

Abstract
The overall neurobiological mechanisms by which lithium and valproate stabilize mood in bipolar disorder patients have yet to be fully defined. The therapeutic efficacy and dissimilar chemical structures of these medications suggest that they perturb both shared and disparate cellular processes. To investigate key pathways and functional clusters involved in the global action of lithium and valproate, we generated interaction networks formed by well-supported drug targets. Striking functional similarities emerged. Intersecting nodes in lithium and valproate networks highlighted a strong enrichment of apoptosis clusters and neurotrophin signaling. Other enriched pathways included MAPK, ErbB, insulin, VEGF, Wnt and long-term potentiation indicating a widespread effect of both drugs on diverse signaling systems. MAPK1/3 and AKT1/2 were the most preponderant nodes across pathways suggesting a central role in mediating pathway interactions. The convergence of biological responses unveils a functional signature for lithium and valproate that could be key modulators of their therapeutic efficacy.
Keywords: Expression, pathways, AKT1/2, MAPK1/3, Cytoscape, MiMI, DAVID, GLay
Introduction
Lithium and valproate salts are popularly prescribed mood stabilizers for patients with recurrent episodes of mania and depression. The therapeutic effect of lithium was serendipitously discovered more than half a century ago1, in later years valproate was also shown to be effective in treating mania2. Such a similar efficacy by agents of dissimilar chemical structures- lithium, an alkali and valproate, a 5- carbon carboxylate- raises the possibility of both shared and distinct mechanisms of action that remain to be fully defined.
Two major mechanisms have been invoked to explain the therapeutic action of lithium. The inositol depletion hypothesis that assigns a major role to the phosphatidylinositol cycle is based on the demonstration that lithium disrupts the activity of enzymes involved in inositol turnover3. Later studies showed that lithium also inhibited GSK3β, thereby implicating the Wnt signaling pathway4. However, these may be just two of the many lithium-responsive biological processes.
Studies have documented diverse cellular effects of lithium in organisms spanning the evolutionary scale. Lithium has been shown to extend the life of the nematode, Caenorhabditis elegans,5 shift the development of the sea urchin, Strongylocentrotus purpuratus, toward vegetalization,6 inhibit fermentation in the yeast Saccharomyces cerevisiae, grown in galactose,7 and disrupt cell fate determination in the social amoeba, Dictyostelium discoideum.4 In various vertebrate model systems lithium has been shown to influence multiple processes including cell survival, neuroprotection, neurogenesis and inhibition of apoptosis.8,9
Valproic acid, an anticonvulsant, found its use in treating bipolar disorder in the 1980s.2 The therapeutic efficacy and side effects of valproate and lithium have been investigated. Valproate has been shown to inhibit histone deacetylases (HDACs),10,11 which may lead to activation of repressed genes. Studies have shown that valproate promotes neurogenesis,12,13 and in cortical neurons, this effect is mediated through a MAPK-dependent pathway.12
A unitary picture has yet to emerge from published studies on the global effects of lithium and valproate. We explored the molecular repertoire that underpins the overall cellular responses to these medications, by selecting well-documented targets that showed consistent differential expression or activity in at least two studies, and using these to build interaction networks. To identify significantly enriched functional clusters and signaling pathways recruited by these networks, functional annotation of nodes was performed. This approach generated drug-responsive interaction networks that integrated diverse pathway interactions and highlighted apoptosis-related functional clusters and multiple pathways, foremost of which was neurotrophin signaling.
Materials and methods
Drug target selection: basic principles
We attempted to capture all molecules that showed consistent lithium- and valproate-induced changes in expression and/or enzyme activity in various studies, including those directed at the transcriptome or proteome, and those that focused on specific molecules in various cells and organisms. We found that the majority of genes across these studies showed inconsistent expression patterns, particularly in microarray-based genome-wide profiling. Experimental differences across studies that could influence the level and pattern of gene expression were apparent. These include the use of various organisms and cells, variability in methods of drug administration, doses and lengths of drug exposure, ages of animals, sacrifice techniques, tissues and brain regions isolated for analysis, microarray chip batches and other divergences in experimental design. It was therefore important to establish a stringent inclusion threshold for drug-regulated molecules in order to create a robust seed for interaction networks.
Assembly of lithium-responsive molecules
After identifying a series of publications that found lithium-induced expression changes in various species, we utilized a multi-pass system to extract as much relevant information as possible from each study’s dataset. The end goal was to convert each study’s data into mouse gene homologs for direct comparison. Accession numbers cited in various studies and relevant databases including eukarYotic OrtholoGY (YOGY) (http://www.sanger.ac.uk/PostGenomics/S_pombe/YOGY/index.shtml) and the Sea Urchin Genome Project (http://annotation.hgsc.bcm.tmc.edu/Urchin/cgi-bin/pubLogin.cgi) were used. A Uniprot ID was established for each differentially-regulated transcript and BLAST was performed against the mouse genome (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). Only evalues <1.00E-5 were considered.
Once a gene name was identified for a transcript, NCBI’s Homologene was queried (http://www.ncbi.nlm.nih.gov/homologene). Using a universal point of reference helped to minimize confusion regarding alternate gene names. If there was no match in Homologene or by protein BLAST, the data point was not considered. A list of mouse homologs was then generated, and compared against all other studies for replication.
Classic targets of lithium
To populate the network seed, priority was assigned to well-documented targets of lithium: GSK3B, IMPA1, AKT1, BCL2 and BDNF (Table 1). Inhibition of GSK3B that leads to the activation of Wnt signaling is one of the best-supported cellular responses to lithium.4,14 A recent report has shown a dose-dependent decrease in Gsk3b mRNA in rat hippocampal cultures.15
Table 1.
Molecules significantly regulated by lithium reported in at least two studies
| Group 1 | Expression/enzyme activity/fold change, organism/cell line |
|---|---|
| AKT1 (PKB) | ↑enzyme activity: rat cerebellar granule cells,19 rat cortex,20 mouse proximal tubular cells;21 ↑protein: human Huh7 cells;22 ↓protein: human Hep40 cells22 ↓protein & transcript: bovine adrenal chromaffin cells23 |
| ALDH1A3 (ALDH6) | ↓transcript = 0.63, human T/C28a cells;38 ↓protein = 0.62, rat kidney44 |
| ATF4 | ↑transcript:1.643, human HepG2 cells;39 1.83, T/C28a cells;38 1.503, mouse brain41 |
| BCL2 | ↑transcript: rat cerebellar cells;25 rat frontal cortex;26 mouse retinal ganglion cells;27 chick cochlear magnocellularis neurons28 |
| BDNF | ↑transcript: rat frontal cortex;29,30 rat hippocampus;31 rat cortical neurons;32 ↓protein: FSL rat frontal cortex;34 ↑protein: FRL rat frontal cortex34 |
| CKB | ↑transcript = 2.2, sea urchin;6 ↑protein = 2.4, rat kidney44 |
| EZR | ↑transcript = 1.5, mouse brain;41 ↑protein = 2.2, rat kidney44 |
| GSK3B | ↓enzyme activity: multiple studies4,14 ↓transcript: rat hippocampal cells15 |
| HIST1H1C | ↓transcript: 0.56, human T/C28a cells;38 0.539, mouse brain41 |
| IMPA1 | ↑transcript: yeast;7 rat brain;17 mouse hippocampus18 |
| MAT2A | ↓transcript: rat brain36,39 |
| PPARGC1A | ↓transcript: human adipocytes;45 0.65, human neuroblastoma cells40 |
| PTGS2 | ↑transcript: 4.11, human T/C28a cells;38 1.855, mouse brain41 |
| PTPRK | ↑transcript: 1.53, rat cortical slices;37 2.23, human neuroblastoma cells40 |
| RGS4 | ↑transcript: 1.66, rat cortical slices;37 2.61, human T/C28a cells38 |
| SYN1 | ↑transcript, rat PC12 cells;46 ↑protein=2.11, rat prefrontal cortex43 |
| TM4SF1 | ↑transcript: 1.71, human T/C28a cells;38 1.876, mouse brain41 |
| TPM1 | ↓transcript: rat brain;36 ↓ protein=0.44, rat kidney44 |
| Group 2 (Group 1 + molecules below) | |
| CALM/CALM2 | ↓transcript = −2.2, rat brain;35 ↓protein = 0.62, rat kidney44 |
| CCNB2 | ↓transcript: yeast;7 0.69, human T/C28a cells38 |
| CRYAB | ↓transcript = 0.7, mouse brain;41 ↓protein = 0.62, rat kidney44 |
| ETFB | ↑transcript: nematode;5 ↑protein=2.7, rat kidney44 |
| MKI67IP | ↓transcript: rat brain;36 yeast7 |
| RCL1 | ↓transcript: rat brain;36 yeast7 |
| SLC3A1 | ↑transcript: yeast;7 nematode5 |
IMPase catalyzes the release of free inositol, and is inhibited by lithium.3,16 Lithium-induced overexpression of Impa1 has been demonstrated in yeast,7 in rat brain17 and mouse hippocampus18 (Table 1).
