

Abstract
BACKGROUND
Our group previously reported that neutrophil gelatinase-associated lipocalin (NGAL) overexpression significantly blocked invasion and angiogenesis of pancreatic ductal adenocarcinoma (PDAC) and also demonstrated a loss of NGAL expression in the advanced stages of PDAC. However, little is known regarding mechanisms of NGAL regulation in PDAC. As EGF-EGFR axis is significantly upregulated in PDAC, we examined EGF-mediated NGAL regulation in these cells.
METHODS
NGAL-positive AsPC-1 and BxPC-3 cells were used as model system. Quantitative RT-PCR, western blot analysis, and immunofluorescence studies were used to investigate EGF-mediated effects on NGAL expression. E-cadherin expression was manipulated using lentiviral overexpression or shRNA constructs. NGAL promoter activity was assessed by luciferase-reporter assay and electrophoretic mobility shift assay (EMSA).
RESULTS
NGAL expression was positively associated with tumor differentiation and was significantly downregulated after EGF treatment along with a concomitant reduction of E-cadherin expression in PDAC cells. E-cadherin downregulation was partly through the EGF receptor (EGFR)-dependent MEK-ERK signaling pathway. In addition, E-cadherin downregulation reduced NGAL expression in PDAC cells, whereas overexpression of E-cadherin led to increased NGAL expression and partly rescued inhibition of NGAL expression by EGF. Furthermore, EGF in part through E-cadherin reduced NGAL promoter activity by blocking NF-κB activation.
CONCLUSIONS
We demonstrated for the first time that EGF potently blocked NGAL expression in PDAC cells. This effect is partly mediated through activation of the EGFR-MEK-ERK signaling pathway, which in turn downregulated E-cadherin with a subsequent reduction in NF-κB activation. Our findings illustrate a novel mechanism by which EGF regulates NGAL expression in PDAC.
Keywords: neutrophil gelatinase associated lipocalin (NGAL), EGF, E-cadherin, nuclear factor-κB (NF-κB) and pancreatic cancer
INTRODUCTION
Neutrophil gelatinase-associated lipocalin (NGAL) also referred as lipocalin 2, is a 25 kD secreted protein that belongs to the lipocalin family 1. NGAL binds to and transports a variety of lipophilic substances through a three-dimensional β-barrel structure that is common to lipocalins 1. It is also considered as an antimicrobial factor because of its ability to deplete iron that is required for bacterial growth 2. In addition, increased NGAL expression is found in a variety of pathological conditions including inflammation 3, 4, acute ischemic renal injury 5, and various human cancers 3, 6, 7.
There is substantial data to support a key role for NGAL in cancer progression. For instance, NGAL expression is upregulated and associated with enhanced tumor growth and invasiveness in breast cancer 8 and esophageal squamous cell carcinoma 9. In contrast, NGAL acts as an antitumor and antimetastatic factor in ovarian 10, colon 11 and pancreatic cancer 12. Moreover, NGAL has emerged as a biomarker for detecting early-stage cancer and monitoring the progression of established cancers including ovarian 10 and pancreatic cancer 12, 13. NGAL is not expressed by normal pancreatic ducts but is aberrantly expressed in the early pre-neoplastic stages of pancreatic ductal adenocarcinoma (PDAC) termed pancreatic intraepithelial neoplasias (PanINs) 13. Further, its expression in established PDAC correlates inversely with the grade of differentiation 13. However, the mechanism by which NGAL expression is regulated in PDAC cells is still unknown.
It is well documented that growth factors, especially epidermal growth factor (EGF) and EGF receptor (EGFR)-driven signaling pathways play an important role in cancer progression 14. Overexpression of EGF and EGFR has been reported in various cancer types, including PDAC 15, 16. EGF treatment in vitro has been found to enhance the invasiveness and metastatic properties of a number of different cancer cells including ovarian 17, cervical 18, epidermoid 19, and breast cancer 20. In PDAC, both EGF and EGFR are overexpressed and associated with increased tumor size, advanced clinical stage and decreased patient survival 21, 22. In addition, EGF has been shown to promote the invasiveness of PDAC cells by activating MMP2 and nuclear factor-κB (NF-κB) 23, 24. However, the downstream signaling that mediates EGF-induced PDAC aggressiveness is still poorly understood.
In the present study, we investigated the possible role of EGF in regulating NGAL expression. We found that EGF downregulates NGAL and E-cadherin expression in PDAC cells in vitro and this was blocked by EGFR or ERK pathway inhibitors. Downregulation and upregulation of E-cadherin resulted in decreased and increased NGAL expression in PDAC cells, respectively. EGF treatment was also associated with a significant decrease in NF-κB -mediated transcription of NGAL mRNA while mutation of the NF-κB binding site (in the NGAL promoter) blocked EGF-induced downregulation of NGAL expression. Thus, our findings reveal that EGF inhibits NGAL expression through an EGFR-driven ERK pathway and involves inhibition of NF-κB mediated transactivation of NGAL. Further, we demonstrate that E-cadherin modulates NGAL expression in PDAC cells, suggesting a possible mechanism for EGF-induced aggressiveness of PDAC cells.
MATERIALS AND METHODS
Cells lines and culture condition
PANC-1, MIA PaCa-2, AsPC-1 and BxPC-3 cells were purchased from ATCC and cultured as described before 12. 293FT cells were obtained from Invitrogen (Carlsbad, CA) and maintained in DMEM supplemented with 10% FBS and 500μg/ml G418.
Reagents and treatments
EGF was purchased from Sigma (St. Louis, MO), PD98059, U0126, E-cadherin and phospho-ERK antibodies from Cell Signaling Technology (Danvers, MA), AG1478 from Calbiochem (Gibbstown, NJ) and mouse monoclonal NGAL antibodies from Antibody Shop (Gentofte, Demark). pLKO-shE-cadherin and its control vectors were gifts from Dr. Sendurai Mani (MD Anderson Cancer Center, Houston, TX). pCMV-SPORT6-E-cadherin was purchased from OPEN Biosystems (Huntsville, AL). The pGL3-luc/NGAL (−900) and pGL3-luc/NGAL (NF-κB mut) were gifts from Dr. Tatsushi Muta (Tohoku university, Sendai, Japan). pGL3-luc/NF-κB was obtained from Dr. Bharat B. Aggarwal (MD Anderson Cancer Center, Houston, TX).
