

Abstract
Background
There are limited data regarding the differences in clinical presentation and outcome of liposarcomas between adult and pediatric patients. The role of adjuvant radiotherapy in the treatment of childhood liposarcoma is unclear.
Procedure
A multi-institutional retrospective analysis of medical records was performed for patients ≤ 21 years of age presenting with a verified histologic diagnosis of liposarcoma.
Results
Thirty-three patients were evaluable for this study, 23 of whom were male. Median age was 17.2 years. Twenty-four cases were myxoid subtype and 7 were pleomorphic subtype. In myxoid cases, 17 (71%) presented with extremity tumors; none had metastases. Eleven of these patients with myxoid subtype were treated with surgery only, 7 with surgery + radiation, 3 with surgery + radiation + chemotherapy. Median radiation therapy dose for patients with myxoid tumors was 60 Gray. At median follow up of 4.2 years (range 0.1–32.2 years), 2 patients relapsed with 1 death from progressive disease. In 7 pleomorphic cases, 4 patients had primary tumors at central axial sites. Six patients (86%) received multimodal therapy, but 6 patients experienced relapse of disease. Four patients died from progressive disease.
Conclusions
Pediatric liposarcoma has a different spectrum of presentation compared to adult cases. Myxoid liposarcoma is the more common subtype, usually occurs in extremities, and has an excellent prognosis. Pleomorphic liposarcoma occurs in axial sites, and despite multimodal therapy, outcome is poor. Further study is needed to identify the optimal therapy for pediatric liposarcoma.
Keywords: Liposarcoma, sarcoma, pediatric, childhood cancer, myxoid
Introduction
Liposarcomas are tumors derived from primitive mesenchymal cells that undergo adipose differentiation and constitute approximately 10% of all soft tissue sarcomas. Liposarcomas are predominantly a disease of adulthood with peak incidence around the 5th to 6th decade of life and with a slight predominance towards males. The extremities are the most common primary site, accounting for about 40% of cases. Nearly one-third of cases arise in visceral spaces, and there is an association between site of primary tumor and histologic subtypes.1–3 Although all subtypes are most common in the extremities, a significant proportion of pleomorphic and well-differentiated tumors arise in the retroperitoneum and in other visceral spaces. Histologic grade is one of the most important predictors of outcome, with low-grade myxoid tumors having significantly improved survival rates compared to the round-cell, pleomorphic, and dedifferentiated subtypes.4–7 Complete surgical resection remains the mainstay of local therapy, but adjuvant radiation therapy is effective at controlling microscopic residual disease after surgical resection.5–9 The use of chemotherapy for treatment of liposarcoma is more controversial, but myxoid tumors appear to respond better than dedifferentiated or well-differentiated tumors.10
Liposarcomas are rare in childhood, representing about 2% of all childhood soft tissue sarcomas, with the peak incidence occurring in the second decade of life.11 Because data on pediatric liposarcoma are limited, it is unclear whether the clinical features and outcomes of pediatric patients are similar to those of adults, and the management of liposarcoma in this age group has been largely based on adult data.
The primary goal of this study was to review the combined clinical experience with childhood liposarcoma at the Children’s Cancer Hospital at the University of Texas M. D. Anderson Cancer Center (UTMDACC), Texas Children’s Hospital (TCH), and St. Jude Children’s Research Hospital (SJCRH) in order to determine the extent to which pediatric liposarcoma mimics its adult counterpart and to identify clinical and biological differences that might mandate a different approach to therapy.
Materials and Methods
An Institutional Review Board – approved retrospective medical record review including all patients less than 22 years of age diagnosed with liposarcoma between 1963 to 2005 was performed at UTMDACC, TCH, and SJCRH. Pathologic samples were re-reviewed by an expert pathologist from the patient’s respective institution to verify the diagnosis and histologic subtype. Baseline clinical data collected included age, gender, race, ethnicity, primary tumor site, tumor size (≤ or > 5 cm), and extent of regional and distant metastases. Treatment information gathered included the extent of surgery at baseline and after neoadjuvant therapy, type of chemotherapy administered, and dose and site(s) of radiotherapy delivered. The site(s) of disease recurrence were identified for patients who experienced tumor recurrence. Vital status, cause of death, and disease status at the time of last clinical follow-up were collected.