Lithium-induced activation of Akt1 accompanied by neuroprotection has been demonstrated in cerebellar granule cells19 (Table 1). Akt1 activation has been shown also in rat brain20 and renal epithelial cells deprived of growth factor.21 Lithium elicited opposite effects on AKT1 protein concentration in Huh7 and Hep40 cells.22 Chronic lithium treatment of bovine adrenal chromaffin cells inhibited Gsk3b activity and reduced Akt1 expression.23
Potent up-regulation of the anti-apoptotic gene, Bcl2, by lithium has been well-documented,24 as illustrated by studies in rat cerebellar cells,25 rat frontal cortex,26 mouse retinal ganglion cells27 and chick cochlear magnocellularis neurons28 (Table 1).
Lithium stimulated the expression of Bdnf, a neurogenesis and neuroprotective factor, in rat frontal cortex,29,30 hippocampus31 and in cortical neurons via activation of the Bdnf promoter IV,32 but failed to elicit neuroprotection in Bdnf deficient mice challenged with glutamate.33 Inconsistent direction of Bdnf brain expression has been reported in lithium-treated FSL and FRL rats.34 The variable effects of lithium on AKT1, BDNF and IMPA1, and possibly other genes seem to depend, at least partly, upon the cellular milieu.
Lithium-responsive expression profiles: transcriptome, proteome and other gene-specific studies
We prepared a list of genes significantly regulated by lithium in whole-transcriptome and proteome screens of organisms spanning a wide spectrum of the evolutionary scale, from yeast to human-derived cell lines (Table 1). Scores of genes whose expression was altered by lithium have been reported in individual transcriptome profiling studies: rat brain,35,36 rat cortical slices,37 human T/C28a cells,38 human HepG2 cells,39 human neuroblastoma cells,40 mouse brain,41,42 yeast,7 sea urchin6 and C. elegans5 (Table 1). Two studies interrogated the effect of lithium on the rat proteome43,44 (Table 1).
Inclusion threshold for drug targets in network seed
In order to build networks that hold biological validity we established a selection threshold for molecules included as seed for interaction networks. First, a significant lithium-induced expression change or change in enzyme activity reported for a particular molecule in one study had to be replicated in at least one other study. Second, for those reported in genome-wide microarray studies we set a fold-change minimum in differential expression and this change had to be confirmed by at least one other study. Assuming 20,000 transcripts sampled in each study, such consistency would rarely occur by chance (<3/10,000).
Molecules that fulfilled the selection criteria were grouped into two hierarchical categories. Group 1 included 18 consisting of the classic targets of lithium and other differentially regulated molecules in at least two studies. Also included in Group 1 were those reported in profiling studies that underwent ≥ 1.5-fold up-regulation or ≤ 0.667-fold down-regulation (Table 1). Group 2 included Group 1 and the stringency was relaxed to include genes up-regulated at ≥ 1.4-fold or down-regulated at ≤ 0.714-fold in profiling studies (Table 1). Fold-change data were not reported in yeast7 and nematode5 mRNA profiles, hence differential expression replicated in these studies were included in Group 2, unless at least two other studies showed support for the expression change, then these molecules were included in Group 1 (Table 1). Transcript profiling in human T/C28a cells38 and proteome analysis in rat kidney44 displayed the highest number (seven) of replicated lithium targets, across studies. These inclusion criteria narrowed the myriad of lithium-regulated molecules to a much smaller number. We believe that this approach helped minimize false positives and increase data reliability.
The five well-documented targets of lithium formed the core of input genes. For all other regulated molecules notable aspects of expression patterns further strengthen the case for inclusion in the network seed (Table 1). Of those genes that met the selection threshold, six showed altered transcript levels in some studies that were replicated by parallel changes in protein concentration in other studies. Significant regulation of the same genes was also found in phylogenetically distant organisms (see below).
In response to lithium, the Ezr transcript was over-expressed in mouse brain41 and was matched by an elevated Ezr protein concentration in rat kidney.44 Also, a significant reduction in transcript levels of ALDH1A3 in T/C28a cells,38 Calm in rat brain,36 and Cryab in mouse brain41 was consistent with decreased levels of their cognate proteins in rat kidney.44 Over-expression of Ckb transcripts in lithium-treated sea urchin6 was supported by an increased abundance of Ckb protein in rat kidney.44 Down-regulation of Ccnb2 was reported in both yeast7 and human T/C28a cells.38
Valproate responsive transcriptome, genes and enzymes
We applied a similar approach detailed in the lithium portion of this study in selecting valproate-responsive molecules as input to construct interaction networks. Expression changes and/or changes in enzyme activity had to be supported by at least two studies. For microarray-based expression profiles we used a threshold of ≥ 1.5 fold significant differential upregulation or downregulation.
Several studies have reported the effect of valproate on the expression or enzyme activity of molecules similarly shown to be regulated by lithium: AKT1,47–51 BCL2,26,49,52 BDNF32,50,53 and GSK3B.26,54,55
The effect of valproate on various transcriptomes has been reported in mammalian cell lines,47,56–60 cortical neurons,50 neural tubes from mouse embryos61,62 and rodent brains63–65 (Table 2). The application of our selection criteria in the aforementioned studies and in other studies that focused on single genes such as GPR78,66–68 yielded 47 valproate targets that comprised the network seed (Table 2). Interestingly, mRNA profiling in rat cortical neurons50 displayed 33 of the 47 valproate targets, producing the highest number of replicated targets across studies.
Table 2.
Molecules significantly regulated by valproate reported in at least two studies
| Gene Name | Expression/enzyme activity change, organism, cell |
|---|---|
| ADAR | ↓transcript: rat brain;64 mouse neural tube58 |
| ADNP | ↓transcript: rat cortical neurons;50 mouse embryo62 |
| AK1 | ↑transcript: human theca cells;47 P19 mouse embryonic cells58 |
| AKT1 | ↓transcript: human theca cells;47 SiHA & HeLa cells;48 rat cortical neurons;50 ↑enzyme activity: mouse muscle;51 human neuroblastoma cells SHSY5Y54 |
| ALDH6A1 | ↑transcript: human theca cells;47 mouse neural tube58 |
| APAF1 | ↑transcript: CLL B cells;59 rat cortical neurons50 |
| BCL2 | ↑transcript: rat cortex;26 human neuroblastoma cells SHSY5Y49 |
| BDNF | ↑transcript: rat brain primary cultures, rat C6 glioma cells; 53 rat cortical neurons; 50 rat cortical neurons32 |
| BOK | ↓transcript: human theca cells;47 rat cortical neurons50 |
| CASP6 | ↑transcript: human theca cells;47 CLL B cells59 |
| CDK5R1 | ↓transcript: mouse brain regions;63 rat cortical neurons50 |
| CDKN1B | ↑transcript: human theca cells;47 rat cortical neurons50 |
| CDKN2C | ↑transcript: rat cortical neurons;50 CLL B cells59 |
| CKB | ↑transcript: P19;58 human lymphoblastoid cell line60 |
| CNKSR2 | ↓transcript: rat brain;64 rat cortical neurons50 |
| DAD1 | ↑transcript: human theca cells;47 rat cortical neurons50 |
| EGR1 | ↑transcript: rat cortical neurons;50 mouse embryo62 |
| FOXO1 | ↑transcript: human theca cells;47 mouse embryo62 |
| GRM3 | ↓transcript: mouse brain regions;63 rat cortical neurons50 |
| GRP78 | ↑transcript: rat cerebral cortex;66 panel of cancer cells;67 panel of colon & pancreatic cell lines68 |
| GSK3B | ↓enzyme activity: human neuroblastoma cells SHSY5Y;26,54 HepG255 |
| GTF2I | ↓transcript: rat cortical neurons;50 mouse neural tubes58 |
| H1F0 | ↑transcript: mouse brain regions;63 rat cortical neurons;50 mouse neural tubes;58 mouse embryo62 |
| H3F3B | ↑transcript: P19 cells;61 mouse embryo62 |
| HDAC1 | ↓enzyme activity: Neuro2A cells;11 hematopoetic cancer cells10 |
| HMGCL | ↑transcript: rat cortical neurons;50 mouse embryo62 |
| HSPA1A | ↑transcript: rat cortical neurons;50 mouse neural tubes58 |
| HTRA1 | ↑transcript: rat cortical neurons;50 P19 cells58 |
| INSIG2 | ↑transcript: rat cortical neurons;50 mouse neural tubes58 |
| LIN7B | ↑transcript: rat cortical neurons;50 mouse neural tubes58 |
| LITAF | ↑transcript: rat cortical neurons;50 P19 cells58 |
| LUM | ↑transcript: HEK293 cells;57 NBFL cells56 |
| LYPLA1 | ↑transcript: mouse brain regions;63 rat cortical neurons50 |
| MARCKS | ↓transcript: hippocampal cells;69 rat cortical neurons50 |
| NRP2 | ↓transcript: human theca cells;47 rat brain64 |
| PLAGL1 | ↑transcript: human theca cells;47 rat cortical neurons50 |
| PTEN | ↑transcript: rat cortical neurons;50 CLL B cells59 |
| PTMS | ↓transcript: human theca cells;47 rat cortical neurons50 |
| RAB7L1 | ↑transcript: human theca cells;47 rat cortical neurons50 |
| RNASE4 | ↑transcript: rat cortical neurons;50 mouse neural tubes58 |
| RPA2 | ↑transcript: rat brain;64 rat cortical neurons50 |
| SRD5A1 | ↑transcript: rat brain;64 rat cortical neurons50 |
| SSR3 | ↑transcript: mouse brain regions;63 rat cortical neurons50 |
| TCF19 | ↑transcript: mouse neural tubes;58 mouse embryo62 |
| TCN2 | ↑transcript: rat cortical neurons;50 mouse neural tubes58 |
| TGFB1I1 | ↓transcript: human theca cells;47 rat cortical neurons50 |
| TPBG | ↑transcript: NBFL cells;56 rat cortical neurons50 |
Interaction networks and pathways
We used the Cytoscape (http://www.cytoscape.org/) plugin tool, MiMI (Michigan Molecular Interactions) 70,71 to generate interaction networks. MiMI retrieves data from a collection of all relevant large databases, and performs “deep merging” to eliminate redundancies. Summary network diagrams are created that can be presented in various layouts. In network terminology, a “node” represents a protein or a gene and an “edge” is the line that connects two nodes.72 Node and edge attributes are detailed in downloadable text files, effectively disentangling the complex network into a comprehensible assortment of connections.