Plasmid construction, lentivirus production, and transduction
Human E-cadherin cDNA was released from pCMV-SPORT6 with EcoRV and NotI and subcloned into the lentiviral vector pCDH-VMV-MCS-EF1-Puro between EcoRI and NotI to create phE-cadherin. The identity and orientation of this construct were confirmed by DNA sequencing. To produce lentivirus that overexpresses shE-cadherin or E-cadherin, we cotransfected pLKO-shE-cadherin, or phE-cadherin and control vectors with their packaging and envelope plasmids into 293FT cells using lipofectamine 2000 reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). 48 hours later, the viral supernatant was collected and centrifuged at 3000 rpm for 15min. For transduction with lentivirus, cells were infected with 2x diluted virus media containing 6 μg/ml polybrene for 16 hours. The expression of target proteins was confirmed by quantitative real-time polymerase chain reaction (Q-PCR) and/or western blot.
Quantitative real-time PCR
RNA isolation and cDNA synthesis were performed as described before 12. Quantitative RT-PCR (qPCR) was performed using SYBR Green on a BioRad thermocycler. PCR primers were designed using Primer3 software. The expression level of a gene was measured using the 2−ΔΔCt method. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control.
Western blots
20 μg of cell lysate were separated on a 10%– 12% SDS-PAGE gel. The separated proteins wereelectrophoretically transferred to Immobilon™ membranes (Millipore, Bedford, MA) and incubated with 5% dry milk in TBST (25 mM Tris-HCl, pH 7.6, 200 mM NaCl, and 0.15% Tween 20), for 1 hour at room temperature. Target protein levels were measured by immunoblotting with corresponding antibodies at 1:1000 dilutions for 2 hours at room temperature as previously described 12.
Immunohistochemical analysis (IHC)
Tissue microarrays made from formalin-fixed, paraffin-embedded tissues were purchased from US Biomax (Rockville, MD, U.S.A.) and comprised normal and PDAC tissue sections (catalog number PA1001). IHC was performed as described previously 13 with the concentration of primary antibody (NGAL rabbit polyclonal) being 1:1000 in PBS. NGAL expression in the tissues was expressed as either positive or negative.
Immunofluorescence staining (IF)
IF was done as described previously 25. NGAL antibody was used at a dilution of 1:100 in PBS. Fluorescent images were viewed under an IX81 confocal microscope (Olympus).
Luciferase assay
To assess the activity of the NGAL promoter, AsPC-1 cells were transfected with a promoter reporter construct containing either an intact NF-κB binding site (pGL3-luc/NGAL [−900]) or a mutation that prevents NF-κB binding (pGL3-luc/NGAL [NF-κB-mut]). After 16 hours serum starvation, these cells were treated with 25 ng/ml EGF. For the NF-κB binding activity assay, cells were transfected with pGL3-luc/NF-κB after 16 hours of infection with lentivirus carrying either shE-cadherin or control lentivrus. pGL4.74 [hRluc/TK] (Promega, WI) was co-transfected as a control for transfection efficiency. 24 hours after transfection, cells were lysed and luciferase activity measured using Dual-luciferase® Reporter Assay System (Promega, WI) according to the manufacturer’s instructions. Firefly luciferase activity was normalized to that of Renilla luciferase.
Electrophoretic mobility shift assay (EMSA)
To determine NF-κB activation, EMSA was performed as described previously 26. The DNA-protein complex that formed was separated from the free oligonucleotide on 6.6% native polyacrylamide gels. The gels were dried and visualized, and the radioactive bands were quantitated using a PhosphorImager (GE Healthcare, Piscataway, NJ) loaded with an ImageQuant software (GE Healthcare).
Statistical analysis
To analyze of the IHC results, each spot was considered as an individual sample. NGAL expression, the type of tissue and PDAC grade were considered as categorical variables. Categorical variables were compared using a chi-square test. Continuous variables were compared using a two-tailed student’s t-test. A p value < 0.05 was considered as significant.
RESULTS
NGAL expression correlates with tumor differentiation
In our previous study 12, we showed that NGAL expression is associated with a well-differentiated phenotype in PDAC cell lines. In the present study, we used a larger pool of primary PDAC samples to analyze NGAL expression. A total of 73 tissue cores were available for IHC. As shown in Figures 1A and 1B, NGAL was not expressed in any of the normal ducts (0 of 10) but was expressed in 2 of 10 (20%) normal tissues adjacent to cancer. Further, NGAL was expressed in 50 of 73 (68%) PDAC tissue sections and its expression was associated with tumor differentiation (p<0.0001). Of the 63 PDAC cases with defined tumor grades, 16 of 20 (80%) well differentiated, 20 of 26 (77%) moderately differentiated and 5 of 21 (24%) poorly differentiated PDAC tissue section expressed NGAL. These results suggest that NGAL promotes differentiation in PDAC cells.
Figure 1. NGAL expression is associated with cell differentiation.
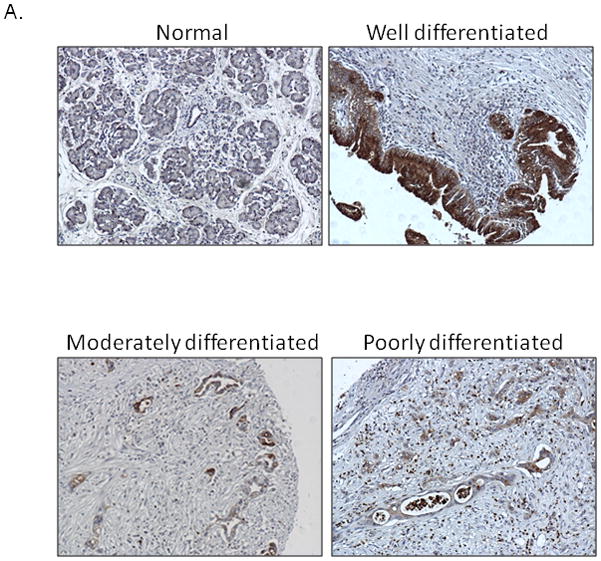
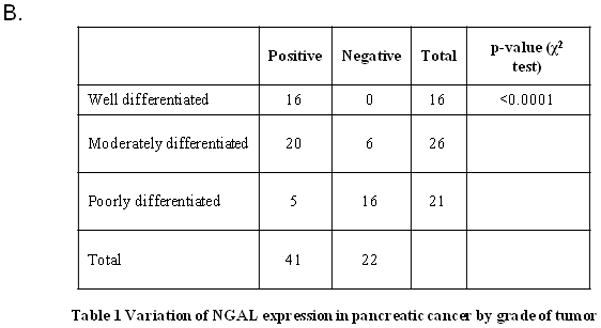
A. IHC analysis revealed NGAL staining at different tumor stages in pancreatic cancer tissues (N: normal, W: well differentiated, M: moderately differentiated and P: poorly differentiated). B. A summary of the results of the IHC analysis. A relationship was found between NGAL expression and cell differentiation.
