged RET alleles. These inhibitory effects paralleled a decrease in RET phosphorylation [57]. Finally, PTC cells carrying the RET/PTC1 rearrangement were found to be more sensitive to Sorafenib than PTC cells carrying a BRAF mutation [70]. There is an ongoing phase II clinical trial using Sorafenib in patients with advanced thyroid cancer [58].
RPI-1 is a 2-indolinone derivative initially shown to inhibit RET/PTC1 activity in an immunokinase assay with an IC50 of 27–42 μM. It selectively inhibited the anchorage-independent growth of NIH3T3-transformed cells expressing the RET/PTC1 gene, and the transformed phenotype of NIH3T3ptc1 cells was reverted to a normal fibroblast-like morphology. In these cells, the constitutive tyrosine phosphorylation of RET/PTC1, of the transducing adaptor protein Shc, and of a series of co-immunoprecipitated peptides was substantially reduced [52]. Activation of JNK2 and AKT was abolished, thus supporting the drug inhibitory efficacy on downstream pathways. In addition, cell growth inhibition was associated with a reduction in telomerase activity by nearly 85% [71].
Sunitinib was initially described as a TKI targeting VEGF and PDGFR receptors [72] and also found to inhibit c-KIT [73]. It is now approved for the treatment of GIST and renal cell carcinoma. In vitro kinase assays showed that Sunitinib inhibited the phosphorylation by RET/PTC3 of a synthetic tyrosine kinase substrate peptide in a dose-dependent manner. RET/PTC-mediated Y705 phosphorylation of STAT3 was inhibited by addition of Sunitinib, and the inhibitory effects of Sunitinib on tyrosine phosphorylation and transcriptional activation of STAT3 very closely correlated with decreased autophosphorylation of RET/PTC. Sunitinib caused a complete morphological reversion of transformed NIH-RET/PTC3 cells and inhibited the growth of TPC-1 cells that have an endogenous RET/PTC1 [53]. Treatment of two patients with progressive metastatic thyroid carcinoma (1 PTC and 1 FTC) demonstrated sustained clinical responses to Sunitinib over a period of four years [54].
Gefitinib was initially approved for nonsmall cell lung cancer since it targets oncogenic EGFR. In vitro data suggests that EGFR contributes to RET kinase activation, signalling, and growth stimulation. Conditional activation of RET/PTC oncoproteins in thyroid PCCL3 cells markedly induced expression and phosphorylation of EGFR, which was mediated in part through mitogen-activated protein (MAP) kinase signalling. RET and EGFR were found to co-immunoprecipitate. Ligand-induced activation of EGFR resulted in phosphorylation of a kinase-dead RET, and this effect was entirely blocked by EGFR kinase inhibitor. Gefitinib also inhibited cell growth induced by various constitutively active mutants of RET in thyroid cancer cells as well as in NIH3T3 cells [55]. These pieces of evidence have provided a biological basis for clinical evaluation of Gefitinib in thyroid cancer. The results obtained in a phase II trial showed no objective responses among the 25 thyroid cancer patients treated with Gefitinib [56].
CEP-701 and CEP-751 are indolocarbazole derivatives that also inhibit RET in MTC cells. Effective inhibition of RET phosphorylation in a dose-dependent manner is achieved at concentrations <100 nM. These compounds also block the growth of MTC cells in culture. CEP-751 and its prodrug, CEP-2563 inhibit tumor growth in MTC cell xenografts [66]. These drugs also potentiate the effects of irinotecan treatment in TT cell culture and xenografts and result in durable complete remission in 100% of the mice. CEP-751 inhibited the induction of the DNA repair program (marked by phospho-H2AX) as well as the checkpoint pathway (marked by the activated Chk1) [74]. Since preclinical models have demonstrated that both CEP-751 and CEP-2563 have antitumor activity in a variety of tumors, phase I trials were undertaken [75].