Statistical Analysis
Descriptive statistics were used to summarize patient characteristics. The Kaplan-Meier product-limit method was used to estimate survival for all patients and to estimate difference in survival based on treatment received. The log-rank test was used to compare survival curves between groups of patients defined by characteristics of interest.
Results
Patient Characterisitics
Thirty-three evaluable patients were identified: UTMDACC (n=24), TCH (n=5) and SJCRH (n=4) (Table I). Median age at diagnosis was 17.6 years (range 11–21.5 years), and 23 patients were male. Fourteen patients were Caucasian, 13 patients were Hispanic/Latino, 3 were black, and 3 were of other ethnicity. Median duration of clinical follow up was 4.2 years (range 0.1–32.2 years).
Table I.
Clinical and demographic characteristics of evaluated patients
| N | % of cohort | |
|---|---|---|
| Gender | ||
| Female | 10 | 30.3 |
| Male | 23 | 69.7 |
| Tumor Histology | ||
| Myxoid | 24 | 72.7 |
| Pleomorphic | 7 | 21.2 |
| Well-differentiated | 2 | 6.1 |
| Primary Site of Tumor | ||
| Upper Extremity | 1 | 3 |
| Lower Extremity | 20 | 60.6 |
| Head/Neck | 2 | 6.1 |
| Thorax | 3 | 9.1 |
| Abdomen/Pelvis | 7 | 21.2 |
| Tumor Size | ||
| ≤ 5 cm | 6 | 18.2 |
| > 5 cm | 16 | 48.5 |
| Unknown | 11 | 33.3 |
| Nodal Involvement | ||
| No | 22 | 66.7 |
| Yes | 2 | 6 |
| Unknown | 9 | 27.3 |
| Metastases | ||
| No | 32 | 97 |
| Yes | 1 | 3 |
Tumor Characteristics
Twenty-four patients had myxoid subtype, 7 had pleomorphic subtype, and 2 had well-differentiated subtype. Three of the pleomorphic cases had histologic features consistent with a pleomorphic myxoid liposarcoma subtype that has been recently described.12 However, these 3 cases were categorized as being pleomorphic subtype when analyzing outcomes. The extremities were the most common primary tumor site with 21 cases total (64%); the retroperitoneum/abdomen/pelvis was the second most common site with 7 cases (21%). Myxoid histology was most common in the extremities (17 of 24; 71%), whereas pleomorphic occurred predominantly in axial sites (4 of 7; 57%).
Size of primary tumor was evaluable for 22 cases. The majority of patients had primary tumors greater than 5 cm at time of diagnosis (16 cases; 73% of evaluable cases). All 4 evaluable patients with pleomorphic subtype and both patients with well-differentiated subtypes had primary tumors greater than 5 cm in size at diagnosis. Metastasis was noted in 1 patient with pleomorphic tumor at initial diagnosis. This patient had a retroperitoneal primary tumor with metastases to the liver and peritoneum. This patient, along with another patient with pleomorphic disease of the left gluteal region, also had locoregional lymph node involvement. None of the myxoid or well-differentiated cases had metastases at diagnosis.
Treatment Approach
Complete surgical resection was the sole treatment modality for 13 patients (39%), with 11 of the cases having myxoid tumors (Table II). The next most common treatment approach was gross resection followed by adjuvant radiation therapy in 8 cases (24%); adjuvant chemotherapy in addition to surgery and radiotherapy was used in 7 cases (21%). Neoadjuvant radiotherapy was administered to only 3 patients, all of whom had myxoid histology and received 50 Gray (Gy); all achieved negative surgical margins at the time of definitive resection.
Table II.