EGF treatment inhibits NGAL expression in PDAC cells
Studies have shown that EGF and EGFR expression is associated with a loss of PDAC cell differentiation, whereas NGAL expression correlates with a well-differentiated phenotype 27, 28. To investigate whether EGF is involved in the downregulation of NGAL in advanced PDAC, we analyzed the effect of EGF on two NGAL expressing PDAC cell lines, AsPC-1 and BxPC-3. Exposure to EGF in vitro significantly reduced the expression of NGAL mRNA (by 48.4% in AsPC-1 and 44.7% in BxPC-3 cells) compared with that in untreated cells as determined by Q-PCR (Figure 2A). Further, the production of NGAL protein was also significantly reduced by EGF treatment as determined by both western blot analysis (Figure 2B) and immunofluorescence (Figure 2C).
Figure 2. EGF reduces NGAL expression and its effect via EGFR.
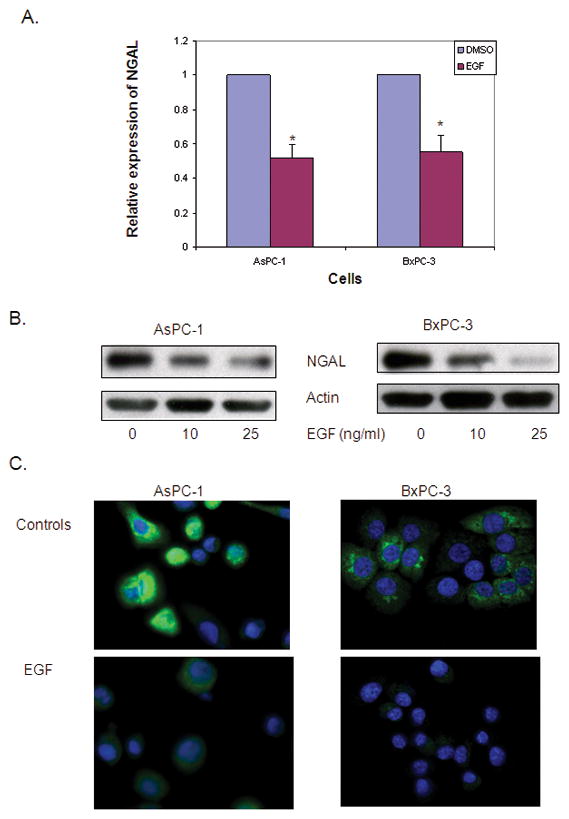
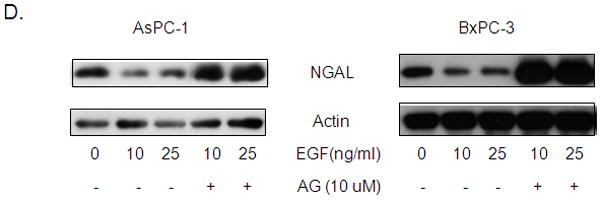
A. Quantitative real-time PCR for NGAL expression using total RNA extracted from cells that were treated with 25 ng/ml of EGF for 24 hours. The NGAL expression was determined using the formula of 2−ΔΔCT and expressed as means ± SE of three independent experiments. * represents p<0.05. B. Western blot analysis revealed NGAL expression in cells treated with the indicated concentration of EGF for 24 hours. C. Immunofluorescent staining of NGAL. Cells were treated with 25 ng/ml of EGF in serum free media. Twenty-four hours later, cells were stained for NGAL (green) and nuclei (DAPI, blue) and photographed using a confocal microscope at ×60 magnification. D. Cells were treated with or without AG1478 for 1 hour and followed by EGF treatment at the indicated dose for 24 hours. NGAL protein levels were determined by western blot analysis.
EGF is a well-known ligand of EGFR. The binding of EGF to EGFR activates downstream signals that stimulate cancer cell invasion and metastasis. To determine whether the EGF-mediated inhibition of NGAL is EGFR dependent, we treated AsPC-1 and BxPC-3 cells with 10 μM of AG1478, an EGFR inhibitor, before stimulating them with EGF. As expected, AG1478 abolished the EGF-mediated down-regulation of NGAL expression (Figure 2D).
EGF exposure leads to downregulation of E-cadherin
Accumulating evidence demonstrates a positive correlation between high EGF and EGFR expression and increased cancer invasion and metastasis in PDAC cells. Loss of E-cadherin has also been demonstrated to be a causal factor that promotes tumor progression. To determine whether E-cadherin contributes to EGF-mediated inhibition of NGAL expression, we examined the alteration in E-cadherin by western blot after exposing cells to EGF. EGF treatment led to remarkably reduced E-cadherin protein levels concurrent with a reduction in NGAL protein levels (Figure 3A). Furthermore, E-cadherin expression positively correlated with NGAL expression in all four cell lines (Figure 3B), suggesting that E-cadherin may involve in the downregulation of NGAL by EGF.
Figure 3. Downregulation of E-cadherin reduces NGAL expression.
