

Abstract
The kibdelones are hexacyclic tetrahydroxanthones and potent anticancer agents isolated from an Australian microbe. Herein, we describe the synthesis of a chiral, non-racemic iodocyclohexene carboxylate EF ring fragment of the kibdelones employing an intramolecular iodo halo-Michael aldol reaction and its merger with an ABCD ring fragment to afford the congener kibdelone C.
The polycyclic xanthone natural products are a diverse family of polyketides characterized by their highly oxygenated, hexacyclic frameworks (Figure 1). Kibdelones A–C (1–3, Figure 1) are hexacyclic tetrahydroxanthone1b natural products which were isolated by Capon and coworkers from the rare Australian microbe Kibdelosporangium sp. along with isomeric metabolites including isokibdelone A 4.1a During their isolation, it was found that methanol solutions of 2 and 3 equilibrated to a mixture of 1–3,1b likely through keto/enol tautomerizations followed by quinone/hydroquinone redox reactions. The kibdelones also display potent nanomolar activity in variety of human tumor cell lines. For example, kibdelone C 3 has a GI50 of <1 nm against a SR (leukemia) tumor cell line and <1 nm (GI50) against SN12C (renal) cell carcinoma. There are numerous related hexacyclic xanthone natural products (cf. 5–9, Figure 1).2 Overall, the class of molecules exhibits a diverse range of biological activities, including potent antimalarial, antibiotic, anticoccidial, and anticancer properties.2
Figure 1.

Kibdelones and Related Hexacyclic Xanthone Natural Products
Despite extensive synthetic efforts towards polycyclic xanthones such as the cervinomycins 5,3c–g there have been limited reports describing synthetic efforts3a,b,h towards hexacyclic tetrahydroxanthone natural products, in part due to the challenge in constructing the polyhydroxylated F ring moiety. In our retrosynthetic analysis, we envisioned that the congener kibdelone C 3 may be obtained from merger of ABCD fragment 124 and 2-iodo-1-cyclohexenecarboxylate fragment 13. In order to access the tetrahydroxanthone ring-system, we envisioned use of an oxa-Michael/retro-Michael5 Friedel-Crafts annulation sequence. This approach has been utilized in the literature for the synthesis of xanthones from biaryl-ethers,3d,6 but has not previously been used to access tetrahydroxanthone ring systems. Fragment 12 may be derived from Pt(IV)-catalyzed arylation of quinone monoketal 10 and hydroxystyrene 11.4 We envisioned that the chiral EF ring fragment 13 may be obtained from diastereoselective, intramolecular halo-Michael aldol reaction of aldehyde ynoate 14, the latter derived from protected diol 15. There are numerous literature reports describing intermolecular reactions of preformed β-iodoallenoates reacting with aldehydes7 including a recent report by Frontier and coworkers describing an intramolecular variant with an alkynone substrate.8
The synthesis of the requisite EF ring fragment 13 began with Dess-Martin-periodinane oxidation of the commercially available, enantiopure alcohol 15.9 Zinc-acetylide, chelation-controlled addition to aldehyde 16, followed by benzylation with NaH in THF and desilylation with TBAF, provided the 1,3 anti-protected propargylic alcohol 17 (60%, 3 steps).10 Alcohol 17 was oxidized to a carboxylic acid in a two step sequence using IBX in ethyl acetate11 followed by Pinnick oxidation. Further treatment with H2SO4 in MeOH provided both the derived methyl ester and deprotected propargylic diol 18 (73%, three steps). Selective benzylation of the secondary alcohol of 18 was achieved through benzylidene formation with benzaldehyde dimethylacetal and p-TsOH in toluene which was followed by reduction with sodium cyanoborohydride and TMSCl in CH3CN to provide bis-benzyl ether 19 (76%, two steps).12 Primary alcohol 19 was finally oxidized with IBX to afford the requisite ynoate aldehyde 14 (90%).11
We next evaluated the intramolecular halo-Michael aldol reaction. Treatment of 14 with of magnesium iodide7f–g (2 equiv.) at −20 °C in CH2Cl2 afforded 2-iodo-1-cyclohexenecarboxylate 20 in 78% yield (19% overall yield from 15) on a multigram scale along with minor diastereomer 21 (9%). The stereochemistry of the major diastereomer was confirmed by X-ray crystal structure analysis of the derived p-bromobenzoate 22.13 A proposed mechanism for the transformation is shown in Figure 3. We envision reversible7f formation β-iodoallenoate diastereomers A and B, the former which may undergo cyclization through transition state C to major diastereomer 20 and the latter which may react thru assembly D to afford minor diastereomer 21. The preference for C vs. D leading to major diastereomer 20 may be due to stabilizing chelation of the α-benzyl ether and aldehyde with MgI2.7a,b,d,g,14 Alternatively, it is possible that the relative thermodynamic stability of products 20 and 21 may be reflected in the product ratio if epimerization by a retro-halo-Michael aldol process15 can occur under the reaction conditions.
