

Abstract
The role of DNA methylation of CpG islands in parathyroid tumorigenesis has not been analyzed in an unbiased, systematic fashion. DNA was isolated from normal and pathologic parathyroid tissues, bisulphite modified and analyzed using the Infinium HumanMethylation27 BeadChip. Distinct hierarchical clustering of genes with altered DNA methylation profiles in normal and pathologic parathyroid tissue was evident. Comparing normal parathyroid tissue with parathyroid adenomas, 367 genes were significantly altered, while 175 genes significantly differed when comparing parathyroid carcinomas and normal parathyroid tissues. A comparison between parathyroid adenomas and parathyroid carcinomas identified 263 genes with significantly distinct methylation levels. Results were confirmed for certain genes in a validation cohort of 40 parathyroid adenomas by methylation-specific PCR. Genes of known or putative importance in the development of parathyroid tumors showed significant and frequent hypermethylation. DNA hypermethylation of CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4 was associated with reduced gene expression in both benign and malignant parathyroid tumors. Treatment with 5-aza-2′-deoxycytidine of primary cell cultures restores expression of hypermethylated genes in benign and malignant parathyroid tumors. In conclusion, the unbiased, genome-wide study of the parathyroid tumor DNA methylome identified a number of genes with altered DNA methylation patterns of putative importance to benign and malignant parathyroid tumorigenesis.
INTRODUCTION
Primary hyperparathyroidism (pHPT) is common, especially in postmenopausal females, and population-based screening studies suggests a prevalence of 0.7 to 2.3% (Lundgren et al., 1997; Siilin et al., 2011). The disease is due to a single parathyroid adenoma in about 85% of cases whereas parathyroid carcinoma is rare (0.5–1% of cases). Apart from surgical therapy, there exist no effective treatment for pHPT and parathyroid carcinoma is associated with significant morbidity and mortality (Fraker, 2008). The molecular pathogenesis of sporadic pHPT has been partly elucidated; inactivating somatic mutations of the tumor suppressor genes MEN1 and HRPT2/CDC73 have been identified in a subset of parathyroid tumors (Heppner et al., 1997; Carling et al., 1998). The CCND1 oncogene, now recognized to have a central role in many forms of human neoplasia, was initially identified at the breakpoint of a parathyroid adenoma DNA rearrangement(Arnold and Kim 1989; Motokura et al., 1991), and cyclin D1 overexpression has been detected in 18–40% of sporadic parathyroid adenomas (Hsi et al., 1996; Yi et al., 2008). Aberrations in the Wnt/β-catenin signaling pathway have been identified in parathyroid tumors and an aberrantly spliced, internally truncated variant of LRP5, a co-receptor for Wnt ligands, results in stabilization and accumulation of β-catenin in a majority of parathyroid tumors of pHPT (Bjorklund et al., 2007). Parafibromin, the protein product of HRPT2/CDC73, is a part of the human polymerase associated factor complex (PAF), required for facilitating transcriptional elongation and histone modification (Rozenblatt-Rosen et al., 2005). Additionally, parafibromin down-regulates MYC, suggesting its role as a tumor suppressor through inhibition of Wnt signaling (Mosimann et al., 2006). Moreover, CCND1 is a target of the Wnt/β-catenin signaling pathway (Shtutman et al., 1999).
Growing evidence shows that acquired epigenetic abnormalities, including DNA methylation, along with genetic alterations lead to altered patterns of gene expression/function in tumorigenesis. Much is now known about the importance of promoter cytosine methylation in cytosine phosphate guanine (CpG) islands and gene silencing (Jones and Baylin, 2007). It has been established that such methylation is intimately involved in cancer development (Jones and Baylin, 2007). Characterization of DNA methylation information, collectively denoted the DNA methylome, has recently been successfully employed to characterize the molecular pathogenesis and epigenetically classify both solid and hematological malignancies (Kulis and Esteller 2010).
Hypermethylation of promoter regions has also been analyzed in parathyroid tumors and frequent hypermethylation of RIZ1/PRDM2, CDKN2A, RASSF1A, and APC has been reported (Carling et al., 2003; Juhlin et al., 2010; Svedlund et al., 2010). However, such studies have focused on individual genes or a small cohort of genes. In contrast to most other neoplastic processes, parathyroid adenomas are less aggressive, homogeneous, and diagnosed at an earlier stage because of related hormone excess. Thus, they represent an interesting model system to study epigenetic aberrations involved in tumor development.