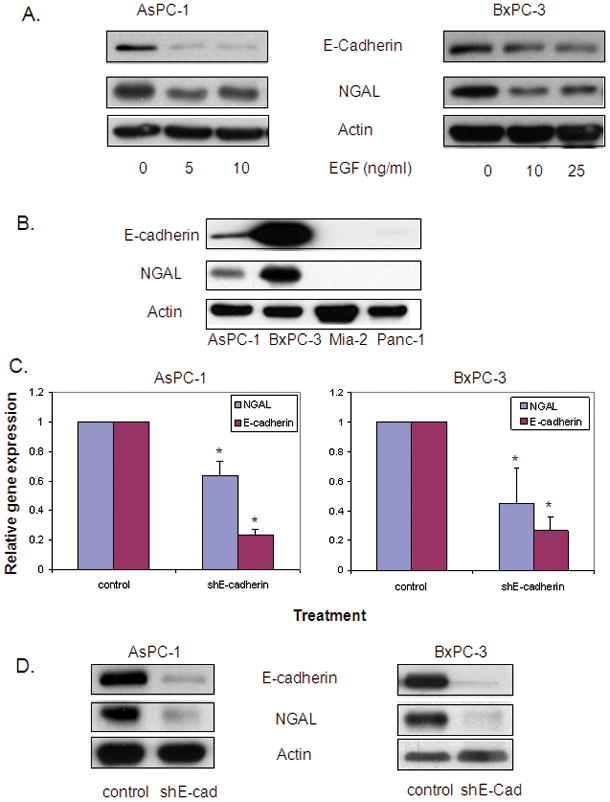
A. Western blot analysis shows NGAL and E-cadherin expression using protein lysate from cells treated with EGF at the indicated doses for 24 hours. B. Western blot analysis shows the relationship between NGAL and E-cadherin expression using protein lysate from selected PDAC cell lines. C. RNA extracted from cells that were infected with lentivirus carrying shE-cadherin or its controls for 48 hours was used for quantitative real-time PCR. Gene expression was calculated using the 2−ΔΔCt method and expressed as the means ± SE of three independent experiments. * represents p<0.05. D. NGAL protein levels were determined by western blot analysis using protein lysate from cells infected with lentivirus carrying shE-cadherin or its controls for 72 hours.
EGF-mediated inhibition of NGAL expression involves E-cadherin
To further confirm the role of E-cadherin in the EGF-mediated inhibition of NGAL expression, we generated lentivirus designed to knockdown or overexpress human E-cadherin. Knock down of E-cadherin by infection with shE-cadherin significantly reduced NGAL mRNA expression in AsPC-1 and BxPC-3 cells (by 36.4% and 44.8% respectively compared to vector control-transected cells) as measured by Q-PCR (Figure 3C). Western blot analysis also demonstrated a reduction in NGAL protein expression in cells infected with shE-cadherin (Figure 3D). Conversely, E-cadherin overexpression led to an increase in NGAL expression (Figure 4A) and abolished the inhibition of NGAL expression by EGF (Figure 4B). Taken together, these observations suggest that E-cadherin positively regulates NGAL expression in PDAC cells and that loss of E-cadherin expression by EGF stimulation may contribute to EGF-mediated inhibition of NGAL expression in PDAC cells.
Figure 4. E-cadherin overexpression leads to increased NGAL expression.
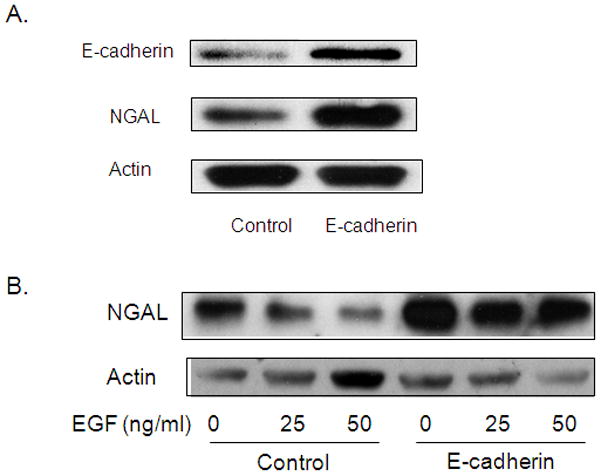
A. Western blot analysis for NGAL protein levels by using protein lysate from cells infected with lentivirus carrying human E-cadherin or its controls for 72 hours. B. Cells were treated with EGF at the indicated doses for 24 hours after being infected with lentivirus carrying human E-cadherin or its controls. NGAL protein levels were then analyzed by western blot analysis.
EGF-mediated inhibition of NGAL and E-cadherin expression involves the ERK pathway
The binding of EGF to EGFR can activate a numbers of downstream signaling pathways including MEK-ERK pathway that in turn regulate the expression of several genes. The ERK pathway has previously been reported to be involved in regulating E-cadherin expression 29, 30. To determine the possible role of the ERK pathway in EGF-mediated inhibition of E-cadherin and NGAL expression, we treated cells with either PD98059 or U0126, inhibitors of ERK for 24 hours, and evaluated NGAL expression by western blot. Both PD98059 and U0126 treatment led to a significant dose-dependent increase in NGAL protein levels (Figures 5A and 5B). In addition, PD98059 or U0126 pretreatment completely abolished the effect of EGF on E-cadherin and NGAL expression (Figure 5C).
Figure 5. Effect of the ERK pathway on NGAL expression.
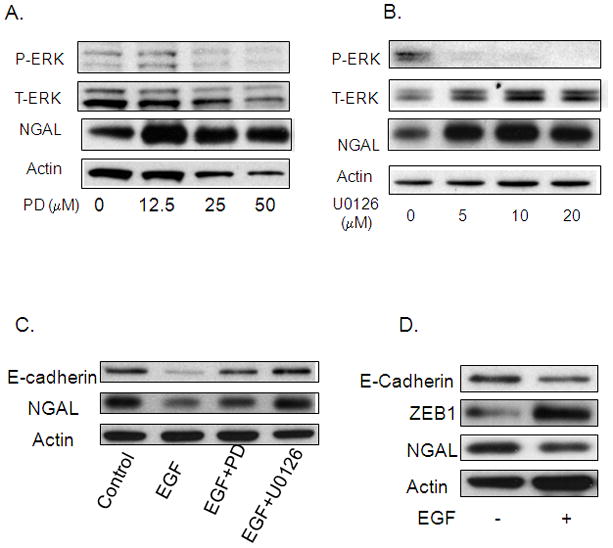
A and B. Western blot analysis for NGAL expression using a cell lysate from cells treated with ERK pathway inhibitor PD98059 or U0126 at the indicated doses for 24 hours. C. Cells were treated with 25 μM of PD98059 or 10 μM of U0126 for 1hour followed by treatment with 25 ng/ml of EGF for 24 hours. The cell lysate was then used for a western blot analysis of E-cadherin and NGAL expression. D. A western blot analysis shows Zeb1 expression after treatment with 25 ng/ml of EGF for 24 hours.