Several other TKI molecules are being evaluated with regard to their efficacy in metastatic MTC treatment with limited published data. Axitinib (AG-013736) [76] was assessed in a phase II study with 60 MTC patients. Eighteen cases (30%) presented partial responses, and 23 (38%) had stable disease [61]. Motesanib (AMG706) [77] was evaluated in differentiated thyroid cancer [59] and in a phase I study in 91 patients with either hereditary (16 cases) or sporadic MTC (75 cases), 2% of the patients showed partial response, and 81% had stable disease [60]. XL184/XL880 is a compound that is rapidly going through the clinical evaluation process. It is a TKI that targets VEGFR2, RET, and also MET and whose efficacy has been demonstrated for several solid tumors, especially thyroid cancer [***]. In patients with hereditary and sporadic MTC very interesting response rates were obtained with 9/17 patients (53%) showing partial remission. Based on these findings, a phase III registration trial of XL184 as a potential treatment for medullary thyroid cancer (MTC) has been initiated.
7. The Influence of Genotype on the Sensitivity to RET-Targeting TkIs and Challenges Ahead
Although a number of patients with refractory MTC have been undergoing treatment with several TKIs in the last few years, it is not yet clear whether clinical response to these drugs is actually influenced by the RET genotype of the tumor cells. At this point, the only reliable source for this type of information comes from in vitro studies. Indeed, some compounds used against RET seem to confirm the paradigm that certain mutations can render RET resistant to inhibition. This was first illustrated by PP1, PP2, and ZD6474 (Vandetanib) which, despite being efficient in inhibiting phosphorylation of most of the MEN2-associated RET mutants (at codons 768, 790, 883, 918, and 634 [50]), were incapable of inhibiting MEN2-associated swap of Valine 804 for bulky hydrophobic Leucine or Methionine within the RET kinase domain. Thus Valine 804 emerged as a structural determinant amino acid mediating resistance to pyrazolopyrimidines and 4-anilinoquinazolines [78, 79]. This was also found to be the case for the V804M/E805K tandem lesion, detected in non-Met918/Ala883 MEN2B, which was shown to also confer resistance to PP1, suggesting a mode of action different from the classical MEN2B mutations [33].
However, inhibition of RET phosphorylation and signaling by mutation of the Val804 gatekeeper residue was not impaired in cells subjected to Sorafenib treatment [80], indicating that this drug could be a potential therapeutic tool for RET Val804 positive thyroid tumors [80].
The fact that using another compound can overcome a mutation-specific primary resistance renders further support to the idea that sensitivity of RET mutants will, in the end, result from mutation-dependent structural determinants of the RET ATP-binding site. However, to support the paradigm of an RET pharmacogenetics, much more needs to be evaluated before we can confirm that this concept is useful for the clinical practice. To start, it would be imperative that the mutation status of the tumors from patients included in clinical trials is ascertained and correlated with clinical response. Until now, none of the clinical studies have published the mutation status of the patients. On the other hand, we must not forget that despite the in vitro data has proven highly informative for genotype/phenotype correlations, it cannot be taken directly to indicate differences in terms of clinical response. In addition, many of these small molecule inhibitors act upon several target RTKs, rendering it difficult to ascertain which of the effects over different RTKs actually accounts for the observed clinical response.
We should also be aware that some of the effects of these compounds may go beyond interference with the ATP-binding pocket and may affect RET expression. For instance, Sorafenib suppresses RET tyrosine kinase activity by direct enzymatic inhibition and also by promoting RET lysosomal degradation independent of proteasomal targeting [80].
At this point, given that a number of molecules are starting to become available, it would be worth to compare these drugs against each other in their efficacy to inhibit the activity of the most frequent RET genotypes. This may come as a means to define and stratify drugs for use as first-line and second-line treatments on the basis of the RET genotype.
As we highlighted before, specific RET mutations may lead stronger induction of specific intracellular signalling targets, many of which have their own dedicated inhibitors under development. In this respect, the information about the specificities in oncogenic signalling of different genotypes might be valuable to design combinatorial therapies employing mutation-specific combinations of inhibitors for treatment.