The current study represents the first comprehensive, unbiased analysis of quantitative DNA methylation alterations in benign and malignant parathyroid tumors.
MATERIALS AND METHODS
Subjects and Tissues
Parathyroid adenomas (n=51) and carcinomas (n=7) were acquired from pHPT patients diagnosed and surgically treated in the clinical routine at Yale-New Haven Hospital, Uppsala University Hospital or Martin-Luther University Hospital, and clinical characteristics are presented in Supplementary Table 1. Inclusion criteria were inappropriate elevation of PTH in relation to serum calcium, normal creatinine levels, no history of familial hyperparathyroidism or exposure to calcimimetic therapy. All tumors were carefully evaluated and dissected by an experienced endocrine pathologist prior to use in the study. The diagnosis of parathyroid carcinoma was unequivocal with all patients demonstrating widely invasive and/or distant metastatic disease. Normal parathyroid tissue (n=3) was obtained from glands inadvertently removed in conjunction with thyroid surgery where auto-transplantation was not required, or as normal parathyroid gland biopsies inpatients subjected to parathyroidectomy. Therefore, a small number of non-pathologic tissues were available for analysis; however, previous parathyroid studies have utilized similar numbers (Juhlin et al., 2010). Tissues were snap-frozen in liquid nitrogen and stored in −80°C or were prepared for primary cultures. Informed consent and approval by institutional review boards at participating institutions were obtained. All cases were analyzed by Sanger sequencing for germline mutations of the MEN1 gene prior to further analysis, and found to be wild-type.
Comprehensive DNA Methylation Profiling
High molecular weight genomic DNA was isolated from normal and pathological parathyroid tissue, as previously described (Carling et al., 1995). Genomic DNA (500 ng) from normal parathyroid tissue (n=3), parathyroid adenoma (n=14), and cancer (n=7) were simultaneously bisulphite modified using the EZ DNA Methylation kit (Zymo Research, Orange, CA) according to the instructions from the manufacturer and analyzed using Infinium HumanMethylation27 BeadChip (Illumina, San Diego, CA). The Infinium HumanMethylation27 BeadChip protocol comprises six steps (whole-genome amplification, fragmentation, hybridization, washing, counterstaining and scanning) (Thirlwell et al., 2010), which were carried out at the Yale Center for Genome Analysis at Yale University according to the manufacturer’s recommendation. The HumanMethylation27 panel targets CpG sites located within the proximal promoter regions of transcription start sites of 14,475 consensus coding sequencing (CCDS) in the NCBI Database (Genome Build 36). In addition, 254 assays cover 110 miRNA promoters. On average, two assays were selected per CCDS gene and from 3–20 CpG sites for >200 cancer-related and imprinted genes. The platform also allows interrogation of 27,578 highly informative CpG sites per sample at single-nucleotide resolution (Bibikova et al., 2006; Killian et al., 2009). The CpG sites are located in promoter regions, up to 1 kb upstream or 0.5 kb downstream of transcription start sites. The BeadChip was scanned on the Illlumina iScan and the resulting files were analyzed with the Beadstudio software (Version 3.2; Illumina). The output of the Beadstudio analysis is a β-value for each CpG site interrogated. This is a continuous value between 0 and 1, where 0 indicates 0% methylation and 1 indicates 100% methylation at a given CpG site. Therefore, this assay provides quantitative methylation measurement at the single CpG site level. The calculation of the β-value is performed as described (Thirlwell et al., 2010).
Methylation-Specific PCR Analysis Using SYBR Green
To verify the findings from the methylation arrays, 6 highly hypermethylated genes were analyzed using methylation-specific PCR (MSP). Methylated and unmethylated specific primers were designed using the Methyl Primer Express software (Applied Biosystems, Foster City, CA, USA) and the primer sequences are presented in supplementary Table 2. Both unmethylated and methylated specific primers displayed an identical target amplicon. Semi-quantitative PCR was performed using SYBR-Green PCR Master Mix (#4309155) and results were analyzed using StepOne Software v2.1 (Applied Biosystems). Human methylated DNA (Epitect Control DNA; Qiagen, Valencia, CA, USA) was utilized as the reference DNA to quantitatively assess the methylation status of the target CpG island, when using methylated specific primers. Similarly, human demethylated DNA (Epitect Control DNA; Qiagen) was used as reference for the unmethylated specific primers. The relative percentage of the values from the methylated and unmethylated measurements was calculated.