Several transcription factors, including Twist, Snail, Slug and Zeb1, are known to be involved in regulating E-cadherin expression. To determine whether EGF inhibits E-cadherin expression through these transcription factors, we evaluated the effect of EGF treatment on Zeb1 expression. Cells exposed to 25 ng/ml of EGF for 24 hours had higher Zeb1 protein levels than control cells (Figure 5D) and this was accompanied by concomitant reduction in E-cadherin and NGAL expression.
EGF inhibits NGAL expression by repressing of NF-κB-mediated transcription
NF-κB is one of the most important transcription factors that regulate NGAL expression. To determine whether it plays a role in EGF-mediated inhibition of NGAL expression, we transfected AsPC-1 cells with a luciferase reporter construct driven by either 900 bp of wild type NGAL promoter or a mutant NGAL promoter carrying an inactivating mutation in its NF-κB binding site. Simulation of the transfected cells with 25 ng/ml of EGF for 24 hours led to a significantly decrease in the activity of the wild-type NGAL promoter-driven luciferase reporter gene compared to the untreated controls. In cells transfected with mutant NGAL promoter, basal transcription at the NGAL promoter was significantly decreased (compared to that in wild-type promoter-transfected cells) and the transcriptional activity did not change with EGF treatment (Figure 6A). In addition, downregulating E-cadherin by infecting AsPC-1 cells with lentivirus carrying shE-cadherin led to significant reduction in luciferase activity in cells co-transfected with wild-type NF-κB promoter reporter construct (Figure 6B). Similar results were observed in cells treated with EGF by EMSA analysis (Figure 6C). Together; these data suggest that EGF downregulates NGAL expression at the transcription levels by inhibiting the binding of NF-κB to the NGAL promoter.
Figure 6. EGF downregulates NGAL expression by inhibition of NF-κB binding activity.
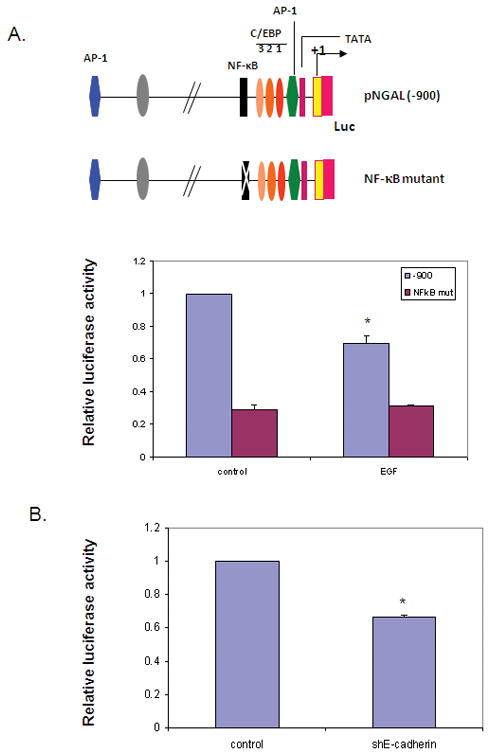
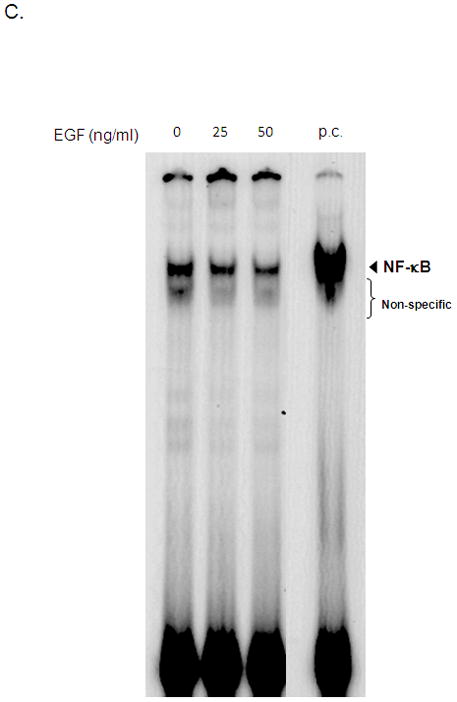
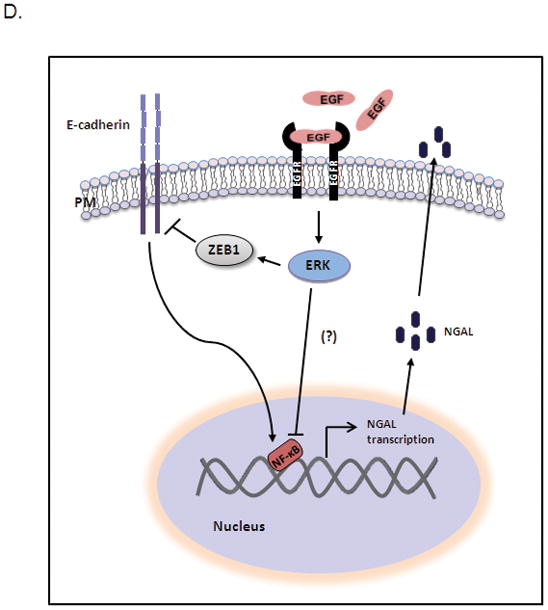
A. AsPC-1 cells were cotransfected with pGL3-luc/NGAL [−900] or pGL3-luc/NGAL [ NF-κB- mut] and pGL4.74 [hRluc/TK] (control for transfection efficiency) for 24 hours after being treated with 25 ng/ml EGF. Cell lysate was then used for the luciferase activity assay. * represents p<0.05. B. AsPC-1 cells were infected with lentivirus carrying shE-cadherin or its controls for 16 hours and followed by cotransfection with pGL3-luc/NF-κB and pGL4.74[hRluc/TK] for 24 hours. Thereafter, the cell lysate was used for a luciferase activity assay. * represents p<0.05. C. Nuclear extract from AsPC-1 cells treated with EGF for 24 hours was analyzed by EMSA as described in the methods section. The p.c. stands for positive control (nuclear extracts from KBM-5 cells stimulated with TNF-α). D. Model illustrating EGF-mediated downregulation of NGAL. EGF activates ERK signaling by activating EGFR, leading to increased Zeb1 expression and reduced E-cadherin expression, which results in decreased NGAL expression by blocking NF-κB binding to the NGAL promoter.