To extract subclusters from the large network we used the Cytoscape plugin, Community Clusters GLay (http://cytoscape.wodaklab.org/wiki/CommunityStructureLayout). 73
Significant Kyoto Encyclopedia of Genes and Genomes, (KEGG; http://www.genome.jp/kegg/) pathways and enriched functional clusters were ascertained using DAVID Bioinformatics Resource (http://david.abcc.ncifcrf.gov/home.jsp, NIAID/NIH).74 Enriched clusters and pathways with false discovery rate (FDR) adjusted p≤0.05 were the only ones considered in this study. PANTHER pathways and reactions by REACTOME were also generated in DAVID.
Results
Interaction networks of lithium and valproate molecular targets
To identify the overall mechanisms involved in lithium and valproate cellular responses we employed a strategy of generating interaction networks for well-selected drug targets and subjecting network nodes to functional annotation. This approach illuminated both convergent and divergent biological effects of lithium and valproate.
Lithium interaction networks
Groups 1 and 2 lithium-responsive molecules created large networks, featuring multi-directional interconnections of hubs and convergence of edges on shared nodes. The use of “query genes + nearest neighbors” option in MiMI70,71 spawned 472 nodes and 3014 edges for Group 1 (Figure 1), and 554 nodes and 3654 edges for Group 2 (data not shown), offering a glimpse of the complexity of lithium-perturbed interactions within the cell.
Figure 1.
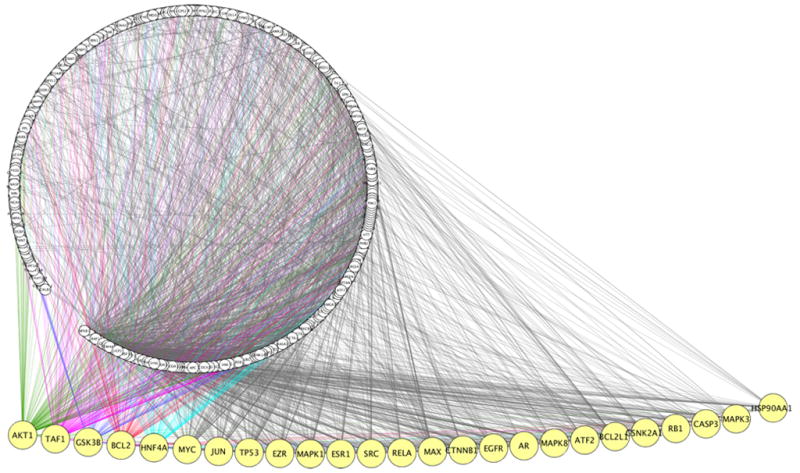
Lithium interaction network. A seed of 18 Group 1 differentially regulated lithium targets was uploaded into MiMI Cytoscape plugin and a network was generated using the “query genes + nearest neighbor” option. The interaction network is presented as the Cytoscape degree sorted circle summary layout; the nodes, shown as circles, form a chain within the periphery of the large circle and the edges are shown as lines linking two nodes. The largest 25 hubs were manually extended away from the network circle to reveal the node labels. Edges for the top 5 hubs are shown in different colors. Lists of 492 nodes and 3101 edges are available upon request.
The scope of interactions formed by a seed of 18 Group 1 molecules depicted multiple hubs; the largest of 25 major hubs was at AKT1, which recruited 131 edges (Figure 1). Other hubs were located at GSK3B, BCL2, EZR, HNF4A, MYC, JUN, TP53, MAPK1 and EZR, and in descending order, were linked to edges ranging from 120 to 63. Connectivities emanated also from families of activating transcription factors (ATFs), caspases, CCAAT/enhancer binding proteins (CEBPs) and mitogen-activated protein kinases (MAPK1, MAPK3 and MAPK8). Several edge intersections between major hubs converged at identical nodes, exemplified by the AP-1 transcription factor JUN which was a frequent hub-to-hub connector. Group 2 produced similar results hence we will focus only on Group 1. Lithium action seems to involve an assortment of molecules that impact various signaling systems.
Valproate interaction networks
The MiMI70,71 interaction network generated by 47 valproate-responsive molecules was an elaborate circuitry of 897 nodes and 7849 edges, about twice the size of the lithium network (Figure 2). The largest hub formed by a single node was at the TATA-box binding protein (TBP)-associated factor (TAF1), which projected 246 edges (Figure 2). Other major hubs were located at HNF4A, HDAC1, MYC, TP53, AKT1, RB1, RPA2, MAX and E2F1, each linked to edges ranging from 99 to 177. Protein families that had connections to >200 edges included cyclin-dependent kinases (CDKs), DNA excision repair ERCC proteins, cyclins (CCNs), mini-chromosome maintenance proteins (MCMs) which make up the pre-DNA replication complex, cell division cycle proteins (CDCs) and the caspases (CASPs). More than 100 edges projected from cyclin-dependent kinase inhibitors (CDKNs) that control cell cycle progression, the cell cycle acting transcription factors E2Fs, general transcription factors (GTFs), DNA polymerases (POLs), and origin recognition complex protein (ORCnLs). Recruitment of this series of proteins to the network suggests a prominent role for the cell cycle, DNA replication, DNA repair, transcription regulation, cell growth and other nuclear processes in valproate response.
Figure 2.
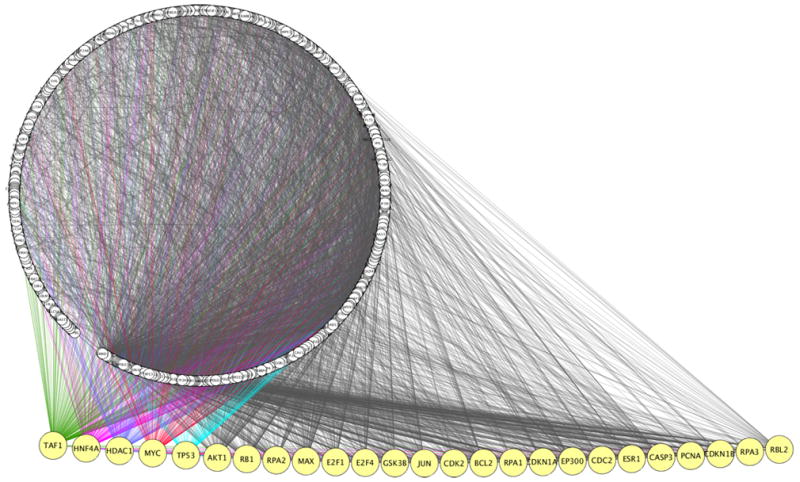
Valproate interaction network. A seed of 47 differentially regulated valproate targets was uploaded into MiMI Cytoscape plugin and a network was generated using the “query genes + nearest neighbor” option. The interaction network is presented as in Figure 1. Lists of 897 nodes and 7849 edges are available upon request.