Quantitative RT-PCR Analysis
cDNA was synthesized using 1 μg of total RNA and iScript cDNA Synthesis Kit (Bio-Rad Laboratories Inc. Hercules, CA). Quantitative real-time PCR was performed on StepOnePlus™ Real-Time PCR systems (Applied Biosystems) using assays for CDKN2B (Hs 00793225_m1), CDKN2A (Hs 00233365_m1), WT1 (Hs 01103754_m1), SFRP4 (Hs 00180066_m1), SFRP1 (Hs 00610060_m1), SFRP2 (Hs 293258_m1) and GAPDH (Hs99999905_m1; all from Applied Biosystems). Each cDNA sample was analyzed in triplicate. Standard curves for each experiment were established by amplifying a purified PCR fragment covering the sites for probes and primers.
Primary Cell Cultures, and Treatment With 5-Aza-2′-Deoxycytidine
Parathyroid carcinoma (n=1) and adenoma cells (n=18) were prepared fresh, directly after operation according to published procedures with minor modifications (Carling et al., 2000). Briefly collagenase digestion was performed for 1 h. Cells were then cultured in 35-mm dishes in DMEM, containing 10% fetal bovine serum and penicillin/fungizone/L-glutamine (Sigma-Aldrich St.Louis, MO) and were treated in triplicates with 5 μM 5-aza-2′-deoxycytidine (Sigma-Aldrich). Cell viability was measured on PC1 cells distributed in triplicates (2×104) onto 96 well plates using the cell proliferation reagent WST-1 (Roche Diagnostics GmbH, Mannheim, Germany) (Svedlund et al., 2010). Fresh 5-aza-2′-deoxycytidine was added every 24 h. Cells were harvested after 24 h and 48 h for RNA extraction based upon previously published literature (Svedlund et al., 2010). Primary cell cultures were utilized due to the lack of either parathyroid adenoma or carcinoma cell lines.
Statistical Analysis
Wilcoxon rank test, Student’s unpaired t-test, and Fisher exact test were used for statistical evaluation, with p<0.05 considered to be significant. All results are expressed as mean ± SEM (standard error of the mean).
RESULTS
Comprehensive DNA Methylation Profiling of Parathyroid Tumors
Using a discovery cohort of normal parathyroid tissue, parathyroid adenomas and parathyroid carcinomas we characterized the DNA methylome of these tumors using the Illumina Infinium HumanMethylation27 BeadChip. Comparing normal parathyroid tissue with parathyroid adenomas, 367 genes were significantly altered, while 175 genes differed comparing parathyroid carcinomas and normal parathyroid tissues (P<0.005 for both). A comparison between parathyroid adenomas and parathyroid carcinomas identified 263 genes with distinct methylation levels (P<0.005). Distinct hierarchical clustering of genes with altered DNA methylation profiles in normal parathyroid tissue, parathyroid adenomas, and parathyroid carcinomas was evident by unsupervised clustering (Fig. 1A). Furthermore, when performing unsupervised examination of the top 100 differentially methylated CpG islands, the normal parathyroid tissue displayed low levels of hypermethylation, parathyroid adenomas intermediate levels of hypermethylation, whereas parathyroid carcinomas displayed hypermethylation of all examined CpG islands (Fig. 1B). When examining the known function of annotated genes showing significantly altered DNA methylation levels, genes involved in regulation of apoptosis and cell cycle control as well as ion channels were frequently altered (Fig. 1C). There was no association between DNA methylation levels and patient characteristics such as age, gender and biochemical indices of the disease in the discovery cohort when analyzing the most frequently hypermethylated genes.
Figure 1.