DISCUSSION
NGAL plays an important role in PDAC cell biology by inhibiting PDAC cell invasion and metastasis. EGF is a critical mitogen involved in PDAC cell invasion and metastasis, but nothing is known about how it regulates NGAL expression to mediate this key biologic event in neoplastic progression. In present study, we demonstrated that EGF downregulated E-cadherin and NGAL by activating EGFR and the MAPK-ERK intracellular signaling pathway in PDAC cells.
Previous studies have demonstrated that the function of EGF is dependent on its binding to and activation of the EGFR. EGFR activation in turn regulates the expression of its target genes that promote cell growth, invasion and metastasis via downstream signaling 31, 32. NGAL appears to play a role opposite to that of EGF and EGFR in modulating PDAC cell invasion and metastasis 12. Given the opposing roles of NGAL and EGF in PDAC, we hypothesize that NGAL is an EGF-regulated gene in PDAC cells. Li et al. 33 have reported that ErbB2 (EGFR type 2) overexpression results in increased NGAL expression in breast cancer cells. In contrast, studies in ovarian cancer cells showed that EGF treatment led to reduce NGAL protein expression 10. These seemingly opposite results are consistent with the different biological functions of NGAL in these two cancer types. In breast cancer, NGAL promotes malignant cell proliferation, invasion and metastasis 8, whereas it is an anti-tumorigenic and anti-metastatic factor in ovarian cancer cells 10. In PDAC cells, exposure to EGF significantly decreased both mRNA and protein levels of NGAL. Blocking EGFR activation with an EGFR antagonist (AG1478) abolished the EGF-mediated NGAL downregulation. These findings suggest that EGF is a negative regulator of NGAL expression in PDAC cells and plays a role in EGF-mediated invasion and metastasis of these cells. In addition, the observed positive association between NGAL expression and differentiation in PDAC tissues may be at least partly due to the negative association between EGF and EGFR expression and PDAC cell differentiation 27.
E-cadherin is widely accepted as playing a critical role in epithelial-mesenchymal transition (EMT), an early event in cancer cell invasion and metastasis 34. E-cadherin expression is frequently lost in metastatic human cancers including PDAC 35, 36. The loss of E-cadherin expression has been associated with activation of EGF/EGFR cascade in a number of cancer types, including breast 19, 37, cervical 18 and PCs 38. Our data confirmed those of earlier reports that described EGF and EGFR mediated downregulation of E-cadherin expression in PDAC cells. In addition, we observed that the downregulation of E-cadherin expression by EGF was paralleled by a downregulation of NGAL expression in PDAC cells, suggesting that E-cadherin is a novel regulator of EGF-mediated NGAL downregulation. Indeed, E-cadherin expression was correlated with NGAL expression in PDAC cells. Furthermore, manipulation of E-cadherin expression by either upregulation or downregulation led to an upregulation or downregulation of NGAL expression, respectively. Moreover, E-cadherin overexpression blocked the EGF-mediated inhibition of NGAL expression, further supporting the role of E-cadherin as a positive regulator of NGAL expression in PDAC cells. Lim et al. 10 have reported that EGF downregulates both E-cadherin and NGAL expression in ovarian cancer cells. In immortalized human breast epithelial cells, E-cadherin downregulation by RNA interference or dominant-negative E-cadherin results in reduced NGAL expression 39. Interestingly, NGAL overexpression in colon cancer cells resulted in altered subcellular localization of E-cadherin, thus suggesting a downstream effect of NGAL on E-cadherin 25. But the detailed mechanism of NGAL-mediated E-cadherin regulation still remains unclear. However, in PDAC cells, we didn’t observe any downstream effects of NGAL overexpression or underexpression on E-cadherin expression 12. Furthermore, we showed that NGAL overexpression significantly reduced invasion/metastasis of PDAC cells both in vitro and in vivo 12. Thus, downstream effects of NGAL could be cell context-dependent. Together, our findings demonstrate that E-cadherin regulates NGAL expression in PDAC cells and contributes to EGF-mediated NGAL downregulation in PDAC cells.
The transcriptional factors Snail, Slug, and Zeb1 are key factors that repress E-cadherin expression 34, 40. Zeb1 expression has also been reported to be inversely correlated with E-cadherin expression in primary PDAC patient tumor samples and PDAC cell lines 41. In the same studies, Zeb1 gene silencing by siRNA led to increased E-cadherin expression. These data suggest that Zeb1 is an important negative regulator of E-cadherin expression in PDAC. We found EGF-associated reciprocal changes in Zeb1 and E-cadherin expression further supporting the role of Zeb1 in EGF-induced E-cadherin downregulation in PDAC cells.
Several of signaling pathways can be triggered by EGF/EGFR activation including the MEK/ERK pathways. These pathways in turn modulate downstream gene expression 31, 42. It has been suggested that E-cadherin expression is regulated by the MEK/ERK signaling cascade in PDAC cells 29. In the present study, we found that treatment with the ERK pathway inhibitors PD98059 or U0126 resulted in increased NGAL expression. Furthermore, pre-treatment with PD98059 and U0126 abolished the inhibition of both NGAL and E-cadherin expression by EGF. Exposure of H1435 lung cancer cells to U0125 has previously been reported to increase Zeb1 expression 43. Together with our results, these data suggest that ERK pathway activation is required for EGF-mediated inhibition of E-cadherin (via Zeb1) and NGAL expression at PDAC cells.
NF-κB is one of the most important transcription factors 33, 44, 45. Its activity may be linked to EGF/EGFR activation. For instance, EGF treatment prevents H2O2-induced NF-κB activity in intestinal Caco-2 cells 46. In contrast, Biswas et al. 47 reported that EGF activates NF-κB activity in estrogen-receptor negative breast cancer cells. Moreover, EGF-induced cyclooxygennase-2 expression was found to be NF-κB independent in human oral squamous carcinoma cells 48. Thus, the relationship between EGF/EGFR activation and NF-κB activity may depend on the cell types and conditions used. Our data suggest that the NF-κB binding site is one of the key cis-elements that regulate NGAL expression in response to EGF stimulation in PDAC cells. This is supported by the observation that a mutation of the NF-κB binding site in the NGAL promoter blocked the inhibition of NGAL promoter activity by EGF. Further, E-cadherin downregulation by an E-cadherin-specific shRNA resulted in decreased binding of NF-κB to its cis cognate element. Moreover, EMSA revealed that EGF stimulation reduces the binding of NF-κB to the corresponding cis-element in a dose-dependent manner. Thus, our data strongly suggests that EGF-mediated NGAL downregulation occurs at the transcriptional level and via the interruption of NF-κB binding to the NGAL promoter in PDAC cells. However, how EGF and E-cadherin interrupt NF-κB’s binding to NGAL promoter needs to be further studied.
