

Abstract
Objective
The origin of the mesenchymal cells responsible for the intimal fibrosis in Systemic Sclerosis (SSc) has not been fully identified. Here, we investigated whether subendothelial mesenchymal cells may emerge through transdifferentiation of endothelial cells (EC) into myofibroblasts via endothelial-mesenchymal transition (EndoMT) in vitro and the signaling pathways involved in this process.
Methods
Primary mouse pulmonary EC isolated by immunomagnetic methods employing sequential anti-CD34 and anti-CD102 antibody selection were cultured in monolayers. Cell morphology and diacetylated LDL uptake assays confirmed their EC characteristics. The induction of EndoMT was assessed by determination of α-smooth muscle actin (α-SMA), collagen type I, and VE-Cadherin expression and the expression of the transcriptional repressor Snail-1 was analyzed. The signaling pathways involved were examined employing small molecule kinase inhibitors and short RNA interference.
Results
TGF-β1 induced α-SMA and type I collagen expression and inhibited VE-Cadherin. These effects were mediated by a marked increase in Snail-1 expression and were abolished by treatment with either the c-Abl tyrosine kinase inhibitor, imatinib mesylate or the protein kinase C δ (PKC-δ) inhibitor, rottlerin. The inihibitory effects of imatinib mesylate and rottlerin were mediated by inhibition of phosphorylation of GSK-3β at residue Ser9. These observations were confirmed employing short interfering RNA specific for c-Abl and PKC-δ.
Conclusion
These results indicate that c-Abl and PKC-δ are crucial for TGF-β-induced EndoMT and that imatinib mesylate and rottlerin or similar kinase inhibitor molecules may be effective therapeutic agents for SSc and other fibroproliferative vasculopathies in which EndoMT plays a pathogenetic role.
Keywords: endothelial-mesenchymal transition, transforming growth factor-β, Snail-1, α-smooth muscle actin, systemic sclerosis, pulmonary artery hypertension, c-Abl tyrosine kinase, imatinib mesylate, protein kinase C δ, rottlerin
Introduction
Fibroproliferative vasculopathies characterized by accumulation of mesenchymal cells and fibrosis in the subendothelial space of medium size and small vessels are the pathological hallmark of a diverse group of disorders including transplant rejection and allograft vasculopathy (1), primary pulmonary hypertension (2), and systemic sclerosis (SSc) (3,4). The origin of the mesenchymal cells and myofibroblasts responsible for subendothelial intimal fibrosis remains elusive. Vessel wall resident fibroblasts and circulating mesenchymal cell progenitors, also known as fibrocytes, migrating into the vessel intima have been considered important sources. However, recent studies have shown that mesenchymal cells and myofibroblasts can also originate from endothelial cells in a process known as endothelial–mesenchymal transition (EndoMT) in which endothelial cells (EC) acquire a myofibroblastic phenotype (5,6). EndoMT has been reported to contribute to the myofibroblastic population in chronic pulmonary hypertension (7) and cardiac, renal, and pulmonary fibrosis (8–12), thus, the possibility that some of the fibrotic tissue-producing subendothelial mesenchymal cells responsible for the severe SSc fibroproliferative vasculopathy may also originate from endothelial cells is attractive.
Transforming growth factor (TGF)-β1 plays a crucial role in tissue fibrosis and is implicated in the pathogenesis of numerous disorders including the fibroproliferative vasculopathies (13–17). Besides causing a potent stimulation of the expression of numerous genes participating in the fibrotic process, TGF-β is involved in the generation of myofibroblasts through epithelial to mesenchymal transition (EMT) (18–21) and EndoMT (7–12). Here, we examined the intracellular signaling pathways involved in TGF-β induced EndoMT in murine pulmonary EC. The results demonstrated that TGF-β1 caused a potent induction of α-SMA expression in murine pulmonary EC. This process was accompanied by a marked increase in the expression of the transcriptional repressor, Snail-1 and was abolished by treatment with the cAbl kinase inhibitor, imatinib mesylate or the PKC-δ inhibitor, rottlerin. The potent inhibition of TGF-β-induced α-SMA expression by small molecule c-Abl and PKC-δ inhibitors suggests that these or similar inhibitor molecules may be effective therapeutic agents for SSc fibroproliferative vasculopathy as well as for other pathologic conditions in which EndoMT plays a role.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM) was from Gibco (Carlsbad, CA). Glutamine, penicillin/streptomycin, sodium pyruvate, HEPES and non-essential amino acids were from Mediatech (Manassas, VA). Fetal bovine serum (FBS) was from Atlanta Biological (Lawrenceville, GA). Anti-CD34 and anti-CD102 antibodies were from BD Biosciences (Bedford, MA). Western blots and indirect immunofluorescence antibodies were: α-SMA (clone SPM332) and GAPDH (Abcam, Cambridge, MA), VE-Cadherin, collagen1A1, Snail-1 (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-GSK-3β (Ser9) and total GSK-3β (Cell Signaling Technology, Danvers, MA). Cy3-conjugated secondary antibodies were from Sigma (St. Louis, MO). TGF-β1 was from R&D Systems (Minneapolis, MN). Endothelial cell growth supplement was from Sigma (St. Louis, MO). The kinase inhibitors used were: c-Abl inhibitor imatinib mesylate from LC Laboratories (Woburn, MA), p38 MAP kinase inhibitor SB 203580, the JNK MAP kinase inhibitor SP 600125 and the Erk1/2 specific upstream inhibitor PD 98059 from Cayman Chemical (Ann Arbor, MI). The PI-3 kinase inhibitor LY294002 was from Cell Signalling (Danvers, MA) and the PKC-δ inhibitor rottlerin was from Alexis Biochemical (San Diego, CA).