Figure 3.

Proposed Mechanisms for Formation of 20 and 21
Initial attempts at fragment coupling of 12 with benzyl-protected iodoacrylate 20 proved to be unsuccessful under a variety of basic, palladium, and copper-catalyzed conditions.16 Accordingly, we decided to target the benzyl-deprotected acetonide 13 (Scheme 2) as reaction partner with the expectation that an unprotected, allylic hydroxyl should impart greater reactivity in oxa-Michael reactions.5b Numerous conditions for debenzylation of 20 were attempted, including hydrogenolysis and various Lewis acids (e.g. AlCl3, ZrCl4, and TMSI).13 Unfortunately, most conditions led to either degradation or formation of complex aggregates and emulsions upon aqueous workup. Fortunately, treatment of 20 with BCl317 effected smooth conversion to a triol which was reprotected by treatment with 2,2-dimethoxypropane under acidic conditions to afford 13 (87%, two steps, Scheme 2).
Scheme 2.

Fragment Coupling
Initial base-catalyzed oxa-Michael reactions of ABCD ring fragment 12 and iodocyclohexene carboxylate 13 included evaluation of NaH, K2CO3, Cs2CO3, (n-Bu)4NOH, and KOtBu as bases in either CH3CN or DMF at temperatures from 25–100 °C. However, these conditions failed to produce any detectable coupling products. We next evaluated a variety of copper and palladium-catalyzed methods, including conditions reported by Buchwald and coworkers for C-O bond formation using Cu(I), picolinic acid, and K3PO4 as base in DMSO (Scheme 2).16c In light of the latter precedent for coupling of sterically hindered and electron rich nucleophiles, we believed that this would be an effective condition for fragment coupling of 12 and 13. Further experimentation revealed that copper (I) was not required for the transformation. Dimethyl sulfoxide proved to be an optimal solvent, presumably due to its ability to dissolve phenolate(s) derived from dihydrophenanthrene 12 at reduced temperatures. The base was also found to be important, as little to no conversion was observed with NaH, K2CO3, Cs2CO3, and KOtBu. Moreover, the workup procedure was found to be crucial due to acid sensitivity of vinylogous carbonate 23. Careful workup at 0° C with 0.5 N KHSO4 (pH = 2) was found to be optimal for protonation of the phenolates and to minimize degradation of the newly formed vinylogous carbonate linkage. 1H NMR comparison of the phenol chemical shifts of dihydrophenanthrene 12 and vinylogous carbonate 23 strongly supported the desired connectivity of the oxa-Michael product.13
Initially we believed that the lack of reactivity seen in iodoacrylate 20 versus 13 may be due to the steric bulk imparted by the allylic, benzyl ether moiety. Accordingly, conformer searches of both 13 and 20 were performed using Spartan ’08 calculated at a semi-empirical level of theory using an AM1 basis set. In the low energy conformer of dibenzyl ether 20, the allylic ether substituent appears to be in an axial orientation. In contrast, in the low energy conformer of successful substrate 13, the hydroxyl moiety resides in an equatorial orientation (Figure 4). Accordingly, the lone-pair of the benzyl ether may raise the LUMO of the acrylate system by an σ-π* interaction which may also contribute to the low reactivity observed for 20.18
Figure 4.