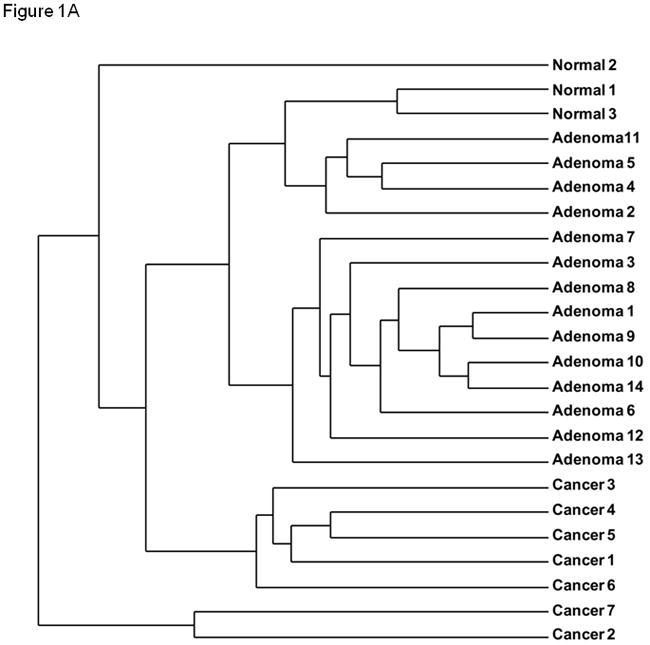
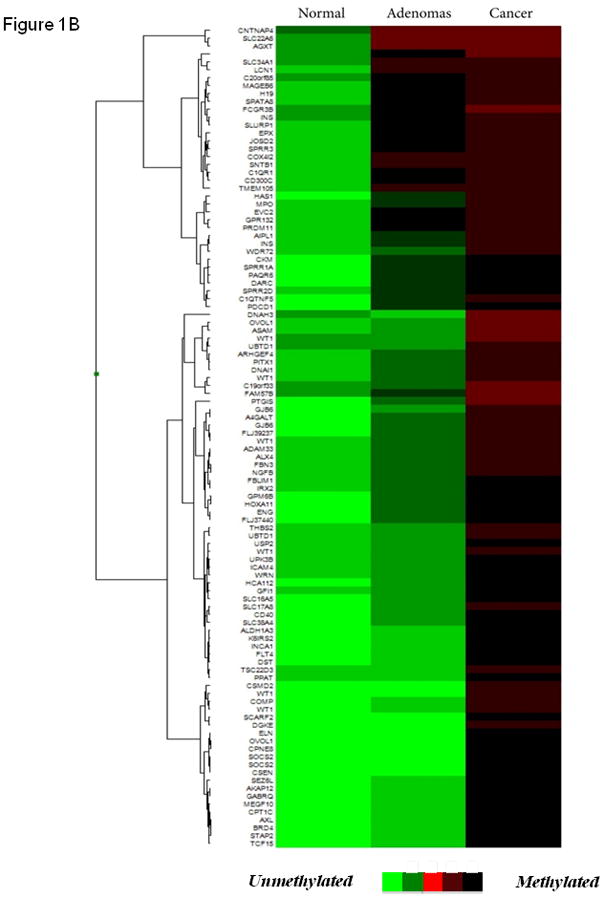
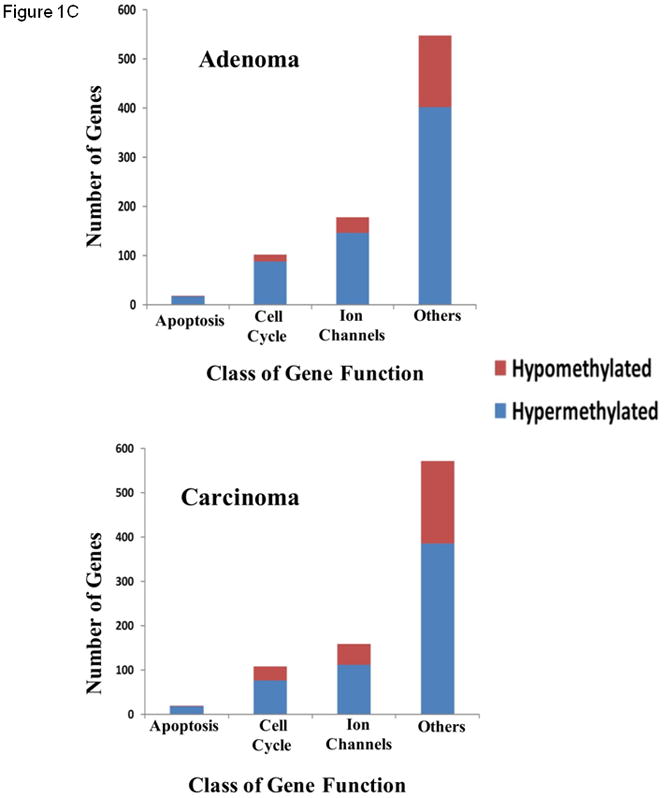
(A) Unsupervised hierarchical clustering of parathyroid specimens analyzed using the genome-wide DNA methylation platform (Illumina Infinium HumanMethylation27 BeadChip). (B) Heat map of unsupervised top 100 differentially methylated CpG islands. Green indicated unmethylated genes (β-value ≤ 0.2), whereas red indicates hypermethylated genes (β-value ≥ 0.6). (C) Functional annotation of genes differentially methylated (diff. β-value ≥ 0.25 and p < 0.05) in parathyroid adenomas (top) and carcinomas (bottom) compared with normal parathyroid tissue.