At present, the clinical use of tyrosine kinase inhibitors in patients with thyroid cancer still does not rely in the genetic background of each tumor [58, 61, 81]. Nonetheless, results from clinical trials suggest that these compounds have a more cytostatic than cytolytic effect, and thus are just adding another step of selective pressure to the progressing tumor (which buys time), but eventually secondary resistance can develop. In models such as ABL/CML (imatinib), EGFR/lung cancer (Gefitinib), or KIT/GIST (imatinib), prolonged therapy with TKIs leads to the acquisition of resistance mutations in the receptors targeted by these drugs, rendering them insensitive to therapy. Although no secondary RET mutations have been described thus far, the experience with patients undergoing clinical trials taught that some patients suddenly fail to respond while on treatment. Most probably, the same underlying resistance mechanisms are at play. This implies that, in order to translate the use of these inhibitors into increased long-term survival, we may need to perform molecular followup of the progressing lesions, in order to predict resistance and eventually change from one inhibitor to another.
Finally, to reduce the biology of MTC to RET activation and signaling boosting is almost certainly a simplistic view. RET mutations do not only determine MTC development (even in hMTC). Likely these tumours also carry mutations in other genes, and possibly one should also know these to think about combinatory therapies. Indeed, data is accumulating regarding alternative pathways that contribute to MTC development from precursor C-cell hyperplasia. This is the case of the WNT pathway activation by RET-mediated tyrosine phosphorylation of β-Catenin [11] and the synergistic effects of p18 and p27, two members of the RB pathway [82, 83]. This may provide additional targets for combination of RET inhibitors with other compounds targeting these pathways. Also relevant to this is the recent recognition of mechanisms of cross-talk between different RTKs. For instance, EGFR may cooperate with RET in activating intracellular signaling pathways [55]. This provides biological basis for combining different RTK inhibitors.
The challenge for the years to come is to use the pools of knowledge generated in RET signaling pathways and MTC progression steps to rationalize combinatory therapies, targeting different molecules and different signaling pathways that are relevant in MTC.
Conflict of Interests
The authors declare that they have no proprietary, financial, professional, or other personal interest of any nature and kind in any product, service, and/or company that could be construed as influencing the position presented in this paper.
Authors' Contribution
H. Prazeres wrote the paper, J. Torres reviewed bibliography on RET signalling pathways, F. Rodrigues provided insight into clinical aspects of MTC management, J. P. Couto reviewed bibliography concerning the tyrosine kinase inhibitors, J. Vinagre composed the figures of the paper, and M. Sobrinho-Simões and P. Soares performed critical reviews of the paper.
Acknowledgments
This study was supported by the Portuguese Foundation for Science and Technology (FCT) through a Project Grant (PTDC/SAU-OBD/101242/2008), the Portuguese Society of Endocrinology and Metabolism (Edward Limber Prize), and the Portuguese Ministry of Health (project 13/2007). The authors would like also to acknowledge FCT for grants to H. Prazeres (Refs. SFRH/BD30041/2006 and SFRH/BPD/72004/2010), Ph.D. grant to J. P. Couto (SFRH/BD/40260/2007), and a BI to J. Torres. IPATIMUP is an associated Laboratory of the Portuguese Ministry of Science, Technology and Higher Education and is partially supported by the Portuguese Foundation for Science and Technology.