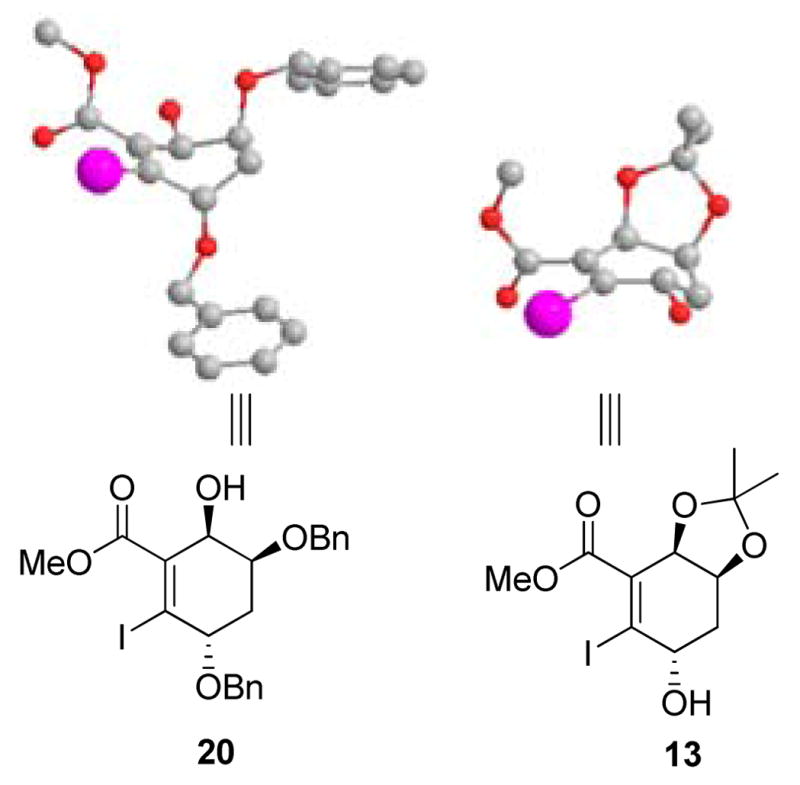
AM1 low energy conformers for 20 and 13
The acid lability of 23 proved to be disappointing, as in our initial plan for formation of the tetrahydroxanthone ring system we envisioned treatment of ester 23 with either sulfuric acid, 19a Eaton’s reagent,19b or PPA (polyphosphoric acid),19c known reagents for intramolecular Friedel-Crafts annulation of esters. A report describing a mild, intramolecular Friedel-Crafts cyclization using cyanuric chloride and AlCl3 led us to target the carboxylic acid derived from ester 23.20 Fortunately, we were able to saponify 23 by treatment with LiOH in dioxane. In this reaction, some cleavage of the vinylogous carbonate linkage to afford 12 was observed (LC/MS). Fortunately, we were able to carry on the crude acid mixture (contained approx. 10% of 12) directly into cyclization via treatment with cyanuric chloride and pyridine in DCE (75° C) which afforded tetrahydroxanthone 24 (39%, two steps, Scheme 3).20 The fact that AlCl3 was not required for the transformation led us to propose the mechanism shown in Figure 5. Activation of the carboxylic acid may afford an intermediate such as E which can further react to form the highly reactive α-oxo-ketene21,22 intermediate F, the latter which may further undergo 6π-electrocyclization and rearomatization to form the requisite tetrahydroxanthone 24. As the reaction was conducted with excess pyridine, it is conceivable that intermediate E is an acyl pyridinium23 rather than an acid chloride.
Scheme 3.

Xanthone Formation and Elaboration to kibdelone C
Figure 5.