We compared the edges linked to the HDACs with those of other large hubs and found multiple common connectors. The HDACs and cyclins showed the highest number of identical linkers that were mostly involved in the cell cycle, replication, transcription, mitosis and DNA methylation.
Enriched functional clusters and canonical pathways
In an effort to uncover functional clusters and pathways central to lithium and valproate cellular response we explored the biological implications of the diverse interactions within the networks, through functional annotation of network nodes.
Lithium functional clusters and pathways
Analysis in DAVID74 of the 492 network nodes recruited by 18 lithium targets revealed that regulation of apoptosis was the most highly enriched functional cluster. Node clusters involved in regulation of apoptosis, positive and negative regulation of apoptosis garnered enrichment scores of 54, 40 and 31 (FDR p = 2.8E-52, 2.6E-37 and 7.2E-29, respectively, under the highest stringency setting) (Supplementary Figure 1). The next sets of enriched clusters consisted of Zn finger containing molecules and nuclear hormone receptors, regulation of synaptic transmission, and activation of caspases and had enrichment scores of 10, 8.6 and 8.4 (FDR p = 4.2E-09, 1.4E-06 and 3E-05, respectively).
Further analysis in DAVID74 showed that the majority (60%) of network nodes were constituents of 46 enriched KEGG signaling pathways (FDR p ≤ 0.05), indicating that lithium triggered a multiplicity of cellular responses. Sixteen pathways were disease-specific, of which 14 were involved in various forms of cancer, one was involved in amyotrophic lateral sclerosis and one in Alzheimer’s disease but discussion of these pathways is beyond the scope of this study. It is important to note however that this finding provides further evidence for the multi-functionality of nodes and the extent of their involvement in a variety of diseases and biological processes.
Lithium induced a strong enrichment of 30 other enriched KEGG pathways, foremost of which was neurotrophin signaling (Figure 3A). Other highly enriched pathways included MAPK, ErbB, insulin, apoptosis, VEGF, Wnt, long-term potentiation and axon guidance, indicating a widespread perturbation of signaling systems. To explore potential pathway interactions, we ascertained the distribution of network nodes across all 30 pathways and noted a marked recurrence of MAPK1 and MAPK3 (henceforth referred to as MAPK1/3) and AKT1 and AKT2 (henceforth referred to as AKT1/2) (Figure 3A). MAPK1/3 was a constituent of 22 pathways, and the related enzyme RAF1 (MAP3K) that phosphorylates MEKs that, in turn, phosphorylate MAPKs was a component of 18 pathways. AKT1/2, PIK3R1 (a regulatory subunit of PI3K, the enzyme involved in AKT1 activation) and GSK3B were constituents of 16, 14 and 10 pathways, respectively (Figure 3A). The preponderance of MAPK1/3 and AKT1/2 suggests a major role for these molecules in mediating pathway crosstalks.
Figure 3.



Figure 3A Enriched pathways recruited by lithium network nodes. Bars are colored to indicate those pathways containing or lacking one or a combination of AKT1/2, MAPK1/3 and GSK3B. Red bars represent pathways that contain AKT1/2, MAPK1/3 and GSK3B; purple bars include AKT1/2 and MAPK1/3; yellow bars contain AKT1/2; green bars contain GSK3B; AKT1/2, MAPK1/3 and GSK3B are absent in pathways represented by black bars and blue bars contain MAPK1/3; sky blue bars contain both GSK3B and MAPK1/3. Abbreviations: AJ=adherens junction; BCR=B cell receptor signaling; ECSHPI=epithelial cell signaling in Helicobacter pylori infection; FA=focal adhesion; FceRI=Fc epsilon RI signaling; FcgRMP=Fc gamma R-mediated phagocytosis; GJ=gap junction; LTD=long term depression; LTM=leukocyte transendothelial migration; LTP=long term potentiation; NKCMC=natural killer cell mediated cytotoxicity; NOD-like R=NOD-like receptor; OM=oocyte meiosis; P-MOM=progesterone mediate oocyte maturation; TCR=T cell receptor signaling; Toll-like R=Toll-like receptor signaling. Lists of nodes populating individual pathways are available upon request.
Figure 3B Enriched pathways recruited by valproate network nodes. Representations are as in Figure 3A.
Figure 3C Enriched pathways recruited by lithium-valproate intersecting network nodes. Representations are as in Figure 3A.
PANTHER placed 43% of lithium nodes in various pathways and disclosed a prominent enrichment of apoptosis (FDR p=1.9E-18). In addition, REACTOME included 51% of lithium nodes in established biological reactions, the most highly represented of which were signaling by NGF and apoptosis.
Valproate functional clusters and pathways
Valproate network nodes were analyzed in the same manner as those of lithium. Functional annotation of 897 valproate nodes showed that under the highest stringency setting, the topmost enrichment scores of 73, 60, 42, 38 and 34 were associated with the nuclear lumen (FDR p=6.7E-77), regulation of cell death (apoptosis) (FDR p=1.4 -4.6E-57), programmed cell death (FDR p=7.45E-42), positive regulation of cell death (FDR p=4.3E-36) and negative regulation of cell death (FDR p=3E-32).
The cell cycle emerged as the most strongly enriched (FDR p=7.4E-49) of the 41 highly supported KEGG pathways (Figure 3B). (Here again, 16 of 41pathways were disease-specific and will not be discussed further). Nucleotide excision repair, DNA replication, mismatch repair and homologous recombination pathways were also significantly enriched. The predominance of cellular functions that included the nuclear lumen cluster, cell cycle, nucleotide excision repair and DNA replication was unique to the valproate network and it exposed a clear distinction between the effects of lithium and valproate.
Conversely, marked similarities between lithium and valproate networks were apparent. Valproate nodes populated 19 of the same pathways significantly recruited by lithium nodes, among which were neurotrophin, ErbB, chemokine, p53, apoptosis, MAPK, VEGF, insulin and Wnt (FDR p = 2.4E-27, 1.5E-20, 1.2E-19, 4E-13, 1E-12, 6.3E-12 and 7.9E-09, respectively) (Figure 3B). We noted also that MAPK1/3 and AKT1/2 were the most predominant constituents of more than half of 25 enriched pathways (Figure 3B). PIK3R1 and RAF1 were both components of 11 pathways and GSK3B was in nine.
Intersection of lithium and valproate network nodes
To further explore similarities in molecular response to lithium and valproate, we examined both networks for the occurrence of intersecting nodes. Of the 492 lithium and 897 valproate nodes, 332 overlapped (list is available upon request). Analysis of these nodes in DAVID74 exposed explicit imprints on cellular function that resembled findings in the lithium-responsive network. Most particularly, node clusters involved in regulation of apoptosis, negative regulation of apoptosis and positive regulation of apoptosis displayed very high enrichment scores of 60 (FDR p=5E-58), 40 (FDR p=5.5E- 38), and 39 (FDR p=1.4E-35), respectively. Similarly, neurotrophin signaling was ranked topmost among 45 significantly enriched KEGG pathways (Figure 3C). Other significantly represented pathways (excluding 16 disease-specific pathways) included MAPK, T cell receptor, insulin, ErbB, Wnt, VEGF and apoptosis (Figure 3C). Cell cycle, the most enriched pathway in valproate, was 22nd in this list.