Treatment and outcome of patients with myxoid versus pleomorphic tumors
| Myxoid (n=24) | Pleomorphic (n=7) | P value | |
|---|---|---|---|
| Median age (years) | 17.6 | 17.1 | NS |
| Treatment | |||
| Surgery only | 11 | 0 | P= 0.048 |
| Surgery + XRT | 7 | 1 | |
| Surgery+chemo+XRT | 3 | 4 | |
| Other | 2 | 2 | |
| Median Radiation Dose (Gy) (range) [n] |
60 (50 – 66) [n = 10] |
64.5 (55.8 – 70) [n = 5] |
NS |
| Final Surgical Margin Status | |||
| Negative | 17 | 1 | P=0.021 |
| Positive | 1 | 3 | |
| Unknown | 4 | 2 | |
| Median Duration of Follow-up (years) (range) |
4.5 (0.1–32.2) | 2.3 (0.8 – 8.6) | |
| Number of Patients who Relapsed | 2 | 6 | |
| Deaths from Disease | 1 | 4 |
XRT = radiation therapy; Chemo = chemotherapy; Gy = Gray; NS = not significant
There was a significant association between histologic subtype and treatment modality in that patients with myxoid tumors were more likely to have only surgical treatment (11 of 23; 48%) compared to patients with pleomorphic tumors (0 of 7; 0%) (P=0.048).
There was also an association between surgical margin status and histologic subtype, with negative margins achieved more often in patients with myxoid histology (P=0.021).
A total of 15 patients received radiotherapy, all of whom had tumors > 5 cm in maximal diameter. Among patients receiving radiotherapy, there was no difference in the median dose between those with myxoid and pleomorphic histologies.
Chemotherapy was administered to 11 patients (5 myxoid, 6 pleomorphic). The most common regimen utilized was doxorubicin with an alkylator, most commonly ifosfamide. The median number of chemotherapy courses was six. Tumor response could not be accurately assessed in the 5 patients receiving neoadjuvant therapy due to variability of available records. One patient with pleomorphic liposarcoma of the left gluteal area received both neoadjuvant and adjuvant chemotherapy. The neoadjuvant chemotherapy consisted of 4 courses of cyclophosphamide, doxorubicin, and dacarbazine, but no radiographic response was demonstrated. The patient then had complete surgical removal of tumor followed by 2 courses of ifosfamide therapy. The patient with retroperitoneal pleomorphic liposarcoma with metastases received 6 courses of doxorubicin with ifosfamide as primary therapy without any demonstrable response. Four patients (2 with myxoid tumors and 2 with pleomorphic tumors) received adjuvant therapy for microscopic residual disease, thus response to chemotherapy could not be ascertained. For patients with recurrent disease, there was a wide variety of salvage chemotherapy combinations utilized with no consistent regimen noted.
Outcome
The median duration of follow-up for the cohort was 4.2 years (range 0.1 –32.2 years). Figure 1 shows the Kaplan-Meier survival plot for the entire cohort. The overall probabilities of 5-year and 10-year survival for the entire cohort are 89% [95% confidence interval (CI) 0.68 to 0.96] and 64% (95% CI 0.25 to 0.86) respectively. When comparing survival for patients who received only surgical treatment versus multimodal therapy, there was no statistical significant difference in outcome (P=0.54). However, patients with myxoid or well-differentiated histology fared significantly better than those with pleomorphic histology (5-year OS 100% vs. 54%; P=0.001; Figure 2). Two of 24 patients (8%) in the myxoid group developed a tumor recurrence, both of which were local recurrences of an abdominal primary tumor. The initial treatment for these 2 patients was surgical resection in 1 patient and surgery with adjuvant chemotherapy in the other patient. One was treated with surgery, radiation therapy and chemotherapy but was lost to follow-up with active disease 8 years after diagnosis. The other patient underwent subtotal surgical resection and received radiation therapy but died from disease progression. Both patients with well-differentiated liposarcoma are alive and without evidence of disease.
Figure 1.