On the bases of the results of this study, we propose a model of the EGF-mediated NGAL expression inhibition in PDAC cells (Figure 6D). EGFR activation by EGF binding triggered the MEK/ERK signaling pathway, which in turn upregulated Zeb1 expression. The upregulated Zeb1 in turn reduces E-cadherin expression. The loss of E-cadherin led to reduced NGAL expression by inhibiting the binding of NF-κB to the NGAL promoter. MEK/ERK pathways activation can also inhibit this binding by pathways other than E-cadherin. Further, being a secreted protein, NGAL can be detected in body fluids including urine, blood, pancreatic juice and saliva. Thus, NGAL may be a potential biomarker for the loss of E-cadherin expression in PDAC.
Supplementary Material
Acknowledgments
Grants support: This work was supported in part by MDACC Physician Scientist Program Award (to SG), Institutional Research Grant, Cyrus Scholar Award (to SG), McNair Foundation Scholar Award (to SG), NIH R01 CA69480 (to RSB), NIH R01 CA78590 (to SKB), and NIH 5P30CA16672 (Cancer Center Support Grant to MDACC).
We would like to thank Ann M. Sutton in the Department of Scientific Publications at MD Anderson Cancer Center for reviewing this manuscript.
Abbreviations
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- NGAL
neutrophil gelatinase associated lipocalin
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen activated protein kinase
- MEK
MAPK and ERK kinase
- NF-κB
nuclear factor-κB
- RT-PCR
real time PCR
- EMSA
electrophoretic mobility shift assay
- PDAC
pancreatic ductal adenocarcinoma
Footnotes
Disclosure of Potential Conflicts of Interest: None
References
- 1.Flower DR. The lipocalin protein family: structure and function. Biochem J. 1996;318 (Pt 1):1–14. doi: 10.1042/bj3180001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–43. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 3.Nielsen BS, Borregaard N, Bundgaard JR, Timshel S, Sehested M, Kjeldsen L. Induction of NGAL synthesis in epithelial cells of human colorectal neoplasia and inflammatory bowel diseases. Gut. 1996;38:414–20. doi: 10.1136/gut.38.3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saiga H, Nishimura J, Kuwata H, et al. Lipocalin 2-dependent inhibition of mycobacterial growth in alveolar epithelium. J Immunol. 2008;181:8521–7. doi: 10.4049/jimmunol.181.12.8521. [DOI] [PubMed] [Google Scholar]
- 5.Mishra J, Mori K, Ma Q, et al. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol. 2004;15:3073–82. doi: 10.1097/01.ASN.0000145013.44578.45. [DOI] [PubMed] [Google Scholar]
- 6.Stoesz SP, Friedl A, Haag JD, Lindstrom MJ, Clark GM, Gould MN. Heterogeneous expression of the lipocalin NGAL in primary breast cancers. Int J Cancer. 1998;79:565–72. doi: 10.1002/(sici)1097-0215(19981218)79:6<565::aid-ijc3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 7.Bartsch S, Tschesche H. Cloning and expression of human neutrophil lipocalin cDNA derived from bone marrow and ovarian cancer cells. FEBS Lett. 1995;357:255–9. doi: 10.1016/0014-5793(94)01303-i. [DOI] [PubMed] [Google Scholar]
- 8.Bauer M, Eickhoff JC, Gould MN, Mundhenke C, Maass N, Friedl A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res Treat. 2008;108:389–97. doi: 10.1007/s10549-007-9619-3. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Xu L, Xiao D, et al. Upregulation of neutrophil gelatinase-associated lipocalin in oesophageal squamous cell carcinoma: significant correlation with cell differentiation and tumour invasion. J Clin Pathol. 2007;60:555–61. doi: 10.1136/jcp.2006.039297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim R, Ahmed N, Borregaard N, et al. Neutrophil gelatinase-associated lipocalin (NGAL) an early-screening biomarker for ovarian cancer: NGAL is associated with epidermal growth factor-induced epithelio-mesenchymal transition. Int J Cancer. 2007;120:2426–34. doi: 10.1002/ijc.22352. [DOI] [PubMed] [Google Scholar]
- 11.Lee H-J, Lee E-K, Lee K-J, Hong S-W, Yoon Y, Kim J-S. Ectopic expression of neutrophil gelatinase-associated lipocalin suppresses the invasion and liver metastasis of colon cancer cells. Int J Cancer. 2006;118:2490–7. doi: 10.1002/ijc.21657. [DOI] [PubMed] [Google Scholar]
- 12.Tong Z, Kunnumakkara AB, Wang H, et al. Neutrophil gelatinase-associated lipocalin: a novel suppressor of invasion and angiogenesis in pancreatic cancer. Cancer Res. 2008;68:6100–8. doi: 10.1158/0008-5472.CAN-08-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moniaux N, Chakraborty S, Yalniz M, et al. Early diagnosis of pancreatic cancer: neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br J Cancer. 2008;98:1540–7. doi: 10.1038/sj.bjc.6604329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 15.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 16.Korc M, Chandrasekar B, Yamanaka Y, Friess H, Buchier M, Beger HG. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J Clin Invest. 1992;90:1352–60. doi: 10.1172/JCI116001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poon SL, Hammond GT, Leung PCK. Epidermal growth factor-induced GnRH-II synthesis contributes to ovarian cancer cell invasion. Mol Endocrinol. 2009;23:1646–56. doi: 10.1210/me.2009-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee M-Y, Chou C-Y, Tang M-J, Shen M-R. Epithelial-mesenchymal transition in cervical cancer: correlation with tumor progression, epidermal growth factor receptor overexpression, and snail up-regulation. Clin Cancer Res. 2008;14:4743–50. doi: 10.1158/1078-0432.CCR-08-0234. [DOI] [PubMed] [Google Scholar]
- 19.Lu Z, Ghosh S, Wang Z, Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4:499–515. doi: 10.1016/s1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
- 20.Kimura FIK, Kawaguchi T, Kaise H, Yamada K, Mukai K, Matsubara O, Ikeda N, Kohno N. Epidermal growth factor-dependent enhancement of invasiveness of squamous cell carcinoma of the breast. Cancer Sci. 2010;101:1133–40. doi: 10.1111/j.1349-7006.2010.01527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamanaka Y, Friess H, Kobrin MS, Buchler M, Beger HG, Korc M. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1993;13:565–9. [PubMed] [Google Scholar]