As shown previously AKT1, BCL2, BDNF and GSK3B activity and/or expression are subject to differential regulation by both lithium and valproate hence these were included in both sets of network seed. This, in itself, would predict substantial overlaps between lithium and valproate networks. To test whether the major effects of these drugs were due to these common targets, we performed a secondary analysis in which all four were deleted from the two sets of network seed. These truncated seeds were then used to generate new networks (data not shown but available upon request). Although this analysis produced a smaller lithium network, the recruited nodes largely recapitulated the functional attributes displayed by the untrimmed lithium seed. Components of ErbB, MAPK and neurotrophin signaling pathways, and node clusters for Zn finger containing molecules/nuclear hormone receptors and regulation of apoptosis were most prevalent. Similarly, the pruned valproate network placed the cell cycle, nucleotide excision repair and DNA replication as the top supported pathways, and nuclear lumen and regulation of cell death as the most enriched functional clusters, replicating prior findings with the full valproate seed. These results confirm the pervasive influence of lithium and valproate on specific biological processes; in the absence of AKT1, BCL2, BDNF and GSK3B, the remaining seed molecules recruited highly enriched pathways and functional clusters similar to those displayed by the primary networks.
To identify molecules that might play a dominant role in mediating pathway interactions we examined the distribution of nodes across pathways. MAPK1/3 and AKT1/2 showed the highest prevalence and were constituents of 22 and 16 pathways, respectively (Figure 3C). In addition, RAF1was a component of 16 pathways, and PIK3R1was present in 15 pathways. Eleven pathways contained GSK3B. Collectively, these nodes potentially coordinate pathway interactions following lithium and valproate treatment.
Subclusters within the large networks
We used the Community Clustering program GLay73 to extract subclusters from the large networks and distill potential core protein-protein and functional complexes. For lithium, the largest subcluster (150 nodes and 738 edges) contained the seed nodes BCL2 and EZR (Figure 4A). Analysis of the BCL2-EZR interacting nodes in DAVID74 tagged varied pathways that were similarly enriched in the large lithium network. Neurotrophin signaling was the best supported pathway. The most highly enriched functional clusters were regulation of apoptosis, positive regulation of apoptosis and negative regulation of apoptosis (with enrichment scores of 42, 30 and 19, respectively). It is interesting that BCL2 and EZR node complexes reproduced functions expressed by the primary lithium network, suggesting that BCL2 and EZR are core subclusters.
Figure 4.


Figure 4A Subclusters extracted from the large lithium network. Smaller complexes were generated by using the Community Clustering algorithm, GLay. Seed molecules included in each cluster are in yellow and subclusters are color coded.
Figure 4B Subclusters extracted from the large valproate network. Smaller complexes were generated by using the Community Clustering algorithm, GLay. Seed molecules included in each cluster are in yellow and subclusters are color coded.
From the large valproate network, GLay73 extracted 12 subclusters, 11 of which contained at least one seed molecule (Figure 4B). Three core subclusters recapitulated the functional characteristics of the large network. The largest (355 nodes and 2135 edges) contained 13 seed molecules that included AKT1 and BCL2. This subcluster exposed neurotrophin signaling as the most enriched pathway, which together with other enriched pathways largely matched those highly represented in the large valproate network. Programmed cell death and regulation of cell death and apoptosis were the top functional clusters, with enrichment scores of >50.
The second largest subcluster (251 nodes, 1036 edges) that was formed by 13 seed nodes including HDAC1 (Figure 4B) featured the cell cycle as the most highly supported pathway. Transcription regulation and nuclear lumen were the most enriched functional clusters, indicating that this complex contained those particular nodes that constitute the highlighted pathways and functional clusters in the large valproate network. The third subcluster (187 nodes, 2222 edges) contained GSK3B and 4 other seed molecules. Nodes in this subcluster showed enrichment of the cell cycle, nucleotide excision repair, DNA replication, mismatch repair, homologous recombination and Wnt signaling, reminiscent of the valproate-associated pathways in the large network. In addition, the nucleoplasm and nuclear lumen showed a large enrichment score of 72.
In sum, subcluster analysis reduced the complexity of the interaction networks into cores of condensed interacting partners that separately recruited pathways and functional clusters associated with the primary networks. Furthermore, constricted sets of drug-regulated targets that convey defined functions were revealed.
Discussion
Interaction networks of well-selected molecular targets of lithium and valproate, revealed some striking similarities and clear differences in the biological processes perturbed by these mood stabilizers. Valproate but not lithium networks were highly enriched for nodes associated with the nuclear lumen functional cluster, cell cycle, nucleotide excision repair and DNA replication, representing a major distinction in the cellular effects of these medications. Clearly, the wide array of edges formed by nuclear proteins (including the cyclins, cell division proteins, excision repair proteins, HDACs and transcription factors) suggests that valproate triggers an extensive perturbation of functions of the nucleus. It is not apparent however whether these protein clusters and pathways are the main determinants of valproate’s effect on mania.
We explored the coincident mechanisms involved in lithium and valproate actions to illuminate a molecular repertoire that might be paramount in stabilizing mood. Remarkably, the lithium-valproate intersecting nodes largely reproduced the enriched functional clusters and canonical pathways associated with lithium. The striking enrichment of apoptosis functional clusters, neurotrophin signaling and a range of signaling pathways points to potential core mechanisms involved in the therapeutic effect of these drugs. The interaction networks thus revealed a shared functional signature for lithium and valproate.
Whether lithium or valproate acts to restore the normal kinetics of programmed cell death in brains of bipolar disorder patients is not known. Nevertheless, these medications seem to provoke a yin-yang effect on apoptosis. Molecules differentially regulated by both drugs including AKT1, BCL2 and BDNF and many recruited nodes are known to exert anti-apoptotic properties (Supplementary Figure 1). In contrast, various pro-apoptotic nodes, including caspases and cyclin-dependent kinase inhibitors are also well represented in the networks (Supplementary Figure 1). Conceivably, lithium and valproate help fine-tune the apoptotic switch, but the precise timing of this switch in specific cells during the course of treatment remains to be determined.
Neurotrophin signaling was the most enriched pathway identified by identical nodes in lithium and valproate networks. The neurotrophin-signaling cascade transduces signals from BDNF and other neurotrophic factors leading to neuronal cell growth, proliferation and survival. Neurotrophin signaling interacts with downstream pathways including MAPK, which in turn, maintains crosstalk with other significantly enriched pathways, including Wnt, apoptosis, ErbB, and insulin (KEGG Pathways). Stimulation MAPK pathway in rat hippocampus and frontal cortex by mood stabilizers has been reported.31 Regulation of neurotrophin signaling, its interactions with other pathways, and regulation of apoptosis may be the basis for the observed neurotrophic and neuroprotective effects of lithium and valproate.
It is noteworthy that some of the most prominent mood stabilizer biological pathway responses we found in this study are consistent with the major pathway categories recruited by differentially expressed genes in a transcriptome analysis of pairs of bipolar I disorder and control postmortem dorsolateral prefrontal cortex (BA46), that included cellular growth and proliferation, nervous system development and function, and cell death.75
Uncovering key molecules that mediate pathway interactions helps simplify an otherwise overwhelmingly complex molecular response to drug treatment. To isolate these factors we ascertained nodes that recurred frequently across diverse enriched pathways. AKT1/2 and MAPK1/3 were the most preponderant, exposing potential pleiotropy and a major role in coordinating cellular response following lithium and valproate treatment. However the spatial and temporal regulation of the balance between repression and activation of diverse pathways has yet to be established.
AKT1 phosphorylates multiple substrates involved in various cellular processes including regulation of neurogenesis, neuroprotection, cell growth, cell proliferation, cell survival and anti-apoptotic processes.76–78 AKT1 is a vital component of the PI3K growth pathway and its inhibitory effect on GSK3B leads to the activation of Wnt signaling.
Animal models for AKT1, MAPK1 (ERK2) and MAPK3 (ERK1) have exhibited characteristics relevant to brain function. Akt1 knockout mice treated with dopaminergic, adrenergic and cholinergic agents displayed altered working memory.79 Homozygous deletion of MAPK1 is embryonic lethal in mice but mice in which MAPK1 expression was knocked down to ≤ 40% exhibited long-term memory deficits.80 MAPK3 null mice displayed behavioral changes such as hyperactivity, region-specific altered synaptic function and increased brain expression of MAPK1.81 It would be interesting to determine whether these animal models recover wild-type behavior and characteristics following either lithium or valproate treatment.
The lingering question of whether molecular targets for mood stabilizers hold any relevance to the genetic etiology of psychiatric disorders remains to be addressed comprehensively.82–86 For lithium, the newly formed international Consortium on Lithium Genetics (ConLiGen) is assembling and conducting genome-wide association analysis on the largest sample scored for lithium response.87 This study would allow us to determine whether variation in node constituents of enriched pathways and functional clusters is associated with lithium response. Could any combination of node variation help predict response and/or clinical outcome? If so, could nodes in the implicated pathway (s) be used as targets to develop new, effective and faster-acting mood stabilizers with lower toxicity?