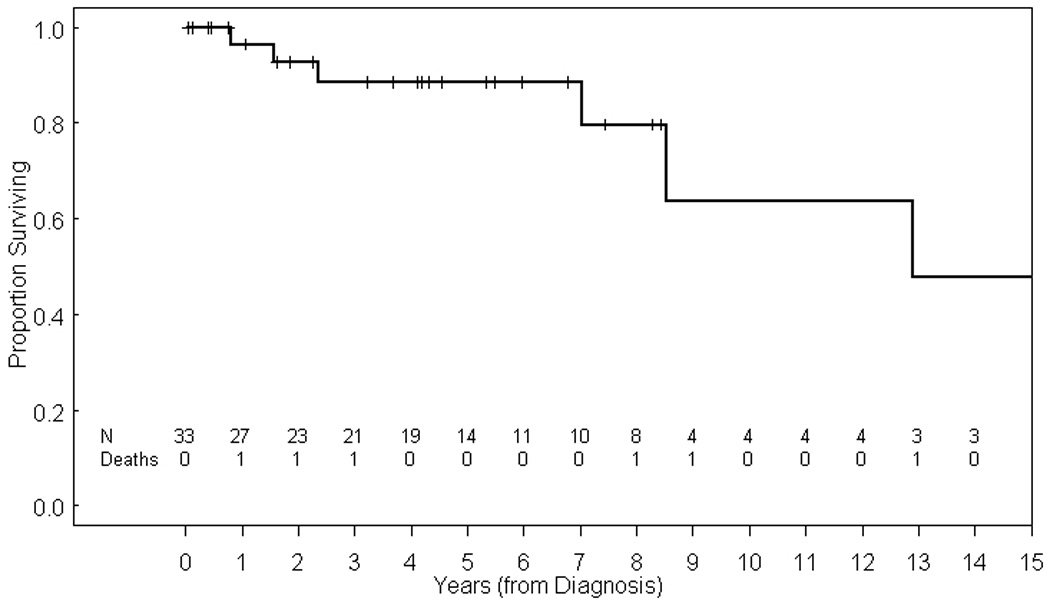
Kaplan-Meier estimate of overall survival (OS) for entire evaluated cohort.
Figure 2.
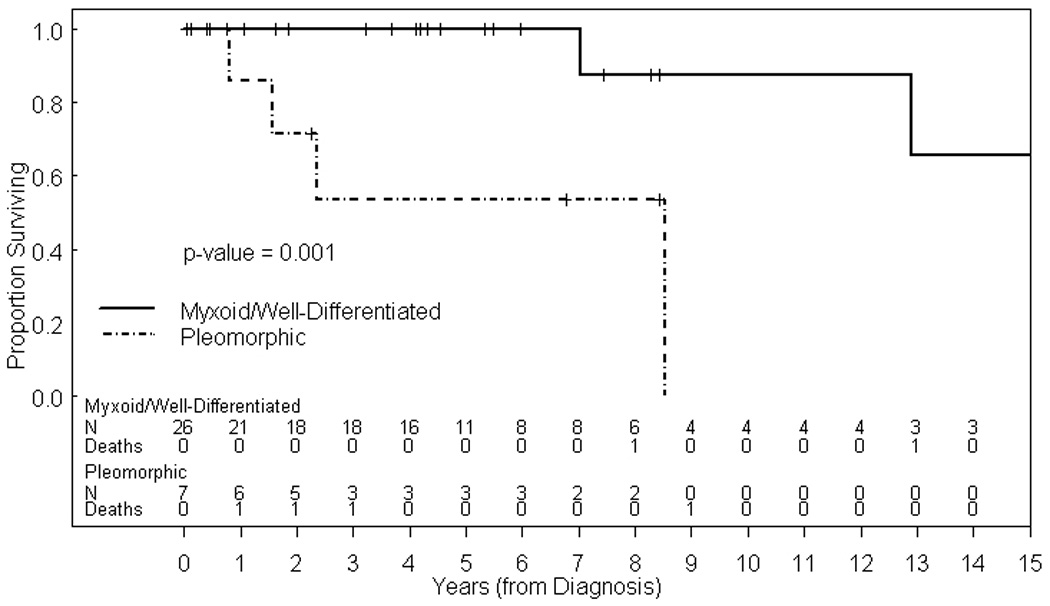
Kaplan-Meier estimate of OS for patients with myxoid and well differentiated liposarcomas versus pleomorphic liposarcomas.
For the pleomorphic subtype, 6 of 7 patients experienced disease recurrence, four of whom had local recurrence. The primary tumor sites for these 4 patients were the head/neck in 2 patients, the retroperitoneum, and the anterior mediastinum. Two of the patients with local recurrence had received radiation therapy following subtotal tumor resection, while final surgical margin was indeterminate in another patient who received adjuvant radiation therapy. Two patients had distant metastatic recurrence in 2 different sites with the lungs being involved for both patients. Four patients died from progressive disease. Two of these patients had multiple local recurrences of a head/neck site and retroperitoneal site respectively, while the other 2 patients had second recurrence of pulmonary metastases.