Proposed Mechanism for Xanthone Formation
In the final stages of the synthesis, we found that compound 24 was a poor substrate for oxidative demethylation.13 Accordingly, the acetonide was removed via treatment with 3N HCl in THF to provide triol 25 in 95% yield (Scheme 4). X-ray crystal analysis of 25 showed two molecules in the asymmetric unit and served to confirm the hexacyclic tetrahydroxanthone framework (Figure 6).13 Due to the known instability of kibdelone B 2,1b we envisioned an oxidative demethylation/in-situ reduction sequence to access kibdelone C.24 Attempted oxidations using ceric ammonium nitrate (CAN) were found to be both pH and temperature sensitive. Reaction of 25 with 2.0 equiv. of CAN in pH 7.0 buffer afforded mixtures of B and D ring-oxidized products, as well a compound showing a mass for both B and D ring hydroquinones. Reaction of 25 with CAN in water led to a 3:2 mixture of B vs. D ring oxidized products. Fortunately, treatment of 25 with CAN (2.2 equiv.) and 10 equivalents of AcOH at rt for 2 minutes, followed by a reductive quench with excess sodium dithionite, provided a ~ 5:1 mixture of kibdelone C and a doubly demethylated product. In this case, we believe that protonation of the xanthone carbonyl with acetic acid may produce a DE ring benzopyrylium species13,25 which should reduce the propensity for oxidation of the D ring. Alternatively, it is conceivable that the more selective oxidant cerium (IV) acetate may be formed under the reaction conditions.26 Proton, carbon, UV/VIS and optical rotation ([α]D23 = +48° synthetic, +49° natural (c = 0.5, CHCl3)) for synthetic 3 were found to be identical in all aspects to the natural product.1 The absolute stereochemical assignment of natural kibdelone C was accordingly assigned as 10R, 11S, 13S as shown in structure 3 (Figure 1).
Figure 6.
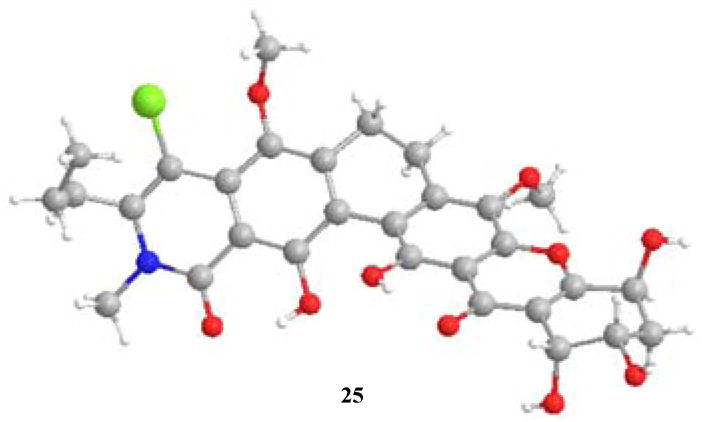
X-Ray Crystal Structure of kibdelone C methyl ether 25
In summary, a convergent total synthesis of kibdelone C has been achieved. A diastereoselective halo Michael/aldol reaction sequence was utilized for construction of the highly functionalized 2-iodo-1-cyclohexenecarboxylate EF ring fragment 13. The ABCD dihydrophenanthrene fragment 12 was reacted through a site-selective oxa-Michael reaction to afford a sensitive vinylogous carbonate precursor 22. The acid lability of 22 was overcome utilizing cyanuric chloride to mildly activate a derived carboxylic acid to afford the tetrahydroxanthone ring system. A precursor to kibdelone C, methyl ether 24, was synthesized and found to be very stable in relation to the highly oxidizable 3. Further studies concerning the synthesis and biological evaluation of additional kibdelone congeners and additional tetrahydroxanthone natural products are currently in progress and will be reported in due course.
Supplementary Material
Figure 2.

Retrosynthetic Analysis for Kibdelone C
Scheme 1.

Synthesis of the EF ring fragment
Acknowledgments
We thank the National Institutes of Health (R01 CA137270) and Merck Research Laboratories for research support, Drs. Emil Lobkovsky (Cornell) and Bruce Noll (Bruker) for X-ray crystal structure analyses and refinement assistance, and Dr. Paul Ralifo (Boston University) for NMR assistance. We thank Andrew Little, Stephen Scully, and Dr. Branko Mitasev (Boston University) for extremely helpful and stimulating discussions. We also thank Prof. Robert J. Capon (University of Queensland) for kindly providing a natural sample and 1H and 13C NMR spectra for kibdelone C.