Association with gene variants disclosed in small sample sizes must be assessed in the context of recent genome-wide association studies in large schizophrenia and bipolar disorder samples that detected significantly associated genetic variations.88–90 The proposed association of AKT1 variants with schizophrenia in a relatively small sample has not been consistently supported.91 A much weaker association with AKT1 variants has been reported in bipolar disorder and selected phenotypes.92,93 Recently, AKT1 activation has been shown to provoke defects in neuronal development that parallels the effect of DISC1 suppression in newborn neurons.94 In turn, GSK3B has been shown to interact with DISC1, suggesting a possible mechanism by which DISC1 promotes proliferation of adult neural progenitor cells.95 Compelling support for DISC1 as a schizophrenia risk gene has accumulated96 since it was found to be disrupted in an extended Scottish pedigree affected with psychiatric disorders including schizophrenia and bipolar disorder.97–99
ErbB signaling, another highly represented pathway in this study, mediates the action of neuregulin1 (NRG1), a proposed schizophrenia gene.100 Also, modest support for association of NRG1 variants with psychotic bipolar disorder has been presented.101 Interestingly, AKT1 and GSK3B are downstream effectors of the ErbB signaling cascade that maintains crosstalk with cell cycle progression, MAPK, mTOR and calcium signaling pathways (KEGG pathways).
As we have indicated we assembled genes that show drug-induced differential expression in diverse studies. Clearly, changes in gene expression would have been influenced by dissimilar experimental designs and conditions across studies performed on diverse organisms and cell lines from various origins. Despite these confounding factors, a limited number of differentially expressed genes survived our selection criteria, which thus provide some validity for the inclusion of those genes in the network seeds.
Finally, we need to point out that the extensive network interactions presented here could include false positives. False negatives are also possible because of the paucity of published studies on some differentially regulated molecules, e.g. IMPA1, the lack of functional data on many of the network nodes, and the lack of supporting experimental data for potential drug targets thus were not included in the network seeds. Understudied nodes would fail to establish substantial connections within the networks and/or would not be included in established canonical pathways. Conversely, the comparatively extensive literature on certain genes/proteins may bias the representation of their interactions within the network. Therefore these expansive, multi-directional matrices may actually represent a constrained view of lithium and valproate actions in the cell. Future advances in functional genomics coupled with improvements in network building tools allowing more precise modeling of reactions and interactions within the cell would permit creation of more robust networks.
In summary, biological interaction networks generated in this study offer a glimpse of the molecular repertoire that underlies the global cellular effect of lithium and valproate. Consistent with their divergent chemical structures, valproate but not lithium induced a highly enhanced recruitment of nuclear lumen functional clusters and nodes enriched for the cell cycle, nucleotide excision repair and DNA replication pathways. Conversely, lithium and valproate intersecting network nodes perturbed convergent cellular functions embodied by a striking enrichment of the regulation of apoptosis clusters, neurotrophin signaling and a series of diverse signaling pathways. This shared effect hints at a unique functional footprint for lithium and valproate that may be salient to their effectiveness as mood stabilizers. The emergence of recurrent pathway constituents implies functional pleiotropy, an idea that may harmonize the complexity of intracellular responses to lithium and valproate. AKT and MAPK seem to be central to the execution of these responses, but a more complete picture will require additional experimental work.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health.
Footnotes
Conflict of interest
The authors declare no competing financial interest in relation to the work described in this manuscript.
References
- 1.Cade JFJ. Lithium salts in the treatment of psychotic excitement. Med J Australia. 1949;2:349–352. doi: 10.1080/j.1440-1614.1999.06241.x. [DOI] [PubMed] [Google Scholar]
- 2.Belmaker RH. Bipolar disorder. N Engl J Med. 2004;351:476–486. doi: 10.1056/NEJMra035354. [DOI] [PubMed] [Google Scholar]
- 3.Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 1989;59:411–419. doi: 10.1016/0092-8674(89)90026-3. [DOI] [PubMed] [Google Scholar]
- 4.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J Biol Chem. 2008;283:350–357. doi: 10.1074/jbc.M705028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poustka AJ, Kühn A, Groth D, Weise V, Yaguchi S, Burke RD, et al. A global view of gene expression in lithium and zinc treated sea urchin embryos: new components of gene regulatory networks. Genome Biol. 2007;8:R85.1–R85.18. doi: 10.1186/gb-2007-8-5-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bro C, Regenberg B, Lagniel G, Labarre J, Montero-Lomelí M, Nielsen J. Transcriptional, proteomic, and metabolic responses to lithium in galactose-grown yeast cells. J Biol Chem. 2003;278:32141–32149. doi: 10.1074/jbc.M304478200. [DOI] [PubMed] [Google Scholar]
- 8.Manji HK, Moore GJ, Chen G. Bipolar disorder: leads from the molecular and cellular mechanisms of action of mood stabilizers. Br J Psychiatry Suppl. 2001;41:s107–s119. [PubMed] [Google Scholar]
- 9.Chuang D-M, Chen R-W, Chalecka-Franaszek E, Ren M, Hashimoto R, Senatorov V, Kanai H, et al. Neuroprotective effects of lithium in cultured cells and animal models of diseases. Bipolar Disord. 2002;4:129–136. doi: 10.1034/j.1399-5618.2002.01179.x. [DOI] [PubMed] [Google Scholar]
- 10.Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 12.Hao Y, Creson T, Zhang L, Li P, Du F, Yuan P, Gould TD, et al. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J Neurosci. 2004;24:6590–6599. doi: 10.1523/JNEUROSCI.5747-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leng Y, Liang M-H, Ren M, Marinova Z, Leeds P, Chuang D-M. Synergistic neuroprotective effects of lithium and valproic acid or histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci. 2008;28:2576–2588. doi: 10.1523/JNEUROSCI.5467-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Brien TW, Klein PS. Validating GSK3 as an in vivo target of lithium action. Biochem Soc Trans. 2009;37:1133–1138. doi: 10.1042/BST0371133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendes CT, Mury FB, Mury, de Sa′ Moreira E, Alberto FL, Forlenza OV, et al. Lithium reduces Gsk3b mRNA levels: implications for Alzheimer Disease. Eur Arch Psychiatry Clin Neurosci. 2009;259:16–22. doi: 10.1007/s00406-008-0828-5. [DOI] [PubMed] [Google Scholar]
- 16.Hallcher LM, Sherman WR. The effects of lithium ion and other agents on the activity of myo-inositol-1-phosphatase from bovine brain. J Biol Chem. 1980;255:10896–10901. [PubMed] [Google Scholar]
- 17.Parthasarathy Latha K, Seelan Ratnam S, Wilson Mark A, Parthasarathy Ranga N. Regional changes in rat brain inositol monophosphatase I (IMPase 1) activity with chronic lithium treatment. Prog Neuro-Psychophamacol Biol Psychiatry. 2002;27:55–60. doi: 10.1016/s0278-5846(02)00315-9. [DOI] [PubMed] [Google Scholar]
- 18.Shamir A, Shaltiel G, Greenberg ML, Belmaker RH, Agam G. The effect of lithium on expression of genes for inositol biosynthetic enzymes in mouse hippocampus; a comparison with the yeast model. Brain Res Mol Brain Res. 2003;115:104–110. doi: 10.1016/s0169-328x(03)00120-7. [DOI] [PubMed] [Google Scholar]
- 19.Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasaki T, Han F, Shioda N, Moriguchi S, Kasahara J, Ishiguro K, et al. Lithium-induced activation of Akt and CaM kinase II contributes to its neuroprotective action in a rat microsphere embolism model. Brain Res. 2006;1108:98–106. doi: 10.1016/j.brainres.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Sinha D, Wang Z, Ruchalski KL, Levine JS, Krishnan S, Lieberthal W, et al. Lithium activates the Wnt and phosphatidylinositol 3-kinase Akt signaling pathways to promote cell survival in the absence of soluble survival factors. Am J Physiol Renal Physiol. 2005;288:F703–F713. doi: 10.1152/ajprenal.00189.2004. [DOI] [PubMed] [Google Scholar]