Genes or members of pathways of known or putative importance in the development of parathyroid tumors showed significant and frequent hypermethylation. Such genes included those involved in regulation of the cell cycle and transcription (CDKN2B, CDKN2A, RB1, WT1, RASSF1A and RIZ1/PRDM2) and members in the Wnt/β-catenin signaling pathway (APC, SFRP1, SFRP2 and SFRP4). Interestingly, significant hypermethylation was seen frequently in parathyroid tumors at multiple examined CpG islands of individual genes, such as CDKN2B, CDKN2A, APC, GATA4, and PYCARD. The top 50 hypermethylated genes in parathyroid adenomas versus normal parathyroid tissue (Table 1), parathyroid carcinomas versus normal parathyroid tissue (Table 2), and parathyroid carcinomas versus parathyroid adenomas (Supplementary Table 3) are presented. For each comparison, the top 25 hypomethylated genes are presented in supplementary Tables 4–6.
TABLE 1.
The Top-50 Most Differentially Hypermethylated Genes in Parathyroid Adenomas Versus Normal Parathyroid Glands.
| Gene symbol | P Value | β-value difference | Function |
|---|---|---|---|
| CDKN2B* | 0.001 | 0.728 | Tumor suppressor |
| RAMP1 | 0.034 | 0.445 | Protein transport |
| KCTD1 | 0.002 | 0.431 | Ion channel |
| SEMA3F | 0.002 | 0.430 | Unknown |
| SOCS3 | 0.006 | 0.417 | Anti- apoptosis |
| ESPN | 0.002 | 0.408 | Cell adhesion |
| SFRP1 | 0.003 | 0.401 | Cell differentiation |
| GJA5 | 0.012 | 0.399 | Connexon channel |
| TNFRSF10C* | 0.011 | 0.381 | Signal transduction, apoptosis |
| NBL1 | 0.003 | 0.0378 | Cell cycle regulator |
| SCARA3 | 0.040 | 0.378 | response to oxidative stress |
| APC* | 0.007 | 0.372 | Tumor suppressor |
| PRDM2 | 0.003 | 0.372 | Transcription factor |
| HCG9* | 0.011 | 0.370 | Unknown |
| CDKN2A | 0.009 | 0.365 | Tumor suppressor |
| MCAM | 0.038 | 0.363 | Cell adhesion |
| SLC2A3 | 0.049 | 0.357 | Glucose transport |
| BHMT2 | 0.001 | 0.355 | Homocysteine S-methyltransferase |
| P8 | 0.007 | 0.354 | Apoptosis |
| PDGFRB | 0.012 | 0.353 | Signal transduction |
| RASSF2 | 0.020 | 0.350 | Signal transduction |
| TMPRSS2 | 0.009 | 0.349 | Proteolysis |
| PIK3R2 | 0.013 | 0.347 | Apoptosis |
| GNB4 | 0.019 | 0.347 | Signal trasduction |
| WT1 | 0.006 | 0.344 | Transcription factor, cell cycle regulator |
| PSKH2 | 0.022 | 0.342 | Kinase, tranferase |
| SLC16A3 | 0.003 | 0.329 | Anion transport |
| CFTR | 0.032 | 0.329 | Ion channel |
| PSTPIP1 | 0.012 | 0.328 | Signal transduction |
| SFRP4 | 0.006 | 0.325 | Cell differentiation |
| GIMAP1 | 0.001 | 0.324 | Immunity associated protein |
| GIMAP5 | 0.015 | 0.320 | Immunity associated protein |
| KCNE3 | 0.005 | 0.320 | Ion transport |
| RASSF1 | 0.004 | 0.319 | Cell cycle regulator |
| ADCY9 | 0.009 | 0.318 | Signal transduction |
| C20orf100 | 0.026 | 0.318 | Transcription |