Footnotes
Supporting Information Available: Experimental procedures, and characterization data for all new compounds described herein, including CIF files for compound 22 and 25. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Ratnayake R, Lacey E, Tennant S, Gill JH, Capon RJ. Org Lett. 2006;8:5267–5270. doi: 10.1021/ol062113e. [DOI] [PubMed] [Google Scholar]; (b) Ratnayake R, Lacey E, Tennant S, Gill JH, Capon RJ. Actinomycete. Chem Eur J. 2007;13:1610–1619. doi: 10.1002/chem.200601236. [DOI] [PubMed] [Google Scholar]
- 2.For isolation and biological studies of polycyclic xanthones, see: albofungin: Gurevich AI, Karapetyan MG, Kolosov MN, Oaelchenko K, Onoprienko V, Petrenko GI, Popravko SA. Tetrahedron Lett. 1972;18:1751–1754.. lysolipin: Drautz H, Keller-Schierlein W, Zaehner H. Arch Microbiol. 1975;106:175–190. doi: 10.1007/BF00446521.Dobler M, Keller-Schierlein W. Helv Chim Acta. 1977;60:177–185. doi: 10.1002/hlca.19770600120.. cervinomycin: Omura S, Nakagawa A, Kushida K, Shimizu H, Lukacs GJ. Antibiot. 1987;40:301–308. doi: 10.7164/antibiotics.40.301.Tanka H, Kawakita K, Suzuki H, Nakagawa PS, Omura SJ. J Antibiot. 1989;42:431–439. doi: 10.7164/antibiotics.42.431.. Actinoplanones: Kobayashi K, Nishino C, Ohya J, Sato S, Mikawa T, Shiobara Y, Kodama M. J Antibiot. 1988;4:741–750. doi: 10.7164/antibiotics.41.741.. Simaomicin α: Lee TM, Carter GT, Borders DB. J Chem Soc Chem Commun. 1989;22:1771–1772.Carter GT, Goodman JJ, Torrey MJ, Borders DB, Gould SJ. J Org Chem. 1989;54(18):4321–4323.Koizumi Y, Tomoda H, Kumagai A, Zhou X-p, Koyota S, Sugiyama T. Cancer Sci. 2009;100:322–326. doi: 10.1111/j.1349-7006.2008.01033.x.. Kigamicin: Kunimoto S, Someno T, Yamazaki Y, Lu J, Esumi H. J Antibiot. 2003;56:1012–1017. doi: 10.7164/antibiotics.56.1012.Someno T, Kunimoto S, Nakamura H, Naganawa H, Ikeda D. J Antibiot. 2005;58:56–60. doi: 10.1038/ja.2005.6.Masuda T, Ohba S, Kawada M, Osono M, Ikeda D, Esumi H, Kunimoto S. J Antibiot. 2006;59:209–214. doi: 10.1038/ja.2006.29.. Sch 56036 Chu M, Truumees I, Mierzwa R, Terracciano J, Patel M, Das PR, Puar MS, Chan TM. Tetrahedron Lett. 1998;39:7649–7652.
- 3.For synthetic work on polycyclic xanthones, see: lysolipin: Duthaler RO, Wegmann UH. Helv Chim Acta. 1984;67:1217–1211.Duthaler RO, Wegmann UH. Helv Chim Acta. 1984;67:1755–1766.. cervinomycin Mehta G, Venkateswarlu Y. J Chem Soc, Chem Commun. 1988:1200–1202.Kelly TR, Jagoe CT, Li Q. J Am Chem Soc. 1989;111:4522–4524.Rao AV, Yadav JS, Reddy K, Upender V. Tetrahedron Lett. 1991;32:5199–5202.Yadav JS. Pure Appl Chem. 1993;65:1349–1356.Mehta G, Shah SR, Venkateswarlu Y. Tetrahedron. 1994;50:11729–11742.. SCH 56036: Barrett AGM. Tetrahedron Lett. 2005;46:6537–6540.. kigamicins: Turner PA, Griffin EM, Whatmore JL, Shipman M. Org Lett. 2011;13:1056–1059. doi: 10.1021/ol103103n.