Discussion
Our analysis suggests that pediatric liposarcoma differs significantly from its adult counterpart. As illustrated in our series, myxoid histology is the most common subtype in children,12–15 whereas in adult patients the well-differentiated or dedifferentiated subtypes are more commonly encountered.4 Similar to other published pediatric single institution studies, our study also found that the lower extremity was the most common primary site,12–15 most have tumors > 5 cm at initial presentation, and metastatic disease at the time of initial diagnosis is uncommon. Our series also confirms the observation that pleomorphic histology has a less favorable prognosis since 6 of our 7 patients experienced tumor recurrence despite aggressive multimodal therapy.
As in other soft tissue sarcomas wide local excision remains the preferred approach for local control of primary disease. In cases where sufficient surgical margins cannot be achieved, the optimal treatment strategy is unclear. There are data that demonstrate that myxoid tumors are radiosensitive, and preoperative, intraoperative, and post-operative radiation approaches have all been effective.5–7, 9, 16 The optimal radiation dose is also unclear. One large study of a primarily adult cohort with myxoid tumors achieved favorable rates of local control using 50 Gy in the neoadjuvant setting and 60 Gy in the adjuvant setting.9 The number of patients in our cohort was too small to yield information about radiation dose selection in pediatric patients. However, we observed no local recurrences in patients with myxoid tumors who received radiation therapy, 3 of whom received 50 Gy in a neoadjuvant approach. Our series also included 4 patients who received radiation therapy despite having undergone a wide local excision at the time of initial diagnosis. This treatment approach may be a reflection of the clinical practice at the time since all 4 patients were treated before 1990. Growing concerns regarding the long-term side-effects of radiation therapy, such as decreased limb growth and functional deficits in joint mobility, and recent evidence from studies in adults suggest that adjuvant therapy is not needed after a wide local excision.17 There are no prospective data yet on the indications for radiotherapy in pediatric patients, but the current Children’s Oncology Group (COG) nonrhabdomyosarcoma soft tissue sarcoma clinical trial is evaluating which patients can be safely managed without adjuvant radiotherapy and is testing a dose of 55.8 Gy for patients requiring adjuvant therapy.
The role of chemotherapy for treatment of pediatric liposarcomas is not well established in pediatric patients. In our series chemotherapy use was primarily limited to myxoid tumors that had microscopic residual disease after surgical intervention and unresectable pleomorphic tumors. Given the adult data that high local control rates can be achieved with adjuvant radiotherapy for microscopic residual liposarcoma, adjuvant chemotherapy appears not to be necessary for facilitation of local tumor control. Chemotherapy does have a role in facilitating tumor resection in patients with unresectable disease, but the optimal therapeutic agents remain unclear. The emerging roles of gemcitabine/docetaxel and trabectedin in adults have been less well investigated in pediatric patients.18–20
Given the recent development of targeted therapies for a variety of malignancies, there is interest in identifying potential targets in liposarcomas. Greater than 90% of myxoid liposarcomas are associated with the t(12;16) (q13;p11) translocation which results in the fusion of the CHOP (DDIT3) and TLS (FUS) genes.3 The CHOP gene is a member of the leucine zipper transcription factor family, but the exact mechanism leading to liposarcoma formation still needs to be elucidated. Also, it is unclear if the presence of the fusion protein offers prognostic value since some studies have not found any correlation between fusion protein status and clinical outcome, while one study indicated that patients with a certain subtype of TLS-CHOP transcript were at increased risk of skeletal metastasis.21–24 Another common finding is amplification of the MDM2 gene in well-differentiated and dedifferentiated liposarcomas.3 However, these histologic subtypes are exceedingly rare in younger patients.
The cytogenetic features of pleomorphic liposarcomas are less consistent. Amplifications of MDM2 and alterations of TP53 have been found in approximately 30–40% of patient tumor samples, but these studies were limited by the small number of samples.25, 26 In limited studies the TLS-CHOP fusion has not been detected in pleomorphic liposarcomas.27 Despite these limited findings, pleomorphic liposarcomas may still be a candidate for therapies that utilize MDM2 antagonists, such as Nutlin-3A.28
Limitations of this study include its retrospective nature pooled from 3 different institutions. While this study represents the largest clinical series of liposarcoma in young patients, the cohort size is still too small to perform more detailed statistical analysis, especially in the pleomorphic subgroup. Collaborative group studies, such as the current COG nonrhabdomyosarcoma soft tissue sarcoma study, may in the future provide a larger cohort for multivariate analysis of potential factors, such as primary site of tumor, histology, and extent of surgical resection.