Isolation and culture of pulmonary endothelial cells
Pulmonary EC were isolated from C57BL/6 mice (8–12 weeks old) following IACUC approval with a modification of the Marelli-Berg method (22, 23). Briefly, lungs were harvested, mechanically minced, and enzymatically digested with collagenase (30 mg/100ml; Worthington, Lakewood, NJ) in 0.1% BSA at 37 °C for 1 h with constant gentle mechanical agitation. The resulting mixture of cells was filtered through a sterile 75 μm cell strainer (BD Biosciences, San Diego, CA). The single cell suspension obtained was incubated with rat anti-mouse CD34 antibody (1:50) at 4 °C for 45 min with gentle shaking, followed by magnetic bead separation using goat anti-rat IgG-conjugated microbeads (1:5, Miltenyi Biotec, Auburn, CA, USA). The isolated EC were cultured in DMEM with 20% FBS, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, 20 mM HEPES, 1% non-essential amino acids, 100 μg/ml endothelial cell growth supplement and 50 U/ml heparin in 2% gelatin pre-coated tissue culture dishes. Following expansion to 70–80% confluence (3–5 days), the cells were resuspended and a second immunologic separation carried out using rat anti-mouse CD102 antibody. The resulting cells were plated and their endothelial phenotype confirmed by cellular uptake of DiI-acetylated LDL (DiI-AcLDL, Biomedical Technologies, Stoughton, MA) and cell morphology. The proportion of cells taking up DiI-AcLDL compared to the number of cell nuclei representing the entire cell population, was used to assess the purity of the isolated EC populations. For TGF-β treatment, pulmonary EC were washed with serum-free medium and then incubated with 10 ng/ml TGF-β1 in endothelial cell culture medium containing 0.5% FBS. The medium was changed and additives replenished every 2 days. Phase-contrast and fluorescent images were obtained using an inverted microscope with SpotBasic software (Sterling Heights, MI).
Immunofluorescence staining
Pulmonary EC were seeded onto glass culture slides and treated as described above. One or 3 days following TGF-β1 treatment, cells were fixed with 3.7% formaldehyde and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 3 min. Slides were washed with PBS and blocked with 1% bovine serum albumin in PBS at room temperature for 1 h and then they were incubated with primary antibodies against α-SMA (1:200) or Snail 1 (1:50). Slides were then incubated with Cy3-conjugated secondary antibodies (1:500).
Protein precipitation from cell culture medium
The conditioned culture media from primary pulmonary EC that were treated with or without TGF-β1 for 3 days was mixed with 9 volumes of 100% ethanol, incubated at −80 °C for 15 min and subsequently centrifuged at 13,000 rpm for 15 min at 4°C. The pellet was resuspended in PBS and protein concentrations were assessed using the BCA assay kit (Bio-Rad, Hercules, CA).
Western blot analysis
Primary pulmonary EC were treated for 72 h with TGF-β1 (10 ng/ml) with or without small molecule kinase inhibitors as indicated. Following treatment, the cells were washed with cold PBS, harvested with a cell lifter, centrifuged and lysed with RIPA lysis buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% Na deoxycholate and 0.1 % SDS) supplemented with a complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Cellular proteins were resolved by SDS-polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes (Invitrogen, Carlsbad, CA). Blots were blocked for 1 h in Tris-buffered saline (TBS)-Tween (10 mmol/L Tris-HCl, pH 8.0, 150 mmol/L NaCl, 0.1% Tween 20) containing 5% nonfat dry milk (BioRad, Hercules, CA). Then, membranes were incubated overnight at 4°C with mouse monoclonal α-SMA antibody (1:200) or rabbit polyconal Snail-1 antibody (1:1000), rabbit phospho-GSK-3β (Ser9) antibody (1:1000) or GAPDH polyclonal rabbit antibody (1:200) primary antibodies in a 5% nonfat dry milk/TBS-Tween solution. Membranes were then washed with TBS-Tween, and incubated for 1 h with the appropriate horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, UK). Signal was detected with an enhanced chemiluminescence detection kit (Pierce, Rockford, IL) and collected on an autoradiography film (Denville Scientific, Metuchen, NJ). The signals were quantified using NIH Image J software.
Treatment with small molecule kinase inhibitors
Primary EC cultures were treated for 72 h with TGF-β1, in the presence or absence of the following kinase inhibitors: p38 MAP kinase inhibitor SB 203580 (5μM), JNK MAP kinase inhibitor SP 600125 (1 μM), Erk1/2 specific upstream inhibitor PD 98059 (5μM), PI-3 kinase inhibitor LY294002 (5 μM), PKC-δ inhibitor rottlerin (0.5 μM), or c-Abl kinase inhibitor imatinib mesylate (2μg/ml).