References
- 1.Takahashi M, Cooper GM. ret Transforming gene encodes a fusion protein homologous to tyrosine kinases. Molecular and Cellular Biology. 1987;7(4):1378–1385. doi: 10.1128/mcb.7.4.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Airaksinen MS, Titievsky A, Saarma M. GDNF family neurotrophic factor signaling: four masters, one servant. Molecular and Cellular Neurosciences. 1999;13(5):313–325. doi: 10.1006/mcne.1999.0754. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda T, Kiuchi K, Takahashi M. Novel mechanism of regulation of Rac activity and lamellipodia formation by RET tyrosine kinase. Journal of Biological Chemistry. 2002;277(21):19114–19121. doi: 10.1074/jbc.M200643200. [DOI] [PubMed] [Google Scholar]
- 4.Pandey A, Liu X, Dixon JE, Di Fiore PP, Dixit VM. Direct association between the Ret receptor tyrosine kinase and the Src homology 2-containing adapter protein Grb7. Journal of Biological Chemistry. 1996;271(18):10607–10610. doi: 10.1074/jbc.271.18.10607. [DOI] [PubMed] [Google Scholar]
- 5.Panta GR, Nwariaku F, Kim LT. RET signals through focal adhesion kinase in medullary thyroid cancer cells. Surgery. 2004;136(6):1212–1217. doi: 10.1016/j.surg.2004.06.048. [DOI] [PubMed] [Google Scholar]
- 6.De Groot JWB, Links TP, Plukker JTM, Lips CJM, Hofstra RMW. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocrine Reviews. 2006;27(5):535–560. doi: 10.1210/er.2006-0017. [DOI] [PubMed] [Google Scholar]
- 7.Murakami H, Iwashita T, Asai N, et al. Enhanced phosphatidylinositol 3-kinase activity and high phosphorylation state of its downstream signalling molecules mediated by Ret with the MEN 2B mutation. Biochemical and Biophysical Research Communications. 1999;262(1):68–75. doi: 10.1006/bbrc.1999.1186. [DOI] [PubMed] [Google Scholar]
- 8.Segouffin-Cariou C, Billaud M. Transforming ability of MEN2A-RET requires activation of the phosphatidylinositol 3-kinase/AKT signaling pathway. Journal of Biological Chemistry. 2000;275(5):3568–3576. doi: 10.1074/jbc.275.5.3568. [DOI] [PubMed] [Google Scholar]
- 9.Chiariello M, Visconti R, Carlomagno F, et al. Signalling of the Ret receptor tyrosine kinase through the c-Jun NH-terminal protein kinases (JNKs): evidence for a divergence of the ERKs and JNKs pathways induced by Ret. Oncogene. 1998;16(19):2435–2445. doi: 10.1038/sj.onc.1201778. [DOI] [PubMed] [Google Scholar]
- 10.Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine and Growth Factor Reviews. 2005;16(4-5):441–467. doi: 10.1016/j.cytogfr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Gujral TS, Van Veelen W, Richardson DS, et al. A novel RET kinase-β-catenin signaling pathway contributes to tumorigenesis in thyroid carcinoma. Cancer Research. 2008;68(5):1338–1346. doi: 10.1158/0008-5472.CAN-07-6052. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985;42(2):581–588. doi: 10.1016/0092-8674(85)90115-1. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe T, Ichihara M, Hashimoto M, et al. Characterization of gene expression induced by RET with MEN2A or MEN2B mutation. American Journal of Pathology. 2002;161(1):249–256. doi: 10.1016/S0002-9440(10)64176-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuringa JJ, Wojtachnio K, Hagens W, et al. MEN2A-RET-induced cellular transformation by activation of STAT3. Oncogene. 2001;20(38):5350–5358. doi: 10.1038/sj.onc.1204715. [DOI] [PubMed] [Google Scholar]
- 16.Asai N, Fukuda T, Wu Z, et al. Targeted mutation of serine 697 in the Ret tyrosine kinase causes migration defect of enteric neural crest cells. Development. 2006;133(22):4507–4516. doi: 10.1242/dev.02616. [DOI] [PubMed] [Google Scholar]