- 22.Erdal E, Ozturk N, Cagatay T, Emel Eksioglu-Demiralp E, Ozturk M. Lithium-mediated downregulation of PKB/Akt and cyclin E with growth inhibition in hepatocellular carcinoma cells. Int J Cancer. 2005;115:903–910. doi: 10.1002/ijc.20972. [DOI] [PubMed] [Google Scholar]
- 23.Nemoto T, Kanai T, Yanagita T, Satoh S, Maruta T, Yoshikawa N, et al. Regulation of Akt mRNA and protein levels by glycogen synthase kinase-3β in adrenal chromaffin cells: Effects of LiCl and SB216763. Eur J Pharmacol. 2008;586:82–89. doi: 10.1016/j.ejphar.2008.02.075. [DOI] [PubMed] [Google Scholar]
- 24.Adams JM, Cory S. The bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 25.Chen R-W, Chuang D-M. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. J Biol Chem. 1999;274:6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- 26.Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH, et al. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem. 1999;72:879–882. doi: 10.1046/j.1471-4159.1999.720879.x. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Wu D-Y, Chen G, Manji H, Chen DF. Support of retinal ganglion cell survival and axon regeneration by lithium through a Bcl-2–dependent mechanism. Invest Ophthalmol Vis Sci. 2003;44:347–354. doi: 10.1167/iovs.02-0198. [DOI] [PubMed] [Google Scholar]
- 28.Bush AL, Hyson RL. Lithium increases Bcl-2 expression in chick cochlear nucleus and protects against deafferentation-induced cell death. Neuroscience. 2006;138:1341–1349. doi: 10.1016/j.neuroscience.2005.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology. 2001;158:100–106. doi: 10.1007/s002130100871. [DOI] [PubMed] [Google Scholar]
- 30.Omata N, Murata T, Takamatsu S, Maruoka N, Mitsuya H, Yonekura Y, et al. Neuroprotective effect of chronic lithium treatment against hypoxia in specific brain regions with upregulation of cAMP response element binding protein and brain-derived neurotrophic factor but not nerve growth factor: comparison with acute lithium treatment. Bipolar Disord. 2008;10:360–368. doi: 10.1111/j.1399-5618.2007.00521.x. [DOI] [PubMed] [Google Scholar]
- 31.Einat H, Yuan P, Gould TD, Li J, Du JH, Zhang L, et al. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23:7311–7316. doi: 10.1523/JNEUROSCI.23-19-07311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51–59. doi: 10.1038/sj.mp.4002099. [DOI] [PubMed] [Google Scholar]
- 33.Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang D-M. Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: An essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology. 2002;43:1173–1179. doi: 10.1016/s0028-3908(02)00217-4. [DOI] [PubMed] [Google Scholar]
- 34.Angelucci F, Aloe L, Jiménez-Vasquez P, Mathé AA. Lithium treatment alters brain concentrations of nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in a rat model of depression. Int J Neuropsychopharmacol. 2003;6:225–231. doi: 10.1017/S1461145703003468. [DOI] [PubMed] [Google Scholar]
- 35.Bosetti F, Seemann R, Bell JM, Zahorchak R, Friedman E, Rapoport SI, et al. Analysis of gene expression with cDNA microarrays in rat brain after 7 and 42 days of oral lithium administration. Brain Res Bull. 2002;57:205–209. doi: 10.1016/s0361-9230(01)00744-4. [DOI] [PubMed] [Google Scholar]
- 36.Fatemi SH, Reutiman TJ, Folsom TD. The role of lithium in modulation of brain genes: relevance for aetiology and treatment of bipolar disorder. Biochem Soc Trans. 2009;37:1090–1095. doi: 10.1042/BST0371090. [DOI] [PubMed] [Google Scholar]
- 37.Brandish PE, Su M, Holder DJ, Hodor P, Szumiloski J, Kleinhanz RR, et al. Regulation of gene expression by lithium and depletion of inositol in slices of adult rat cortex. Neuron. 2005;45:861–872. doi: 10.1016/j.neuron.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 38.Zhang WV, Jüllig M, Connolly AR, Stott NS. Early gene response in lithium chloride induced apoptosis. Apoptosis. 2005;10:75–90. doi: 10.1007/s10495-005-6063-x. [DOI] [PubMed] [Google Scholar]
- 39.Zellmer S, Schmidt-Heck W, Gaunitz F, Baldysiak-Figie A, Guthke R, Gebhardt R. Dynamic network reconstruction from gene expression data describing the effect of LiCl stimulation on hepatocytes. J Integr Bioinform. 2005;2:11–26. [Google Scholar]
- 40.Seelan RS, Khalyfa A, Lakshmanan J, Casanova MF, Parthasarathy RN. Deciphering the lithium transcriptome: microarray profiling of lithium-modulated gene expression in human neuronal cells. Neuroscience. 2008;151:1184–1197. doi: 10.1016/j.neuroscience.2007.10.045. [DOI] [PubMed] [Google Scholar]
- 41.McQuillin A, Rizig M, Gurling HM. A microarray gene expression study of the molecular pharmacology of lithium carbonate on mouse brain mRNA to understand the neurobiology of mood stabilization and treatment of bipolar affective disorder. Pharmacogenet Genomics. 2007;17:605–617. doi: 10.1097/FPC.0b013e328011b5b2. [DOI] [PubMed] [Google Scholar]
- 42.Chetcuti A, Adams LJ, Mitchell PB, Schofield PR. Microarray gene expression profiling of mouse brain mRNA in a model of lithium treatment. Psychiatr Genet. 2008;18:64–72. doi: 10.1097/YPG.0b013e3282fb0051. [DOI] [PubMed] [Google Scholar]
- 43.Corena-McLeod MdP, Oliveros A, Charlesworth C, Madden B, Liang YQ, Boules M, et al. Paliperidone as a mood stabilizer: a pre-frontal cortex synaptoneurosomal proteomics comparison with lithium and valproic acid after chronic treatment reveals similarities in protein expression. Brain Res. 2008;1233:8–19. doi: 10.1016/j.brainres.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci USA. 2008;105:3634–3639. doi: 10.1073/pnas.0800001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodriguez ML, Yubero P, Iglesias R, Giralt M, Villarroya F. Lithium inhibits brown adipocyte differentiation. FEBS Letters. 2005;579:1670–1674. doi: 10.1016/j.febslet.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 46.Cordeiro ML, Umbach JA, Gundersen CB. Lithium ions up-regulate mRNAs encoding dense-core vesicle proteins in nerve growth factor-differentiated PC12 cells. J Neurochem. 2000;75:2622–2625. doi: 10.1046/j.1471-4159.2000.0752622.x. [DOI] [PubMed] [Google Scholar]
- 47.Wood JR, Nelson-Degrave VL, Jansen E, McAllister JM, Mosselman S, Strauss JF., III Valproate-induced alterations in human theca cell gene expression: clues to the association between valproate use and metabolic side effects. Physiol Genomics. 2005;20:233–243. doi: 10.1152/physiolgenomics.00193.2004. [DOI] [PubMed] [Google Scholar]
- 48.Chen J, Ghazawi FM, Bakkar W, Li Q. Valproic acid and butyrate induce apoptosis in human cancer cells through inhibition of gene expression of Akt/protein kinase B. Molecular Cancer. 2006;5:71. doi: 10.1186/1476-4598-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Creson TK, Yuan P, Manji HK, Chen G. Evidence for the involvement of ERK, PI3K, and RSK in induction of Bcl-2 by valproate. J Mol Neurosci. 2009;37:123–134. doi: 10.1007/s12031-008-9122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukuchi M, Nii T, Ishimaru N, Minamino A, Hara D, Takasaki I, Tabuchi A, Tsuda M. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci Res. 2009;65:35–43. doi: 10.1016/j.neures.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 51.Gurpur PV, Liu J, Burkin DJ, Kaufman SJ. Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am J Pathol. 2009;174:999–1008. doi: 10.2353/ajpath.2009.080537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen WT, Wong TS, Chung W-Y, Wong MG, Kebebew E, Duh Q-Y, Clark OH. Valproic acid inhibits growth, induces apoptosis, and modulates apoptosis-regulatory and differentiation gene expression in human thyroid cancer cells. Surgery. 2005;138:979–985. doi: 10.1016/j.surg.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 53.Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang C-C, et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase-3β phosphorylation by sodium valproate and lithium. Neuropharmacol. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 55.Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J Cell Sci. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- 56.Jurata LW, Bukhman YV, Charles V, Capriglione F, Bullard J, Lenire AL, et al. Comparison of microarray-based mRNA profiling technologies for identification of psychiatric disease and drug signatures. J Neurosci Methods. 2004;138:173–188. doi: 10.1016/j.jneumeth.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 57.Milutinovic S, D’Alessio AC, Detich N, Szyf M. Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis. 2007;28:560–571. doi: 10.1093/carcin/bgl167. [DOI] [PubMed] [Google Scholar]