RNA interference
Transfection of short interfering RNAs (siRNAs) was performed in 6-well tissue culture plates employing the HiPerFect reagent (Qiagen, Valencia, CA). Two different sequences were targeted and the final concentration of the siRNAs used was 5 nM. Following siRNA transfection, 10 ng/ml TGF-β1 was added to the media, and incubation was continued for 72 h. The siRNA target sequences were: murine Snail-1: 5′-CAG TTT ATT GAT ATT CAA TAA-3′ and 5′-ATG GTT AAT TTTA TAT ACT AAA-3′; murine c-Abl: 5′-CTC GAT GGA ACT CCA AGG AAA-3′ and 5′-CAG GCC CAC ACT GAT CAC CTA-3′; murine PKC-δ: 5′-AGG GAA GAC ACT GGT ACA GAA-3′ and 5′-CCG ATT CAA GGT TTA TAA CTA-3′. Control siRNA was obtained from Qiagen and was transfected employing the same conditions.
Semi-quantitative reverse transcription (RT)-PCR
Treated primary pulmonary EC were harvested, washed in cold PBS, and processed for RNA extraction (RNeasy kit; Qiagen). Total RNA (1 μg) was reverse-transcribed using Superscript II reverse transcriptase (Invitrogen) to generate first strand cDNA. The primers for RT-PCR were designed using Vector NTI 5 software (Invitrogen) and validated for specificity. The following primers were used: Snail-1; forward, 5′-ACC TTC CAG CAG CCC TAC GA-3′, reverse, 5′-AGC GTG TGG CTT CGG ATG TG-3′. Snail-2; forward, 5′-ACT ACA GCG AAC TGG ACA CA-3′, reverse, 5′-CTG GAG AAG GTT TTT GGA GCA-3′. GAPDH; forward, 5′-TGT GGA TGG CCC CTC TGG AA-3′, reverse, 5′-CGG CAT CGA AGG TGG AAG AG-3′. RT-PCR was performed following a standard amplification protocol. The products were electrophoresed on 1% agarose gels and stained with ethidium bromide. The reaction products were visualized with a FluorImager (Molecular Dynamics, Sunnyvale, CA). Images were quantitated using NIH Image J software and the signals were normalized to those obtained for loading controls. The values for the untreated control samples were set as 1 and all the experimental values were expressed as a multiple thereof.
Quantitative real time PCR
Real-time PCR was performed using SYBR green employing a standard amplification protocol on a ABI 7900 HT Fast Real-time PCR system. The following primers were used: α-SMA, forward, 5′-GTG GCC CCT GAA GAG CAT C-3′, reverse, 5′-AAT CTG GGT CAT TTT CTC CCG-3′. Collagen I, forward, 5′-GCA TGG CCA AGA AGA CAT CC-3′, reverse, 5′-TCC ACG TCT CAC CAT TGG G-3′. 18S, forward, 5′-CGG CTA CCA CAT CCA AGG AA-3′, reverse, 5′-GGG CCT CGA AAG AGT AAT GT-3′. The primers for Snail-1 amplification were the same as those used for RT-PCR (see above). To confirm the amplification specificity, the PCR products were subjected to melting temperature dissociation curve analysis. No amplification and no template controls were examined in parallel. The cycle number (CT) at which amplification entered the exponential phase was determined and used as an indicator of the amount of target RNA produced in the reactions. The control experiment levels were set as 1 and all the other values expressed as a multiple thereof.
Statistical analysis
Data are expressed as means ± SD. Means of different groups were compared using one-way analysis of variance. Statistical analysis was performed by using unpaired Student’s t test. A value of p < 0.05 was considered to be statistically significant.
Results
Isolation and characterization of murine pulmonary EC
To ensure that there were no other contaminating cells, we performed a sequential immunomagnetic isolation procedure using an initial purification with anti-CD34 antibody followed by expansion in monolayer cell culture and a second immunomagnetic purification with CD-102 antibodies. These two antibodies are reported to be specific for endothelial cells. This procedure yielded 1–1.5 million pure EC from each pair of lungs with 95% viability. The pulmonary EC monolayer cultures showed classical EC cobblestone morphology and following confluency displayed the formation of characteristic capillary tubes (Figures 1A and 1B). The endothelial phenotype of the cultured cells was confirmed by uptake of DiI-Ac-LDL (Figure 1C), a specific procedure to identify EC in culture (24). The proportion of DiI-AC-LDL- positive cells indicated that greater than 99% of cells in the preparation were EC (Figure 1C and 1D).
Figure 1. Characterization of pulmonary EC.

A. Phase contrast of pulmonary EC in culture isolated employing sequential anti-CD34 and anti-CD102 antibody selection. B. Characteristic tube-formation at confluence. C. Ac-LDL uptake assay. D. Cells in the same field as IC stained with DAPI for nuclear staining. Pulmonary EC display cobblestone morphology, form multicellular tubes (arrow) and express high levels of Ac-LDL confirming their endothelial phenotype. Size bar = 100μm
TGF-β1 induction of a myofibroblast phenotype in primary pulmonary EC
Primary pulmonary EC cultures treated with TGF-β1 for 72 h lost their characteristic cobblestone-like morphology and expressed the myofibroblast-specific protein α-SMA (Figure 2A and 2B). Western blot analysis confirmed induction of α-SMA in these cells, whereas in cells cultured without TGF-β1 there was no detectable α-SMA expression (Figure 2C). We next examined whether TGF-β1-treated EC also displayed expression of the fibroblast phenotype-specific marker type I collagen. In the untreated cells there was no evidence of collagen I expression, whereas treatment with TGF-β1 induced significant (p<0.05 when compared with control cells) collagen I expression (Figure 2D). The TGF-β1-treated EC also lost the endothelial cell marker VE-cadherin. As expected, untreated cells displayed high expression of VE-cadherin, whereas treatment with TGF-β1 caused a marked and significant (p<0.05 compared with control cells) reduction in VE-cadherin expression (Figure 2E). We next studied the time course of α-SMA and collagen I mRNA expression following TGF-β exposure by quantitative real-time PCR. The results showed a marked increase in α-SMA mRNA expression at 12 h (p<0.05 when compared with control) with a further increase at 24 h. In contrast, collagen I mRNA displayed a significant increase only after 24 h (p<0.05 when compared with control, Figure 2F).