- 4.Sloman DL, Mitasev B, Scully SS, Beutler JA, Porco JA., Jr Angew Chem Int Ed. 2011;50:2511–2515. doi: 10.1002/anie.201007613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For a review on oxa-Michael reactions, see: Nising CF, Bräse S. Chem Soc Rev. 2008;37:1218–1228. doi: 10.1039/b718357g.. For oxa-Michael reactions of 4-hydroxycyclohexenone, see: Nising CF, Ohnemüller UK, Bräse S. Angew Chem Int Ed. 2006;45:307–309. doi: 10.1002/anie.200502913.
- 6.For xanthone synthesis through diaryl ether formation followed by intramolecular Friedel Crafts annulations, see refs. 3m-p and: Sousa ME, Pinto MMM. Curr Med Chem. 2005;12:2447–2479. doi: 10.2174/092986705774370736.Zewge D, Chen CY, Deer C, Dormer PG, Hughes DL. J Org Chem. 2007;72:4276. doi: 10.1021/jo070181o.Han C, Wang L, Chen Z, Liu G. J Med Chem. 2008;51:1432–1446. doi: 10.1021/jm701405p.
- 7.(a) Taniguchi M, Kobayashi S, Nakagawa M, Hino T, Kishi Y. Tetrahedron Lett. 1986;27:4763–4766. [Google Scholar]; (b) Taniguchi M, Hino T, Kishi Y. Tetrahedron Lett. 1986;27:4767–4770. [Google Scholar]; (c) Zhang C, Lu XY. Synthesis. 1996:586–588. [Google Scholar]; Org Lett. 2001;3:823–826. doi: 10.1021/ol000377+. [DOI] [PubMed] [Google Scholar]; (d) Wei HX, Gao JJ, Li G, Pare PW. Tetrahedron Lett. 2002;43:5677–5680. [Google Scholar]; (e) Wei HX, Chen D, Xu X, Li G, Pare PW. Tetrahedron: Asymmetry. 2003;14:971–974. [Google Scholar]; (f) Wei HX, Timmons C, Farag MA, Pare PW, Li G. Org Biomol Chem. 2004;2:2893–2896. doi: 10.1039/B409056J. [DOI] [PubMed] [Google Scholar]; (g) Wei HX, Hu J, Jasoni RL, Li G, Pare PW. Helv Chim Acta. 2004;87:2359–2363. [Google Scholar]; (h) Chen D, Guo L, Kotti SRSS, Li G. Tetrahedron: Asymmetry. 2005;16:1757–1762. [Google Scholar]; (i) Timmons C, Kattuboina A, Banerjee S, Li G. Tetrahedron. 2006;62:7151–7154. [Google Scholar]
- 8.Ciesielski J, Canterbury DP, Frontier AJ. Org Lett. 2009;11:4374–4377. doi: 10.1021/ol901721y. [DOI] [PubMed] [Google Scholar]
- 9.Lebar MD, Baker BJ. Tetrahedron Lett. 2007;48:8009–8010. [Google Scholar]
- 10.(a) Shimada BK, Kaburagi Y, Fukuyama T. J Am Chem Soc. 2003;125:4048–4049. doi: 10.1021/ja0340679. [DOI] [PubMed] [Google Scholar]; (b) Hayashi Y, Yamaguchi H, Toyoshima M, Okado K, Toyo T, Shoji M. Org Lett. 2008;10:1405–1408. doi: 10.1021/ol800195g. [DOI] [PubMed] [Google Scholar]
- 11.More JD, Finney NS. Org Lett. 2002;4:3001–3003. doi: 10.1021/ol026427n. [DOI] [PubMed] [Google Scholar]
- 12.Rolf Johansson R, Samuelsson B. J Chem Soc, Chem Commun. 1984:2371–2374. [Google Scholar]
- 13.See Supporting Information for complete experimental details.