- 17.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nature Reviews Cancer. 2005;5(7):505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 18.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 19.Borrello MG, Alberti L, Fischer A, et al. Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(41):14825–14830. doi: 10.1073/pnas.0503039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwahashi N, Murakami H, Nimura Y, Takahashi M. Activation of RET tyrosine kinase regulates interleukin-8 production by multiple signaling pathways. Biochemical and Biophysical Research Communications. 2002;294(3):642–649. doi: 10.1016/S0006-291X(02)00528-4. [DOI] [PubMed] [Google Scholar]
- 21.Shinohara S, Rothstein JL. Interleukin 24 is induced by the RET/PTC3 oncoprotein and is an autocrine growth factor for epithelial cells. Oncogene. 2004;23(45):7571–7579. doi: 10.1038/sj.onc.1207964. [DOI] [PubMed] [Google Scholar]
- 22.Guarino V, Castellone MD, Avilla E, Melillo RM. Thyroid cancer and inflammation. Molecular and Cellular Endocrinology. 2010;321(1):94–102. doi: 10.1016/j.mce.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Puxeddu E, Knauf JA, Sartor MA, et al. RET/PTC-induced gene expression in thyroid PCCL3 cells reveals early activation of genes involved in regulation of the immune response. Endocrine-Related Cancer. 2005;12(2):319–334. doi: 10.1677/erc.1.00947. [DOI] [PubMed] [Google Scholar]
- 24.Muzza M, Degl'Innocenti D, Colombo C, et al. The tight relationship between papillary thyroid cancer, autoimmunity and inflammation: clinical and molecular studies. Clinical Endocrinology. 2010;72(5):702–708. doi: 10.1111/j.1365-2265.2009.03699.x. [DOI] [PubMed] [Google Scholar]
- 25.Borrello MG, Degl’Innocenti D, Pierotti MA. Inflammation and cancer: the oncogene-driven connection. Cancer Letters. 2008;267(2):262–270. doi: 10.1016/j.canlet.2008.03.060. [DOI] [PubMed] [Google Scholar]
- 26.Mulligan LM, Kwok JBJ, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363(6428):458–460. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 27.Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Human Molecular Genetics. 1993;2(7):851–856. doi: 10.1093/hmg/2.7.851. [DOI] [PubMed] [Google Scholar]
- 28.Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific ret proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: international RET mutation consortium analysis. Journal of the American Medical Association. 1996;276(19):1575–1579. [PubMed] [Google Scholar]
- 29.Santoro M, Carlomagno F, Romano A, et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science. 1995;267(5196):381–383. doi: 10.1126/science.7824936. [DOI] [PubMed] [Google Scholar]
- 30.Castellone MD, Verrienti A, Magendra Rao D, et al. A novel de novo germ-line V292M mutation in the extracellular region of RET in a patient with phaeochromocytoma and medullary thyroid carcinoma: functional characterization. Clinical Endocrinology. 2010;73(4):529–534. doi: 10.1111/j.1365-2265.2009.03757.x. [DOI] [PubMed] [Google Scholar]
- 31.Lesueur F, Cebrian A, Cranston A, et al. Germline homozygous mutations at codon 804 in the RET protooncogene in medullary thyroid carcinoma/multiple endocrine neoplasia type 2A patients. Journal of Clinical Endocrinology and Metabolism. 2005;90(6):3454–3457. doi: 10.1210/jc.2004-1622. [DOI] [PubMed] [Google Scholar]
- 32.Pigny P, Bauters C, Wemeau JL, et al. A novel 9-base pair duplication in RET exon 8 in familial medullary thyroid carcinoma. Journal of Clinical Endocrinology and Metabolism. 1999;84(5):1700–1704. doi: 10.1210/jcem.84.5.5665. [DOI] [PubMed] [Google Scholar]
- 33.Cranston AN, Carniti C, Oakhill K, et al. RET is constitutively activated by novel tandem mutations that alter the active site resulting in multiple endocrine neoplasia type 2B. Cancer Research. 2006;66(20):10179–10187. doi: 10.1158/0008-5472.CAN-06-0884. [DOI] [PubMed] [Google Scholar]
- 34.Soares P, Trovisco V, Rocha AS, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. 2003;22(29):4578–4580. doi: 10.1038/sj.onc.1206706. [DOI] [PubMed] [Google Scholar]