Figure 2. TGF-β1 induced α-SMA expression in primary pulmonary EC.
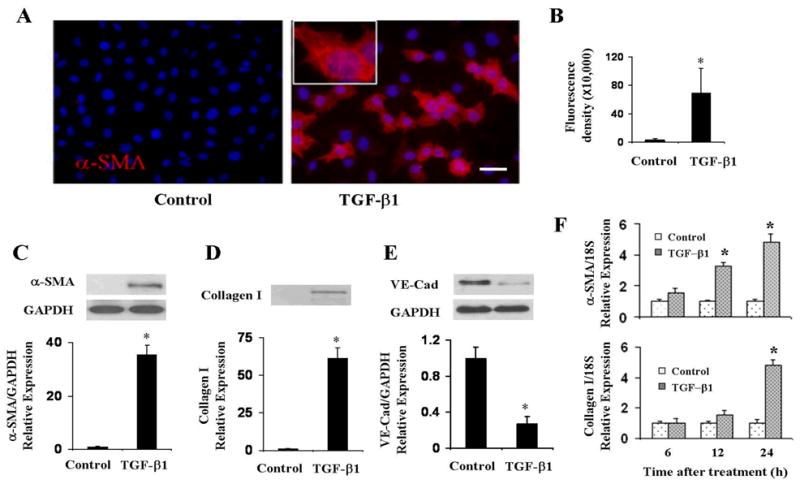
A, B. Primary pulmonary EC were treated with TGF-β1 for 72 h, fixed and stained with α-SMA. Note the phenotypic change of EC which acquire a fibroblast-like appearance and initiate expression of α-SMA. C, D, E. Primary lung EC were treated with TGF-β1 for 72 h, and cell lysates were electrophoresed and probed in Western blots with specific antibodies to α-SMA (C), collagen I (D) and VE-cadherin (E). GAPDH was used as loading control. Fluorescence density of α-SMA expression and quantitative densitometry of Western blots were analyzed using NIH Image J software. F. Quantitative real-time PCR analysis of TGF-β1 induction of α-SMA and collagen I mRNA expression in primary pulmonary EC. Amplification of 18S RNA was used as internal control. Data represent the mean ± SD of one of two independent experiments in which each sample was analyzed in triplicate. *p < 0.05 when compared with controls. Size bar = 50μm.
TGF-β1 induction of Snail-1 expression in primary pulmonary EC
The family of Snail proteins has been shown to regulate EMT and cell behavior during a variety of developmental processes (25–27). Therefore, we tested whether Snail-1 also participates in EndoMT in pulmonary EC. Snail-1 expression examination by immunofluorescence staining showed a very low level of expression in untreated cells that was confined to the nuclei. In contrast, TGF-β1 treatment significantly increased Snail-1 expression (p<0.05 when compared with control cells) in pulmonary EC (Figure 3A and 3B). The results were confirmed by assessment of Snail-1 mRNA levels by RT-PCR. In untreated cells we observed a very low level of basal expression of Snail-1, however, TGF-β1 treatment caused a potent stimulation of its expression. In contrast, Snail-2 showed higher constitutive levels of expression, however, there was no stimulation of its expression following TGF-β1 treatment (Figure 3C). Further quantification of mRNA expression by quantitative real-time PCR showed that following 24 h TGF-β1 treatment, there was a significant increase in Snail-1 mRNA expression (p<0.05 when compared with control cells; Figure 3D).
Figure 3. TGF-β1 induced Snail-1 expression in primary pulmonary EC.

A, B. Primary pulmonary EC were treated with TGF-β1 for 24 h, fixed and stained by indirect immunofluorescence with Snail-1. Note a marked increase in expression of Snail-1 following treatment with TGF-β. B. Fluorescence density of α-SMA expression analyzed using NIH Image J software. C. Primary pulmonary EC were treated with TGF-β1 for 72 h. RNA was purified and reverse transcribed and assessed by PCR for Snail-1 and Snail-2 expression. GAPDH was used as control. Note very low level basal expression of Snail-1. Treatment with TGF-β1 increased Snail-1 expression. In contrast, Snail-2 levels displayed higher constitutive expression and did not show any change following TGF-β treatment. D. Quantitative real-time PCR analysis of TGF-β1 induction of Snail-1 mRNA expression. The data represent the mean ± SD of one of two independent experiments in which each sample was analyzed in triplicate (*p < 0.05 when compared with controls). Size bar = 50μm
Effects of kinase inhibitors on TGF-β1-induced α-SMA and Snail-1 expression in primary pulmonary EC
To examine the intracellular signaling pathways that may be involved in the induction of α-SMA and Snail-1 expression by TGF-β1, we treated the cells with a variety of small molecule kinase inhibitors. The results employing a p38 MAP kinase inhibitor (SB 203580), a JNK MAP kinase inhibitor (SP 600125), or an Erk1/2 specific upstream inhibitor (PD 98059) with or without TGF-β1 are shown in Figure 4A. Treatment with either the p38 MAP kinase inhibitor or the JNK MAP kinase inhibitor decreased but did not abrogate TGF-β1-induced α-SMA and Snail-1 expression. Treatment with the Erk1/2 inhibitor had no significant effects. Studies with the PI3K inhibitor (LY294002) or the PKC-δ inhibitor (rottlerin) with or without TGF-β1 are shown in Figure 4B. Treatment with the PKC-δ inhibitor almost completely abrogated TGF-β1-induced α-SMA and Snail-1 expression, whereas treatment with the PI-3K inhibitor had no detectable effects.