In conclusion, myxoid liposarcoma is the most common histologic subtype encountered in children and young adults, and the overall prognosis for myxoid tumors is excellent, generally with surgical treatment alone. Despite aggressive multimodal therapy, our data suggest that the pleomorphic subtype portends a poorer prognosis, but further study is needed in a larger cohort to validate this observation and to develop better therapies for this subtype. Since liposarcoma is uncommon in childhood and may differ from the same disease in adults, pediatric patients with liposarcoma should be encouraged to enroll on clinical trials to identify the optimal therapeutic approach.
Acknowledgments
This work was supported in part by the Cancer Center Support Grant (NCI Grant P30 CA016672 and CA21765) and the American Lebanese Syrian Associated Charities (ALSAC).
The authors would like to acknowledge Eric Chang, MD, for his assistance in preparing this manuscript.
References
- 1.National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch; National Cancer Institute: Surveillance, Epidemiology, and End Results (SEER) Program Public-Use Data (1973–2006) released April 2009, based on the November 2008 submission. www.seer.cancer.gov.
- 2.Mack TM. Sarcomas and other malignancies of soft tissue, retroperitoneum, peritoneum, pleura, heart, mediastinum, and spleen. Cancer. 1995;75:211–244. doi: 10.1002/1097-0142(19950101)75:1+<211::aid-cncr2820751309>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 3.Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: liposarcoma. Cancer Genet Cytogenet. 2004;155:1–24. doi: 10.1016/j.cancergencyto.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Dalal KM, Kattan MW, Antonescu CR, et al. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244:381–391. doi: 10.1097/01.sla.0000234795.98607.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engstrom K, Bergh P, Gustafson P, et al. Liposarcoma. Outcome based on the Scandinavian Sarcoma Group Register. Cancer. 2008;113:1649–1656. doi: 10.1002/cncr.23784. [DOI] [PubMed] [Google Scholar]
- 6.Linehan DC, Lewis JJ, Leung D, et al. Influence of biologic factors and anatomic site in completely resected liposarcoma. J Clin Oncol. 2000;18:1637–1643. doi: 10.1200/JCO.2000.18.8.1637. [DOI] [PubMed] [Google Scholar]
- 7.Fiore M, Grosso F, Lo Vullo S, et al. Myxoid/ round cell and pleomorphic liposarcomas. Prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2007;109:2522–2531. doi: 10.1002/cncr.22720. [DOI] [PubMed] [Google Scholar]
- 8.Neuhaus SJ, Barry P, Clark MA, et al. Surgical management of primary and recurrent retroperitoneal liposarcoma. Br J Surg. 2005;92:246–252. doi: 10.1002/bjs.4802. [DOI] [PubMed] [Google Scholar]
- 9.Guadagnolo BA, Zagars GK, Ballo MT, et al. Excellent local control rates and distinctive patterns of failure in myxoid liposarcoma treated with conservative surgery and radiotherapy. Int J Radiat Oncol Biol Phys. 2008;70:760–765. doi: 10.1016/j.ijrobp.2007.07.2337. [DOI] [PubMed] [Google Scholar]
- 10.Jones RL, Fisher C, Al-Muderis O, et al. Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur J Cancer. 2005;41:2853–2860. doi: 10.1016/j.ejca.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Wong CK, Edwards AT, Rees BI. Liposarcoma: a review of current diagnosis and management. Br J Hosp Med. 1997;58:589–591. [PubMed] [Google Scholar]
- 12.Alaggio R, Coffin CM, Weiss SW, et al. Liposarcoma in young patients. A study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol. 2009;33:645–658. doi: 10.1097/PAS.0b013e3181963c9c. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari A, Casanova M, Spreafico F, et al. Childhood liposarcoma: a single-institutional twenty-year experience. Pediatr Hematol Oncol. 1999;16:415–421. doi: 10.1080/088800199276967. [DOI] [PubMed] [Google Scholar]