- 35.Hofstra RMW, Landsvater RM, Ceccherini I, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367(6461):375–376. doi: 10.1038/367375a0. [DOI] [PubMed] [Google Scholar]
- 36.Gimm O, Marsh DJ, Andrew SD, et al. Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. Journal of Clinical Endocrinology and Metabolism. 1997;82(11):3902–3904. doi: 10.1210/jcem.82.11.4508. [DOI] [PubMed] [Google Scholar]
- 37.Miyauchi A, Futami H, Hai N, et al. Two germline missense mutations at codons 804 and 806 of the RET proto-oncogene in the same allele in a patient with multiple endocrine neoplasia type 2B without codon 918 mutation. Japanese Journal of Cancer Research. 1999;90(1):1–5. doi: 10.1111/j.1349-7006.1999.tb00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morrison PJ, Nevin NC. Multiple endocrine neoplasia type 2B (mucosal neuroma syndrome, Wagenmann-Froboese syndrome) Journal of Medical Genetics. 1996;33(9):779–782. doi: 10.1136/jmg.33.9.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bethanis S, Koutsodontis G, Palouka T, et al. A newly detected mutation of the RET protooncogene in exon 8 as a cause of multiple endocrine neoplasia type 2A. Hormones. 2007;6(2):152–156. doi: 10.14310/horm.2002.1111011. [DOI] [PubMed] [Google Scholar]
- 40.Berndt I, Reuter M, Saller B, et al. A new hot spot for mutations in the ret protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. Journal of Clinical Endocrinology and Metabolism. 1998;83(3):770–774. doi: 10.1210/jcem.83.3.4619. [DOI] [PubMed] [Google Scholar]
- 41.Pinna G, Orgiana G, Riola A, et al. RET proto-oncogene in Sardinia: V804M is the most frequent mutation and may be associated with FMTC/MEN-2A phenotype. Thyroid. 2007;17(2):101–104. doi: 10.1089/thy.2006.0198. [DOI] [PubMed] [Google Scholar]
- 42.Jimenez C, Habra MA, Huang SCE, et al. Pheochromocytoma and medullary thyroid carcinoma: a new genotype-phenotype correlation of the RET protooncogene 891 germline mutation. Journal of Clinical Endocrinology and Metabolism. 2004;89(8):4142–4145. doi: 10.1210/jc.2004-0041. [DOI] [PubMed] [Google Scholar]
- 43.Raue F, Frank-Raue K. Multiple endocrine neoplasia type 2: 2007 update. Hormone Research. 2007;68(supplement 5):101–104. doi: 10.1159/000110589. [DOI] [PubMed] [Google Scholar]
- 44.Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19(6):565–612. doi: 10.1089/thy.2008.0403. [DOI] [PubMed] [Google Scholar]
- 45.Dvorakova S, Vaclavikova E, Sykorova V, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinomas. Molecular and Cellular Endocrinology. 2008;284(1-2):21–27. doi: 10.1016/j.mce.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 46.Moura MM, Cavaco BM, Pinto AE, et al. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. British Journal of Cancer. 2009;100(11):1777–1783. doi: 10.1038/sj.bjc.6605056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Vega QC, Decker RA, Pandey A, Worby CA, Dixon JE. Oncogenic RET receptors display different autophosphorylation sites and substrate binding specificities. Journal of Biological Chemistry. 1996;271(10):5309–5312. doi: 10.1074/jbc.271.10.5309. [DOI] [PubMed] [Google Scholar]
- 48.Salvatore D, Melillo RM, Monaco C, et al. Increased in vivo phosphorylation of ret tyrosine 1062 is a potential pathogenetic mechanism of multiple endocrine neoplasia type 2B. Cancer Research. 2001;61(4):1426–1431. [PubMed] [Google Scholar]
- 49.Menacho IP, Koster R, Van Der Sloot AM, et al. RET-familial medullary thyroid carcinoma mutants Y791F and S891A activate a Src/JAK/STAT3 pathway, independent of glial cell line-derived neurotrophic factor. Cancer Research. 2005;65(5):1729–1737. doi: 10.1158/0008-5472.CAN-04-2363. [DOI] [PubMed] [Google Scholar]
- 50.Carlomagno F, Vitagliano D, Guida T, et al. The kinase inhibitor PP1 blocks tumorigenesis induced by RET oncogenes. Cancer Research. 2002;62(4):1077–1082. [PubMed] [Google Scholar]
- 51.Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Research. 2002;62(24):7284–7290. [PubMed] [Google Scholar]