Figure 4. Effect of kinase inhibitors on TGF-β1-induced α-SMA and Snail-1 expression in primary pulmonary EC.

Primary pulmonary EC were treated with either 5μM p38 MAP kinase inhibitor SB 203580, 1μM JNK MAP kinase inhibitor SP 600125, or 5μM Erk1/2 specific upstream inhibitor PD 98059 (A), or with 5μM PI3-kinase3 inhibitor LY294002 or 0.5μM PKC-δ inhibitor rottlerin (B) in the presence or absence of TGF-β1 for 72 h, then cell lysates were probed in Western blots for α-SMA. GAPDH was used as loading control. Note that SB 203580 and SP 600125 decrease TGF-β1-induced α-SMA expression, treatment with the PKC-δ inhibitor rottlerin almost completely abolished TGF-β1-induced α-SMA expression, whereas treatment with either the Erk1/2 inhibitor or the PI-3 kinase inhibitor had no significant effects. The quantitative densitometry of α-SMA was analyzed using NIH Image J software and the data represent one of two independent experiments *p < 0.05 when compared with the TGF-β1-treated group.
Imatinib mesylate abrogates TGF-β1-inducedα-SMA expression in primary pulmonary EC
To examine the participation of c-Abl on TGF-β1-induced α-SMA expression pulmonary EC were treated with TGF-β1 in the presence or absence of imatinib mesylate for 72 h. As shown in Figure 5A, imatinib mesylate almost completely abolished TGF-β1-induced α-SMA expression. Moreover, in the few cells that displayed α-SMA staining following treatment with imatinib, α-SMA was distributed diffusely in the cells and did not organize into any typical α-SMA stress fibres. Western blot assay confirmed that imatinib mesylate caused a potent decrease in TGF-β1-induced α-SMA expression (p<0.05 compared with control cells; Figure 5B).
Figure 5. Effect of imatinib mesylate on TGF-β1-induced α-SMA and Snail-1 expression in primary pulmonary EC.

A. Primary pulmonary EC were treated with imatinib with or without TGF-β1 for 72 h, fixed and stained with α-SMA. Imatinib significantly decreased TGF-β1-induced α-SMA expression. Also note that in imatinib-treated cells, α-SMA was distributed homogenously and did not organize into α-SMA stress fibers. B, C. Cell lysates were probed by Western blots for α-SMA (B) and Snail-1 (C). GAPDH was used as loading control. Quantitative densitometry was analyzed using NIH Image J software and the data represent one of two independent experiments in which each sample was analyzed in triplicate. D. Primary pulmonary EC were treated with imatinib with or without TGF-β1 for 72 h. Transcript levels were assessed by PCR. GAPDH was used as control. The quantitative densitometry was analyzed using NIH Image J software. Note that imatinib significantly downregulated Snail-1 protein levels (5C) and mRNA expression (5D). E. Primary EC were transfected with Snail-1 siRNA or with control siRNA followed by TGF-β1 treatment for 72 h, cell lysate proteins were probed with α-SMA. GAPDH was used as loading control. The data represent one of two independent experiments. *p < 0.05 when compared with TGF-β1-treated group. Size bar = 50μm.
Effects of imatinib mesylate on TGF-β1-induced Snail-1 mRNA and protein expression in primary pulmonary EC
To examine whether the effects of imatinib mesylate on TGF-β1-induced α-SMA expression were accompanied by parallel changes in Snail-1 expression, we examined Snail-1 transcript levels using reverse transcription PCR. As shown in Figure 5C, TGF-β1 induced a marked increase in Snail-1 expression, and treatment with imatinib mesylate reduced significantly (p<0.05) these levels to almost baseline values. The effects of imatinib mesylate on TGF-β1-induced Snail-1 expression were confirmed by Western blots (p<0.05 compared with control cells; Figure 5D). To determine whether the potent inhibition of TGF-β induced EndoMT by imatinib mesylate was mediated through blockade of the canonical Smad2/3 cascade, we measured phospho-Smad2 levels. Although TGF-β1 significantly induced phospho-Smad2 levels, this effect was not suppressed by imatinib mesylate (data not shown), indicating that the canonical Smad pathway does not participate in imatinib inhibition of TGF-β1-induced Snail-1 expression.
Effects of Snail-1 knockdown on TGF-β1 induced-SMA expression in primary pulmonary EC
To determine whether Snail-1 is required for TGF-β1 induced α-SMA expression, we transfected Snail-1 siRNA into primary pulmonary EC followed by treatment with TGF-β1. As control, we transfected siRNA consisting of a sequence that has no known homology to any mammalian gene. As shown in Figure 5E, transfection of Snail-1 siRNA caused a significant (p<0.05) inhibition of TGF-β1-inducedα-SMA expression.