| ANXA6 | 0.021 | 0.316 | Calcium ion binding |
| SFRP2 | 0.001 | 0.315 | Cell differentiation |
| C12orf34 | 0.003 | 0.315 | Unknown |
| FZD2 | 0.017 | 0.315 | Signal transduction |
| CA3 | 0.020 | 0.314 | Metal ion binding |
| KIF17 | 0.021 | 0.311 | Protein transport |
| TGIF | 0.001 | 0.310 | Transcription |
| WDR72 | 0.023 | 0.309 | Unknown |
| RB1 | 0.006 | 0.309 | Cell cycle regulator, transcription |
| GPR132 | 0.010 | 0.308 | Signal transduction |
| HIST1H4I | 0.016 | 0.306 | Maintenance of chromatin architecture |
| ANGPTL2 | 0.008 | 0.303 | Development |
| DNAJB8 | 0.029 | 0.303 | Protein folding |
| RRP22 | 0.016 | 0.301 | Transduction |
indicates genes where multiple CpG islands were differentially hypermethylated. Genes in bold were analyzed in the validation cohort of parathyroid tumors. β - Value Difference is the difference between the raw β – Values parathyroid adenomas and normal parathyroid tissue.
TABLE 2.
The Top-50 Most Differentially Hypermethylated Genes in Parathyroid Carcinomas Versus Normal Parathyroid Glands.
| Gene symbol | P Value | β - Value difference | Function |
|---|---|---|---|
| CDKN2B* | 0.001 | 0.728 | Tumor suppressor |
| KIAA0323 | 0.023 | 0.679 | Catalytic activity |
| CLDN6 | 0.004 | 0.679 | Cell adhesion |
| CDKN2A* | 0.008 | 0.675 | Tumor suppressor |
| CFTR | 0.001 | 0.664 | Ion transport |
| CMTM2 | 0.002 | 0.659 | Cytokine |
| PYCARD* | 0.018 | 0.653 | Cell cycle regulation, apoptosis |
| UBTD1* | 0.002 | 0.649 | Protein modification |
| BNC1 | 0.027 | 0.643 | Transcription regulation |
| GPC2 | 0.024 | 0.625 | Galactosylceramide sulfotransferase |
| AJAP1 | 0.031 | 0.615 | Unknown |
| LYPD3 | 0.028 | 0.606 | Unknown |
| HIST1HEJ | 0.001 | 0.602 | Maintinence of chromatin structure |
| HOXC11 | 0.005 | 0.598 | Transcription factor |
| SFRP1 | 0.001 | 0.596 | Cell differentiation |
| DGKI | 0.016 | 0.595 | Intracellular signaling |
| SFRP4 | 0.002 | 0.595 | Cell differentiation |
| ABCA3 | 0.001 | 0.595 | Transporter |
| CTPS | 0.007 | 0.585 | Nucleotide metabolism |
| C100rf116 | 0.035 | 0.584 | Unknown |
| WT1* | 0.047 | 0.574 | Transcription factor |
| TCL1A | 0.016 | 0.570 | Development |
| ALS2CR11 | 0.002 | 0.570 | Calcium ion binding |
| DUSP8 | 0.026 | 0.569 | Phosphatase |
| GATA4 | 0.044 | 0.567 | Transcription regulation |
| RRP22 | 0.006 | 0.567 | Signal transduction |
| SPIB* | 0.007 | 0.562 | Transcription factor |
| BTG3 | 0.010 | 0.558 | Cell cycle regulation |
| VILL | 0.048 | 0.552 | Calcium ion binding |
| TAL1 | 0.003 | 0.545 | Transcription regulation |
| WNK4 | 0.027 | 0.542 | Ion transport |
| NHMT | 0.034 | 0.541 | S- methyltransferase |
| SFRP2 | 0.007 | 0.537 | Cell differentiation |
| PSKH2 | 0.018 | 0.536 | Kinase |
| HIST1H4I | 0.006 | 0.536 | Maintinence of chromatin structure |