- 14.(a) Corey EJ, Li W, Reichard GA. J Am Chem Soc. 1998;120:2330–2336. [Google Scholar]; (b) Wei Han-Xun, Jasoni Richard L, Shao Huawu, Hu Jiali, Paré PW. Tetrahedron. 2004;60:11829–11835. [Google Scholar]
- 15.For retro aldolization using Mg(II), see: Evans DA, Tedrow JS, Shaw JT, Downey WA. J Am Chem Soc. 2002;124:392–393. doi: 10.1021/ja0119548.
- 16.For Pd-catalyzed Ullmann coupling, see: Burgos CH, Barder TE, Huang T, Buchwald SL. Angew Chem Int Ed. 2006;45:4321–4326. doi: 10.1002/anie.200601253.For a review on Cu – catalyzed Ullmann coupling, see: Monnier F, Taillefer M. Angew Chem, Int Ed. 2009;48:6954–6971. doi: 10.1002/anie.200804497.For diaryl ether synthesis using picolonic acid/CuI, see: Maiti D, Buchwald SL. J Org Chem. 2010;75:1791–1794. doi: 10.1021/jo9026935.
- 17.Xie J, Ménanda M, Valéry JM. Carbohydr Res. 2005;340:481–487. doi: 10.1016/j.carres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 18.For σ-π* interactions in osmylations, see: Johnson CR, Tait BD, Cieplak AS. J Am Chem Soc. 1987;109:5875–5876.Halterman RL, McEvoy MA. J Am Chem Soc. 1992;114:980–985.
- 19.Simoneau B, Brassard P. J Chem Soc Perkin Trans I. 1984:1507–1510. [Google Scholar]; (b) Zewge D, Chen CY, Deer C, Dormer PG, Hughes DL. J Org Chem. 2007;72:4276. doi: 10.1021/jo070181o. [DOI] [PubMed] [Google Scholar]; (c) Han C, Wang L, Chen Z, Liu G. J Med Chem. 2008;51:1432–1446. doi: 10.1021/jm701405p. [DOI] [PubMed] [Google Scholar]
- 20.For a recent review on cyanuric chloride, see: Blotny G. Tetrahedron. 2006;62:9507–9522.Kangani CO, Day BW. Org Lett. 2008;10:2645–2648. doi: 10.1021/ol800752v.
- 21.For a review on α-oxoketenes, see: Wentrup C, Heilmayer W, Kollenz G. Synthesis. 1994:1219–1248.
- 22.Briehl H, Lukosch A, Wentrup C. J Org Chem. 1984;49:2772–2779. [Google Scholar]; (b) Fillion E, Fishlock D. Org Lett. 2003;5:4653–4656. doi: 10.1021/ol035817m. [DOI] [PubMed] [Google Scholar]; (c) Xu F, Armstrong JD, III, Zhou GX, Simmons B, Hughes D, Ge Z, Grabowski EJ. J Am Chem Soc. 2004;126:13002–13009. doi: 10.1021/ja046488b. [DOI] [PubMed] [Google Scholar]
- 23.(a) Olah GA, Szilay PJ. J Am Chem Soc. 1969;91:2949. [Google Scholar]; (b) Lohse C, Hollenstein S, Laube T. Angew Chem Int. 1991;103:1656–1658. [Google Scholar]
- 24.(a) Luly JR, Rapoport H. J Org Chem. 1981;46:2745. [Google Scholar]; (b) Heckrodt TJ, Mulzer J. J Am Chem Soc. 2003;125:4680–4681. doi: 10.1021/ja034397t. [DOI] [PubMed] [Google Scholar]
- 25.For studies on protonation of xanthones, see: Ireland JF, Wyatt PAH. J Chem Soc, Faraday Trans 1. 1972;68:1053–1058.Mizutani K, Miyazaki K, Kimiko I, Hosoya H. Bull Chem Soc Jpn. 1974;47:1596–1603.
- 26.Baciocchi E, Rol C, Sebastiani GV. J Chem Res Synop. 1983:232–3. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.