- 14.La Quaglia MP, Spiro SA, Ghavimi F, et al. Liposarcoma in patients younger than or equal to 22 years of age. Cancer. 1993;72:3114–3119. doi: 10.1002/1097-0142(19931115)72:10<3114::aid-cncr2820721037>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 15.Shmookler BM, Enzinger FM. Liposarcoma occurring in children. An analysis of 17 cases and review of the literature. Cancer. 1983;52:567–574. doi: 10.1002/1097-0142(19830801)52:3<567::aid-cncr2820520332>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Pollack A, Zagars GK, Goswitz MS, et al. Preoperative versus postoperative radiotherapy in the treatment of soft tissue sarcomas: a matter of presentation. Int J Radiat Oncol Biol Phys. 1998;42:563–572. doi: 10.1016/s0360-3016(98)00277-6. [DOI] [PubMed] [Google Scholar]
- 17.Paulino AC. Late effects of radiotherapy for pediatric extremity sarcomas. Int J Radiat Oncol Biol Phys. 2004;60:265–274. doi: 10.1016/j.ijrobp.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Maki RG. Gemcitabine and docetaxel in metastatic sarcoma: past, present, and future. Oncologist. 2007;12:999–1006. doi: 10.1634/theoncologist.12-8-999. [DOI] [PubMed] [Google Scholar]
- 19.Demetri GD, Chawla SP, von Mehren M, et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase II study of two different schedules. J Clin Oncol. 2009;27:4188–4196. doi: 10.1200/JCO.2008.21.0088. [DOI] [PubMed] [Google Scholar]
- 20.Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol. 2007;8:595–602. doi: 10.1016/S1470-2045(07)70175-4. [DOI] [PubMed] [Google Scholar]
- 21.Gebhard S, Coindre JM, Michels JJ, et al. Pleomorphic liposarcoma: clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases. A study from the French Federation of Cancer Centers Sarcoma Group. Am J Surg Pathol. 2002;26:601–616. doi: 10.1097/00000478-200205000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Antonescu CR, Tschernyavsky SJ, Decuseara R, et al. Prognostic impact of P53 status, TLS-CHOP fusion transcript structure, and histological grade of myxoid liposarcoma: a molecular and clinicopathologic study of 82 cases. Clin Cancer Res. 2001;7:3977–3987. [PubMed] [Google Scholar]
- 23.ten Heuvel SE, Hoekstra HJ, van Ginkel RJ, et al. Clinicopathologic prognostic factors in myxoid liposarcoma: a retrospective study of 49 patients with long-term follow-up. Ann Surg Oncol. 2007;14:222–229. doi: 10.1245/s10434-006-9043-7. [DOI] [PubMed] [Google Scholar]
- 24.Schwab JH, Boland P, Guo T, et al. Skeletal metastases in myxoid liposarcoma: an unusual pattern of distant spread. Ann Surg Oncol. 2007;14:1507–1514. doi: 10.1245/s10434-006-9306-3. [DOI] [PubMed] [Google Scholar]
- 25.Schneider-Stock R, Walter H, Radig K, et al. MDM2 amplification and loss of heterozygosity at Rb and p53 genes: no simultaneous alterations in the oncogenesis of liposarcomas. J Cancer Res Clin Oncol. 1998;124:532–540. doi: 10.1007/s004320050211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nilbert M, Rydholm A, Willen H, et al. MDM2 gene amplification correlates with ring chromosome in soft tissue tumors. Genes Chromosomes Cancer. 1994;9:261–265. doi: 10.1002/gcc.2870090406. [DOI] [PubMed] [Google Scholar]
- 27.Meis-Kindblom JM, Sjogren H, Kindblom LG, et al. Cytogenetic and molecular genetic analyses of liposarcoma and its soft tissue stimulators: recognition of new variants and differential diagnosis. Virchows Arch. 2001;439:141–151. doi: 10.1007/s004280100423. [DOI] [PubMed] [Google Scholar]
- 28.Muller CR, Paulsen EB, Noordhuis P, et al. Potential for treatment of liposarcomas with the MDM2 antagonist Nutlin-3A. Int J Cancer. 2007;121:199–205. doi: 10.1002/ijc.22643. [DOI] [PubMed] [Google Scholar]