- 22.Pryczynicz A, Guzinska-Ustymowicz K, Kemona A, Czyzewska J. Expression of EGF and EGFR strongly correlates with metastasis of pancreatic ductal carcinoma. Anticancer Res. 2008;28:1399–404. [PubMed] [Google Scholar]
- 23.Binker MG, Binker-Cosen AA, Richards D, Oliver B, Cosen-Binker LI. EGF promotes invasion by PANC-1 cells through Rac1/ROS-dependent secretion and activation of MMP-2. Biochem Biophys Res Commun. 2009;379:445–50. doi: 10.1016/j.bbrc.2008.12.080. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Ma G, Dong M, et al. Epidermal growth factor promotes invasiveness of pancreatic cancer cells through NF-kappaB-mediated proteinase productions. Pancreas. 2006;32:101–9. doi: 10.1097/01.mpa.0000191644.94301.be. [DOI] [PubMed] [Google Scholar]
- 25.Hu L, Hittelman W, Lu T, et al. NGAL decreases E-cadherin-mediated cell-cell adhesion and increases cell motility and invasion through Rac1 in colon carcinoma cells. Lab Invest. 2009;89:531–48. doi: 10.1038/labinvest.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaturvedi MM, Mukhopadhyay A, Aggarwal BB. Assay for redox-sensitive transcription factor. Methods Enzymol. 2000;319:585–602. doi: 10.1016/s0076-6879(00)19055-x. [DOI] [PubMed] [Google Scholar]
- 27.Ueda S, Ogata S, Tsuda H, et al. The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas. 2004;29:e1–8. doi: 10.1097/00006676-200407000-00061. [DOI] [PubMed] [Google Scholar]
- 28.Tobita K, Kijima H, Dowaki S, et al. Epidermal growth factor receptor expression in human pancreatic cancer: Significance for liver metastasis. Int J Mol Med. 2003;11:305–9. [PubMed] [Google Scholar]
- 29.Kumei S, Motomura W, Yoshizaki T, Takakusaki K, Okumura T. Troglitazone increases expression of E-cadherin and claudin 4 in human pancreatic cancer cells. Biochem Biophys Res Commun. 2009;380:614–9. doi: 10.1016/j.bbrc.2009.01.134. [DOI] [PubMed] [Google Scholar]
- 30.Honma N, Genda T, Matsuda Y, et al. MEK/ERK signaling is a critical mediator for integrin-induced cell scattering in highly metastatic hepatocellular carcinoma cells. Lab Invest. 2006;86:687–96. doi: 10.1038/labinvest.3700427. [DOI] [PubMed] [Google Scholar]
- 31.Furuse J. Growth factors as therapeutic targets in HCC. Crit Rev Oncol Hematol. 2008;67:8–15. doi: 10.1016/j.critrevonc.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Gibbs JB. Anticancer drug targets: growth factors and growth factor signaling. J Clin Invest. 2000;105:9–13. doi: 10.1172/JCI9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S-H, Hawthorne VS, Neal CL, et al. Upregulation of neutrophil gelatinase-associated lipocalin by ErbB2 through nuclear factor-kappaB activation. Cancer Res. 2009;69:9163–8. doi: 10.1158/0008-5472.CAN-09-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 35.von Burstin J, Eser S, Paul MC, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology. 2009;137:361–71. 71.e1–5. doi: 10.1053/j.gastro.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 36.Joo Y-E, Rew J-S, Park C-S, Kim S-J. Expression of E-cadherin, alpha- and beta-catenins in patients with pancreatic adenocarcinoma. Pancreatology. 2002;2:129–37. doi: 10.1159/000055903. [DOI] [PubMed] [Google Scholar]
- 37.Lo H-W, Hsu S-C, Xia W, et al. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–76. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang F, Sloss C, Zhang X, Lee SW, Cusack JC. Membrane-bound heparin-binding epidermal growth factor like growth factor regulates E-cadherin expression in pancreatic carcinoma cells. Cancer Res. 2007;67:8486–93. doi: 10.1158/0008-5472.CAN-07-0498. [DOI] [PubMed] [Google Scholar]
- 39.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–54. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 40.Hollier BG, Evans K, Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009;14:29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
- 41.Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–8. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frolov A, Schuller K, Tzeng C-WD, et al. ErbB3 expression and dimerization with EGFR influence pancreatic cancer cell sensitivity to erlotinib. Cancer Biol Ther. 2007;6:548–54. doi: 10.4161/cbt.6.4.3849. [DOI] [PubMed] [Google Scholar]
- 43.Yang L, Amann JM, Kikuchi T, et al. Inhibition of epidermal growth factor receptor signaling elevates 15-hydroxyprostaglandin dehydrogenase in non-small-cell lung cancer. Cancer Res. 2007;67:5587–93. doi: 10.1158/0008-5472.CAN-06-2287. [DOI] [PubMed] [Google Scholar]
- 44.Iannetti A, Pacifico F, Acquaviva R, et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc Natl Acad Sci U S A. 2008;105:14058–63. doi: 10.1073/pnas.0710846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolignano D, Donato V, Lacquaniti A, et al. Neutrophil gelatinase-associated lipocalin (NGAL) in human neoplasias: a new protein enters the scene. Cancer Lett. 2010;288:10–6. doi: 10.1016/j.canlet.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 46.Banan A, Zhang LJ, Farhadi A, Fields JZ, Shaikh M, Keshavarzian A. PKC-beta1 isoform activation is required for EGF-induced NF-kappaB inactivation and IkappaBalpha stabilization and protection of F-actin assembly and barrier function in enterocyte monolayers. Am J Physiol Cell Physiol. 2004;286:C723–38. doi: 10.1152/ajpcell.00329.2003. [DOI] [PubMed] [Google Scholar]
- 47.Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: A major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:8542–7. doi: 10.1073/pnas.97.15.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Husvik C, Bryne M, Halstensen TS. Epidermal growth factor-induced cyclooxygenase-2 expression in oral squamous cell carcinoma cell lines is mediated through extracellular signal-regulated kinase 1/2 and p38 but is Src and nuclear factor-kappa B independent. Eur J Oral Sci. 2009;117:528–35. doi: 10.1111/j.1600-0722.2009.00669.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.