- 52.Lanzi C, Cassinelli G, Pensa T, et al. Inhibition of transforming activity of the ret/ptc1 oncoprotein by a 2-indolinone derivative. International Journal of Cancer. 2000;85(3):384–390. [PubMed] [Google Scholar]
- 53.Kim DW, Jo YS, Jung HS, et al. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. Journal of Clinical Endocrinology and Metabolism. 2006;91(10):4070–4076. doi: 10.1210/jc.2005-2845. [DOI] [PubMed] [Google Scholar]
- 54.Dawson SJ, Conus NM, Toner GC, et al. Sustained clinical responses to tyrosine kinase inhibitor sunitinib in thyroid carcinoma. Anti-Cancer Drugs. 2008;19(5):547–552. doi: 10.1097/CAD.0b013e3282fc6cf7. [DOI] [PubMed] [Google Scholar]
- 55.Croyle M, Akeno N, Knauf JA, et al. RET/PTC-induced cell growth is mediated in part by epidermal growth factor receptor (EGFR) activation: evidence for molecular and functional interactions between RET and EGFR. Cancer Research. 2008;68(11):4183–4191. doi: 10.1158/0008-5472.CAN-08-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pennell NA, Daniels GH, Haddad RI, et al. A phase II study of gefitinib in patients with advanced thyroid cancer. Thyroid. 2008;18(3):317–323. doi: 10.1089/thy.2007.0120. [DOI] [PubMed] [Google Scholar]
- 57.Carlomagno F, Anaganti S, Guida T, et al. BAY 43-9006 inhibition of oncogenic RET mutants. Journal of the National Cancer Institute. 2006;98(5):326–334. doi: 10.1093/jnci/djj069. [DOI] [PubMed] [Google Scholar]
- 58.Gupta-Abramson V, Troxel AB, Nellore A, et al. Phase II trial of sorafenib in advanced thyroid cancer. Journal of Clinical Oncology. 2008;26(29):4714–4719. doi: 10.1200/JCO.2008.16.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sherman SI, Wirth LJ, Droz JP, et al. Motesanib diphosphate in progressive differentiated thyroid cancer. New England Journal of Medicine. 2008;359(1):31–42. doi: 10.1056/NEJMoa075853. [DOI] [PubMed] [Google Scholar]
- 60.Schlumberger MJ, Elisei R, Bastholt L, et al. Phase II study of safety and efficacy of motesanib in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. Journal of Clinical Oncology. 2009;27(23):3794–3801. doi: 10.1200/JCO.2008.18.7815. [DOI] [PubMed] [Google Scholar]
- 61.Cohen EEW, Rosen LS, Vokes EE, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. Journal of Clinical Oncology. 2008;26(29):4708–4713. doi: 10.1200/JCO.2007.15.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eder JP, Appleman L, Heath E, et al. A phase I study of a novel spectrum selective kinase inhibitor (SSKI), XL880, administered orally in patients (pts) with advanced solid tumors (STs) Journal of Clinical Oncology. 2006;24(18S):p. 3041. ASCO Annual Meeting Proceedings Part I. [Google Scholar]
- 63.LoRusso P, Appleman L, Zhu AX, et al. Pharmacodynamics of XL880, a novel spectrum selective kinase inhibitor (SSKI) administered orally in patients with advanced solid tumors (AST). In: Proceedings of the 18th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; November 2006; Prague, Czech Republic. Abstract 404. [Google Scholar]
- 64.Ross RW, Stein M, Sarantopoulos J, et al. A phase II study of the c-Met RTK inhibitor XL880 in patients (pts) with papillary renal-cell carcinoma (PRC) Journal of Clinical Oncology. 2007;25(18S):p. 15601. ASCO Annual Meeting Proceedings. [Google Scholar]
- 65.Salgia R, Hong DS, Camacho LH, et al. A phase I dose-escalation study of the safety and pharmacokinetics (PK) of XL184, a VEGFR and MET kinase inhibitor, administered orally to patients (pts) with advanced malignancies. Journal of Clinical Oncology. 2007;25(18S):p. 14031. ASCO Annual Meeting Proceedings. [Google Scholar]
- 66.Strock CJ, Park JI, Rosen M, et al. CEP-701 and CEP-751 inhibit constitutively activated RET tyrosine kinase activity and block medullary thyroid carcinoma cell growth. Cancer Research. 2003;63(17):5559–5563. [PubMed] [Google Scholar]