Effects of MAP kinase, c-Abl, and PKC-δ inhibition on TGF-β1-induced GSK-3β phosphorylation
Numerous previous studies have demonstrated that GSK-3β phosphorylation at residue Ser9 plays a crucial role in the regulation of EMT (27). To examine whether GSK-3β phosphorylation is also important in EndoMT we studied the changes in TGF-β1-induced GSK-3β Ser9 phosphorylation following treatment with various kinase inhibitors. As shown in Figure 6A and 6B, TGF-β1 caused potent stimulation of Ser9 GSK-3β phosphorylation and treatment with the MAP kinase inhibitors did not have any effect. In contrast, both the c-Abl inhibitor imatinib mesylate and the PKC-δ inhibitor rottlerin almost completely abrogated the TGF-β1-induced GSK-3β Ser9 phosphorylation (p<0.05 for both compounds when compared with the TGF-β1 treated group).
Figure 6. Effect of MAP kinase, c-Abl, and PKC-δ inhibition on TGF-β1-induced GSK-3β phosphorylation.

A, B. Primary pulmonary EC were treated with 5μM p38 MAP kinase inhibitor SB 203580, 5 μM JNK inhibitor SP 600125 (A), or 2μg/ml c-Abl inhibitor imatinib mesylate or 0.5 μM PKC-δ inhibitor rottlerin (B) for 72 h then cell lysates were probed in Western blots with phospho-GSK-3β (Ser9) and total GSK-3β antibodies. Note that the MAP kinase inhibitor did not have any effect on TGF-β1-induced GSK-3β Ser9 phosphorylation (A) whereas, both the c-Abl inhibitor imatinib and the PKC-δ inhibitor rottlerin almost completely abrogated TGF-β1-induced Ser9 GSK-3β phosphorylation (B). C, D. Primary EC were transfected with either c-Abl or PKC-δ specific siRNA or with control siRNA followed by TGF-β1 treatment for 72 h. Cell lysates were probed in Western blots for α-SMA and Snail-1. GAPDH was used as loading control. The data represent one of two independent experiments. The quantitative densitometry was analyzed using NIH Image J software. *p < 0.05 when compared with the TGF-β1-treated group.
Effects of c-Abl, and PKC-δ knockdown on TGF-β1 induced-SMA and Snail-1 expression in primary pulmonary EC
To further confirm the role of c-Abl and PKC-δ in TGF-β1 induced SMA expression, we transfected c-Abl and PKC-δ siRNA into primary pulmonary EC followed by treatment with TGF-β1. Preliminary experiments (data not shown) indicated that transfection of the selected siRNA caused nearly complete inhibition of expression of the corresponding genes. As shown in Figure 6C and 6D, knockdown of either c-Abl or PKC-δ employing siRNA caused a potent inhibition of TGF-β1-induced α-SMA and Snail-1 expression.
Discussion
One of the most characteristic histopathologic alterations in SSc is a severe fibroproliferative vasculopathy affecting the microvasculature as well as some larger vessels. The proliferative vasculopathy of SSc has two distinct components. The first one is a marked proliferation of smooth muscle cells in the media of medium size and small size arterioles, a process which plays a crucial role in SSc-associated pulmonary hypertension. The second component is most prominent in the small arterioles of parenchymal organs such as the lungs and is characterized by the subendothelial accumulation of activated fibroblasts or myofibroblasts and the production of abundant fibrotic tissue. The origin of mesenchymal cells responsible for the fibrotic microvascular occlusion in SSc is not known but recent studies have suggested that at least some of these cells may result from EndoMT, i.e., the transdifferentiation of EC into subintimal fibroblasts induced by locally-secreted cytokines and growth factors.
The purpose of our study was to examine the transdifferentiation of pulmonary EC into mesenchymal cells in vitro and the signaling pathways involved in this process. Our results have shown that: (1) primary murine pulmonary EC undergo EndoMT in response to TGF-β with initiation of expression of α-SMA and assembly of typical intracellular α-SMA stress fibers, and loss of VE-cadherin in vitro; (2) TGF-β induction of EndoMT was associated with a strong upregulation in the expression of the transcriptional repressor Snail-1 indicating that Snail-1 is directly involved TGF-β-induced α-SMA expression; and (3) induction of α-SMA expression in pulmonary EC was mediated by c-Abl and PKC-δ, as specific inhibition of their kinase activity with imatinib mesylate and rottlerin, respectively, or by knockdown of their corresponding transcripts with specific siRNA abrogated the marked increase in TGF-β induced α-SMA and Snail-1 expression and protein levels.
EndoMT is a recently recognized process of non-malignant cellular transdifferentiation in which highly differentiated endothelial cells acquire the expression of specific mesenchymal cell markers such as α-SMA, vimentin, and type I procollagen, lose the expression of VE-cadherin, and change their typical endothelial cell morphology becoming elongated and acquiring the ability to migrate into surrounding tissues. This process, initially identified during development of the cardiac atrioventricular cushion and the cardiac septum in embryonic life, has more recently been demonstrated to occur in adult tissues and to play an important role during the development of pathologic cardiac, pulmonary and renal fibrosis (5–12). Indeed, by using endothelial lineage tracing in transgenic mice, it was shown that 30–50% of all myofibroblasts in renal fibrosis arise from endothelial cells via EndoMT (9,10) and that a significant amount of fibroblasts in bleomycin-induced lung fibrosis are EC derived (11).