- 58.Jergil M, Kultima K, Gustafson A-L, Dencker L, Stigson M. Valproic acid-induced deregulation in vitro of genes associated with neural tube defects. Toxicol Sci. 2009;108:132–148. doi: 10.1093/toxsci/kfp002. [DOI] [PubMed] [Google Scholar]
- 59.Stamatopoulos B, Meuleman N, De Bruyn, Mineur P, Martiat P, Bron D, Lagneaux L. Antileukemic activity of valproic acid in chronic lymphocytic leukemia B cells defined by microarray analysis. Leukemia. 2009;23:2281–2289. doi: 10.1038/leu.2009.176. [DOI] [PubMed] [Google Scholar]
- 60.Sugawara H, Iwamoto K, Bundo M, Ishiwata M, Ueda J, Kakiuchi C, et al. Effect of mood stabilizers on gene expression in lymphoblastoid cells. J Neural Transm. 2010;117:155–164. doi: 10.1007/s00702-009-0340-8. [DOI] [PubMed] [Google Scholar]
- 61.Kultima K, Nyström A-M, Scholz B, Gustafson A-L, Dencker L, Stigson M. Valproic acid teratogenicity: a toxicogenomics approach. Environ Health Persp. 2004;112:1225–1234. doi: 10.1289/txg.7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kultima K, Jergil M, Salter H, Gustafson A-L, Dencker L, Stigson M. Early transcriptional responses in mouse embryos as a basis for selection of molecular markers predictive of valproic acid teratogenecity. Reprod Toxicol. 2010;30:457–468. doi: 10.1016/j.reprotox.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 63.Ogden CA, Rich ME, Schork NJ, Paulus MP, Geyer MA, Lohr JB, et al. Candidate genes, pathways and mechanisms for bipolar disorder (manic-depressive) and related disorders: an expanded convergent functional genomics approach. Mol Psychiatry. 2004;9:1007–1029. doi: 10.1038/sj.mp.4001547. [DOI] [PubMed] [Google Scholar]
- 64.Bosetti F, Bell KM, Manickam P. Microarray analysis of rat brain gene expression after chronic administration of sodium valproate. Brain Res Bull. 2005;65:331–338. doi: 10.1016/j.brainresbull.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 65.Chetcuti A, Adams LJ, Mitchell PB, Schofield PR. Altered gene expression in mice treated with the mood stabilizer sodium valproate. Int J Neuropsychopharmacol. 2005;9:267–276. doi: 10.1017/S1461145705005717. [DOI] [PubMed] [Google Scholar]
- 66.Wang J-F, Bown C, Young LT. Differential display PCR reveals novel targets for the mood-stabilizing drug valproate including the molecular chaperone GRP78. Mol Pharmacol. 1999;55:521–527. [PubMed] [Google Scholar]
- 67.Baumeister P, Dong D, Fu Y, Lee AS. Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor-induced apoptosis. Mol Cancer Ther. 2009;8:1086–1094. doi: 10.1158/1535-7163.MCT-08-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venkataramani V, Rossner C, Iffland L, Schweyer S, Tamboli IY, Walter J, et al. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the Alzheimer amyloid precursor protein. J Biol Chem. 2010;285:10678–10689. doi: 10.1074/jbc.M109.057836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watson DG, Watterson JM, Lenox RH. Sodium valproate down-regulates the myristoylated alanine-rich C kinase substrate (MARCKS) in immortalized hippocampal cells: a property of protein kinase C-mediated mood stabilizers. J Pharmacol Exptl Therapeutics. 1998;285:307–316. [PubMed] [Google Scholar]
- 70.Jayapandian M, Chapman A, Tarcea VG, Yu C, Elkiss A, Ianni A, et al. Michigan Molecular Interactions (MiMI): putting the jigsaw puzzle together. Nucl Acids Res. 2007;35:D566–571. doi: 10.1093/nar/gkl859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao J, Ade AS, Tarcea VG, Weymouth TE, Mirel BR, Jagadish HV, States DJ. Integrating and Annotating the Interactome using the MiMI plugin for Cytoscape. Bioinformatics. 2009;25:137–138. doi: 10.1093/bioinformatics/btn501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferrell JE., Jr Q&A: Systems biology. J Biol. 2009;8:2. doi: 10.1186/jbiol107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Su G, Kuchinsky A, Morris JH, States DJ, Meng F. GLay: community structure analysis of biological networks. Bioinformatics. 2010;26:3135–3137. doi: 10.1093/bioinformatics/btq596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 75.Nakatani N, Hattori E, Ohnishi T, Dean B, Iwayama Y, Matsumoto I, et al. Genome-wide expression analysis detects eight genes with robust alterations specific to bipolar I disorder: relevance to neuronal network perturbation. Hum Mol Genet. 2006;15:1949–1962. doi: 10.1093/hmg/ddl118. [DOI] [PubMed] [Google Scholar]
- 76.Gonzalez E, McGraw TE. The Akt kinases. Cell Cycle. 2009;8:2502–2508. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee HJ, Kim MK, Hee J, Kim HJ, Kim SU. Human neural stem cells genetically modified to overexpress Akt1 provide neuroprotection and functional improvement in mouse stroke model. PLoS ONE. 2009;4:e5586. doi: 10.1371/journal.pone.0005586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beaulieu J-M, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- 79.Lai W-S, Xu B, Westphal KGC, Paterlini M, Olivier B, Pavlidis, et al. Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc Natl Acad Sci USA. 2006;103:16906–16911. doi: 10.1073/pnas.0604994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, et al. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, et al. Knockout of ERK1/MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 82.Detera-Wadleigh SD. Lithium-related genetics of bipolar disorder. Ann Med. 2001;33:272–285. doi: 10.3109/07853890108998756. [DOI] [PubMed] [Google Scholar]
- 83.Alda M, Grof P, Rouleau GA, Turecki G, Young LT. Investigating responders to lithium prophylaxis as a strategy for mapping susceptibility genes for bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1038–45. doi: 10.1016/j.pnpbp.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 84.Cruceanu C, Alda M, Turecki G. Lithium: a key to the genetics of bipolar disorder. Genome Medicine. 2009;1:79. doi: 10.1186/gm79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Detera-Wadleigh SD, Yoshikawa T. Decoding the genetics and underlying mechanisms of mood disorders. Molecular Biology of Psychiatric Disorders. In: Wildernauer D, Gross HJ, editors. Nucleic Acids and Molecular Biology. Vol. 23. Springer-Verlag; Berlin Heidleberg: 2009. pp. 1–50. [Google Scholar]
- 86.Perlis RH, Smoller JW, Ferreira MA, McQuillin A, Bass N, Lawrence J, et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry. 2009;166:718–725. doi: 10.1176/appi.ajp.2009.08111633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schulze TG, Alda M, Adli M, Akula N, Ardau R, Chillotti C, et al. The international consortium on lithium genetics (ConLigen)–an initiative by the NIMH and IGSLI to study the genetic basis of response to lithium treatment. Neuropsychobiology. 2010;62:72–78. doi: 10.1159/000314708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ferreira MAR, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McMahon FJ, Akula N, Schulze TG, Muglia P, Tozzi F, Detera-Wadleigh SD, et al. Meta-analysis of genome-wide association data identifies a risk locus for major mood disorders on 3p21.1. Nat Genet. 2010;42:128–131. doi: 10.1038/ng.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 92.Toyota T, Yamada K, Detera-Wadleigh SD, Yoshikawa T. Analysis of a cluster of polymorphisms in AKT1 gene in bipolar pedigrees: a family-based association study. Neuroscience Lett. 2003;339:5–8. doi: 10.1016/s0304-3940(02)01428-3. [DOI] [PubMed] [Google Scholar]
- 93.Pietiläinen OP, Paunio T, Loukola A, Tuulio-Henriksson A, Kieseppä T, Thompson P, et al. Association of AKT1 with verbal learning, verbal memory, and regional cortical gray matter density in twins. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:683–692. doi: 10.1002/ajmg.b.30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim JY, Duan X, Liu CY, Jang MH, Guo JU, Pow-anpongku N, et al. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron. 2009;63:761–773. doi: 10.1016/j.neuron.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brandon NJ, Millar JK, Korth C, Sive H, Singh KK, Sawa A. Understanding the role of DISC1 in psychiatric disease and during normal development. J Neurosci. 2009;29:12768, 12775. doi: 10.1523/JNEUROSCI.3355-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- 98.Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- 99.Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders-cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goes FS, Willour VL, Zandi PP, Belmonte PL, MacKinnon DF, Mondimore FM, et al. Family-based association study of Neuregulin 1 with psychotic bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:693–702. doi: 10.1002/ajmg.b.30895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.