- 67.Cohen MS, Hussain HB, Moley JF. Inhibition of medullary thyroid carcinoma cell proliferation and RET phosphorylation by tyrosine kinase inhibitors. Surgery. 2002;132(6):960–967. doi: 10.1067/msy.2002.128562. [DOI] [PubMed] [Google Scholar]
- 68.Skinner MA, Moley JA, Dilley WG, Owzar K, DeBenedetti MK, Wells SA. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. New England Journal of Medicine. 2005;353(11):1105–1113. doi: 10.1056/NEJMoa043999. [DOI] [PubMed] [Google Scholar]
- 69.Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocrine-Related Cancer. 2001;8(3):219–225. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- 70.Henderson YC, Ann SH, Kang Y, Clayman GL. Sorafenib potently inhibits papillary thyroid carcinomas harboring RET/PTC1 rearrangement. Clinical Cancer Research. 2008;14(15):4908–4914. doi: 10.1158/1078-0432.CCR-07-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lanzi C, Cassinelli G, Cuccuru G, et al. Inactivation of Ret/Ptc1 oncoprotein and inhibition of papillary thyroid carcinoma cell proliferation by indolinone RPI-1. Cellular and Molecular Life Sciences. 2003;60(7):1449–1459. doi: 10.1007/s00018-003-2381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mendel DB, Douglas Laird A, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clinical Cancer Research. 2003;9(1):327–337. [PubMed] [Google Scholar]
- 73.Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Molecular Cancer Therapeutics. 2003;2(5):471–478. [PubMed] [Google Scholar]
- 74.Strock CJ, Park JI, Rosen DM, et al. Activity of irinotecan and the tyrosine kinase inhibitor CEP-751 in medullary thyroid cancer. Journal of Clinical Endocrinology and Metabolism. 2006;91(1):79–84. doi: 10.1210/jc.2005-1882. [DOI] [PubMed] [Google Scholar]
- 75.Undevia SD, Vogelzang NJ, Mauer AM, Janisch L, Mani S, Ratain MJ. Phase I clinical trial of CEP-2563 dihydrochloride, a receptor tyrosine kinase inhibitor, in patients with refractory solid tumors. Investigational New Drugs. 2004;22(4):449–458. doi: 10.1023/B:DRUG.0000036687.26604.8c. [DOI] [PubMed] [Google Scholar]
- 76.Choueiri TK. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Current Opinion in Investigational Drugs. 2008;9(6):658–671. [PubMed] [Google Scholar]
- 77.Polverino A, Coxon A, Starnes C, et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Research. 2006;66(17):8715–8721. doi: 10.1158/0008-5472.CAN-05-4665. [DOI] [PubMed] [Google Scholar]
- 78.Carlomagno F, Guida T, Anaganti S, et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene. 2004;23(36):6056–6063. doi: 10.1038/sj.onc.1207810. [DOI] [PubMed] [Google Scholar]
- 79.Knowles PP, Murray-Rust J, Kjær S, et al. Structure and chemical inhibition of the RET tyrosine kinase domain. Journal of Biological Chemistry. 2006;281(44):33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- 80.Plaza-Menacho I, Mologni L, Sala E, et al. Sorafenib functions to potently suppress RET tyrosine kinase activity by direct enzymatic inhibition and promoting RET lysosomal degradation independent of proteasomal targeting. Journal of Biological Chemistry. 2007;282(40):29230–29240. doi: 10.1074/jbc.M703461200. [DOI] [PubMed] [Google Scholar]
- 81.Pfister DG, Fagin JA. Refractory thyroid cancer: a paradigm shift in treatment is not far off. Journal of Clinical Oncology. 2008;26(29):4701–4704. doi: 10.1200/JCO.2008.17.3682. [DOI] [PubMed] [Google Scholar]
- 82.Joshi PP, Kulkarni MV, Yu BK, et al. Simultaneous downregulation of CDK inhibitors p18 and p27 is required for MEN2A-RET-mediated mitogenesis. Oncogene. 2007;26(4):554–570. doi: 10.1038/sj.onc.1209811. [DOI] [PubMed] [Google Scholar]
- 83.Van Veelen W, Van Gasteren CJR, Acton DS, et al. Synergistic effect of oncogenic RET and loss of p18 on medullary thyroid carcinoma development. Cancer Research. 2008;68(5):1329–1337. doi: 10.1158/0008-5472.CAN-07-5754. [DOI] [PubMed] [Google Scholar]