Given the crucial role of TGF-β in the development of tissue fibrosis and its participation in the pathogenesis of numerous fibrotic diseases, we examined the mechanisms involved in the induction of EndoMT by this pleotropic growth factor and studied the intracellular transduction pathways involved in this process employing primary pulmonary EC in culture. We confirmed the occurrence of EndoMT following TGF-β and demonstrated that the downstream signaling pathway initiated by TGF-β ligation resulted in strong upregulation of the transcriptional repressor Snail-1. Snail-1 is a zinc-finger transcription factor that causes potent inhibition of the E-cadherin gene transcription in cultured cells (28,29) and plays an important in the process of EMT (26,27). Our observation that TGF-β1 also induced Snail-1 expression in EC confirm previous studies (30) and indicate that EndoMT and EMT very likely share similar molecular mechanisms.
The intracellular signaling studies described here showed that TGF-β1-induced EndoMT involving the participation of c-Abl and PKC-δ. The potent inhibitory effects of imatinib mesylate and rottlerin were shown to be accompanied by inhibition of phosphorylation of the Ser9 residue in GSK-3β. These results are in agreement with recent studies demonstrating that phosphorylation of this specific Ser9 residue in GSK-3β inactivates the kinase which in turn induces the nuclear accumulation of Snail-1 as well as a profound increase in the expression of its corresponding gene and in stabilization of the protein (31–33). These events result in a marked increase in the transcriptional effects of Snail-1 and eventually in the increased expression of mesenchymal cell proteins such as α-SMA and collagen I.
Imatinib mesylate, a small-molecule tyrosine kinase inhibitor widely used for the treatment of chronic myelogenous leukemia has recently been proposed as a novel antifibrotic therapy (34–36). It has been shown to have anti-fibrotic effects in mice models of systemic fibrosis (37–38), and it also can attenuate bleomycin-induced pulmonary fibrosis (34,39). On the other hand, several studies have shown that PKC-δ participates in the pathogenesis of tissue fibrosis in SSc (40,41). Here, by using primary pulmonary EC, we showed that imatinib mesylate can inhibit TGF-β1-induced EndoMT and Snail-1 expression. Furthermore, our studies showed that inhibition of PKC-δ with the highly selective inhibitor rottlerin also abrogated EndoMT induction by TGF-β and blocked the increased expression of α-SMA and Snail-1 indicating that this kinase is also intimately improved in this process. The role of PKC-δ was confirmed employing siRNA knockdown which caused similar abrogation of α-SMA and Snail-1 expression as rottlerin (Figure 6). These latter results indicated that the effects of rottlerin were related to specific inhibition of PKC-δ and were not the result of other non-specific effects.
In conclusion, the results described here identify c-Abl and PKC-δ as two important components of the process of TGF-β-induced EndoMT and suggest that targeting these components may be a feasible therapeutic goal to modify crucial steps in the development of SSc fibroproliferative vasculopathy and may also be of value in the treatment of other fibrotic disorders in which EndoMT plays a pathogenetic role. Obviously, subsequent preclinical studies employing suitable animal models will be required to further support the potential therapeutic role of c-Abl and PKC-δ inhibition for the proliferative vasculopathy of SSc and other disorders.
Acknowledgments
We are grateful to Drs. Sonsoles Piera, Jolanta Fertala for advice and helpful discussions and to Melissa Marcucci and Carol Kelly for their assistance in the preparation of this manuscript.
Funding: The work was supported by the National Institutes of Health grants 5RO1AR019606 and 9RO1 AR055660 to S.A.J.
Abbreviations
- α-SMA
α-smooth muscle actin
- TGF-β
transforming growth factor-β
- EndoMT
endothelial-to-mesenchymal transition
- FCS
fetal calf serum
- PBS
phosphate-buffered saline
- PI3K
phosphoinositol 3 kinase
- PKC-δ
protein kinase C-delta
- GSK-3β
glycogen synthase kinase
- c-Abl
c-Abelson kinase
Footnotes
Conflict of Interest: none declared.
Contributor Information
Zhaodong Li, Email: zhaodong.li@jefferson.edu.
Sergio A. Jimenez, Email: Sergio.jimenez@jefferson.edu.
References
- 1.Avery RK. Cardiac-allograft vasculopathy. N Engl J Med. 2003;349:829–830. doi: 10.1056/NEJMp038124. [DOI] [PubMed] [Google Scholar]
- 2.Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43:25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 3.Fleming JN, Nash RA, Mahoney WM, Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep. 2009;11:103–10. doi: 10.1007/s11926-009-0015-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kahaleh MB, LeRoy EC. Autoimmunity and vascular involvement in systemic sclerosis (SSc) Autoimmunity. 1999;31:195–214. doi: 10.3109/08916939908994064. [DOI] [PubMed] [Google Scholar]
- 5.Arciniegas E, Neves CY, Carrillo LM, Zambrano EA, Ramírez R. Endothelial-mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium. 2005;12:193–200. doi: 10.1080/10623320500227283. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri V, Zhou L, Karasek M. Inflammatory cytokines induce the transformation of human dermal microvascular endothelial cells into myofibroblasts: a potential role in skin fibrogenesis. J Cutan Pathol. 2007;34:146–53. doi: 10.1111/j.1600-0560.2006.00584.x. [DOI] [PubMed] [Google Scholar]
- 7.Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1–8. doi: 10.1152/ajplung.00378.2006. [DOI] [PubMed] [Google Scholar]
- 8.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–61. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 9.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–7. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kizu A, Medici D, Kalluri R. Endothelial-mesenchymal transition as a novel mechanism for generating myofibroblasts during diabetic nephropathy. Am J Pathol. 2009;175:1371–3. doi: 10.2353/ajpath.2009.090698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, Shimokata K, Hasegawa Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;43:161–72. doi: 10.1165/rcmb.2009-0031OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Qu X, Bertram JF. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol. 2009;175:1380–8. doi: 10.2353/ajpath.2009.090096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goumans MJ, van Zonneveld AJ, ten Dijke P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis? Trends Cardiovasc Med. 2008;18:293–8. doi: 10.1016/j.tcm.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Branton MH, Kopp JB. TGF-beta and fibrosis. Microbes Infect. 1999;1:1349–65. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 15.Prud’homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest. 2007;87:1077–91. doi: 10.1038/labinvest.3700669. [DOI] [PubMed] [Google Scholar]
- 16.Rosenbloom J, Castro SV, Jimenez SA. Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Ann Intern Med. 2010;152:159–66. doi: 10.7326/0003-4819-152-3-201002020-00007. [DOI] [PubMed] [Google Scholar]
- 17.Varga J, Whitfield ML. Transforming growth factor-beta in systemic sclerosis (scleroderma) Front Biosci. 2009;1:226–35. doi: 10.2741/s22. [DOI] [PubMed] [Google Scholar]
- 18.Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, et al. A SNAIL-1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11:943–50. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–74. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 20.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–72. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelialMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–8. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 22.Marelli-Berg FM, Peek E, Lidington EA, Stauss HJ, Lechler RI. Isolation of endothelial cells from murine tissue. J Immunol Methods. 2000;244:205–15. doi: 10.1016/s0022-1759(00)00258-1. [DOI] [PubMed] [Google Scholar]
- 23.Larrivee B, Karsan A. Isolation and culture of primary endothelial cells. Methods Mol Biol. 2005;290:315–29. doi: 10.1385/1-59259-838-2:315. [DOI] [PubMed] [Google Scholar]
- 24.Voyta JC, Via DP, Butterfield CE, Zetter BR. Identification and isolation of endothelial cells based on their increased uptake of acetylated-low density lipoprotein. J Cell Biol. 1984;99:2034–40. doi: 10.1083/jcb.99.6.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol. 1997;137:1403–19. doi: 10.1083/jcb.137.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–61. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 27.Nieto MA. Epithelial-Mesenchymal Transitions in development and disease: old views and new perspectives. Int J Dev Biol. 2009;53:1541–7. doi: 10.1387/ijdb.072410mn. [DOI] [PubMed] [Google Scholar]
- 28.Zhou BP, Deng J, Xia W, et al. Dual regulation of Snail by GSK-3s-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–40. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 29.Doble BW, Woodgett JR. Role of glycogen synthase kinase-3 in cell fate and epithelial-mesenchymal transitions. Cells Tissues Organs. 2007;185(1–3):73–84. doi: 10.1159/000101306. [DOI] [PubMed] [Google Scholar]
- 30.Peinado H, Portillo F, Cano A. Switching on-off Snail: LOXL2 versus GSK3beta. Cell Cycle. 2005;4:1749–52. doi: 10.4161/cc.4.12.2224. [DOI] [PubMed] [Google Scholar]
- 31.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 32.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 33.Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci. 2008;121:3317–24. doi: 10.1242/jcs.028282. [DOI] [PubMed] [Google Scholar]
- 34.Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF- and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–16. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bibi Y, Gottlieb AB. A potential role for imatinib and other small molecule tyrosine kinase inhibitors in the treatment of systemic and localized sclerosis. J Am Acad Dermatol. 2008;59:654–8. doi: 10.1016/j.jaad.2008.04.034. [DOI] [PubMed] [Google Scholar]
- 36.Distler JH, Distler O. Tyrosine kinase inhibitors for the treatment of fibrotic diseases such as systemic sclerosis: towards molecular targeted therapies. Ann Rheum Dis. 2010;69:i48–51. doi: 10.1136/ard.2009.120196. [DOI] [PubMed] [Google Scholar]
- 37.Distler JH, Jungel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 2007;56:311–22. doi: 10.1002/art.22314. [DOI] [PubMed] [Google Scholar]
- 38.Akhmetshina A, Venalis P, Dees C, Busch N, Zwerina J, Schett G, Distler O, Dister JH. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheum. 2009;60:219–224. doi: 10.1002/art.24186. [DOI] [PubMed] [Google Scholar]
- 39.Aono Y, Nishioka Y, Inayama M, Ugai M, Kishi J, Uehara H, et al. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2005;171:1279–85. doi: 10.1164/rccm.200404-531OC. [DOI] [PubMed] [Google Scholar]
- 40.Jimenez SA, Gaidarova S, Saitta B, Sandorfi N, Herrich DJ, Rosenbloom JC, et al. Role of protein kinase C-delta in the regulation of collagen gene expression in scleroderma fibroblasts. J Clin Invest. 2001 Nov;108:1395–403. doi: 10.1172/JCI12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gore-Hyer E, Pannu J, Smith EA, Grotendorst G, Trojanowska M. Selective stimulation of collagen synthesis in the presence of costimulatory insulin signaling by connective tissue growth factor in scleroderma fibroblasts. Arthritis Rheum. 2003 Mar;48:798–806. doi: 10.1002/art.10953. [DOI] [PubMed] [Google Scholar]