| hCAP-D3 | 0.028 | 0.534 | Unknown |
| PTPN20B* | 0.003 | 0.530 | Phosphatase |
| C120rf34 | 0.031 | 0.527 | Unknown |
| OTOP3 | 0.025 | 0.524 | Unknown |
| TCF21 | 0.002 | 0.518 | Transcription factor |
| KIAA1822 | 0.049 | 0.517 | Scavenger receptor |
| SAMD4A | 0.036 | 0.516 | Unknown |
| COG4 | 0.017 | 0.516 | Protein transport |
| SCARA3 | 0.011 | 0.514 | Scavenger receptor |
| SLC22A16 | 0.016 | 0.511 | Ion transport |
| SOCS3 | 0.010 | 0.511 | Apoptosis |
| DLEC1 | 0.060 | 0.511 | Cell proliferation |
| C20orf100 | 0.001 | 0.510 | Transcription regulation |
| GEFT | 0.032 | 0.509 | Guanyl- nucleotide exchange factor |
| KCNG3 | 0.001 | 0.507 | Ion transport |
indicates genes where multiple CpG islands were differentially hypermethylated. Genes in bold were analyzed in the validation cohort of parathyroid tumors. β - Value Difference is the difference between the raw β – Values parathyroid carcinomas and normal parathyroid tissue
Verification of Hypermethylation of Selected Genes in Benign and Malignant Parathyroid Tumors
Based on data from the comprehensive DNA methylation profiling of parathyroid tumors, six genes (CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4) showing frequent and significant hypermethylation were selected for further analysis. Validation of promoter hypermethylation by semi-quantitative MSP (SYBR green) was analyzed in the selected genes. CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4 were found to be significantly hypermethylated in both parathyroid adenomas and carcinomas compared to normal parathyroid tissue (Supplementary Fig. 1). Validation of the array results corroborated with highly reproducible results from bisulphite converted DNA allowed us to negate potential batch effect.
Hypermethylated Genes Show Reduced/Lack of Expression in Primary Benign and Malignant Parathyroid Tumors
Next we evaluated whether the hypermethylation of CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4 was related to gene expression in parathyroid tumors. Total RNA was extracted from primary parathyroid tissues and gene expression was analyzed using quantitative RT-PCR. Gene expression of CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4 were significantly reduced in both parathyroid adenomas and carcinomas compared to normal parathyroid tissue (Fig. 2B).
Figure 2.
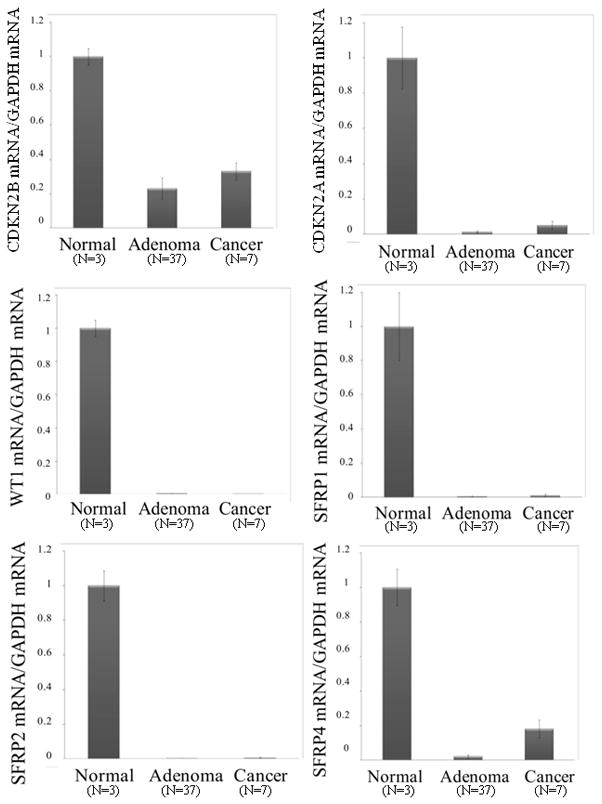
Quantitative RT-PCR of CDKN2A, CDKN2B, WT1, SFRP1, SFRP2 and SFRP4 mRNA in parathyroid tumors of the validation cohort, showing reduced gene expression of all six selected genes in parathyroid adenomas (n=37) and carcinomas (n=7). Reduction of gene expression was significant versus normal parathyroid (p<0.01). Values represent normalized target gene/GAPDH mRNA ratio.
Treatment with 5-Aza-2′-Deoxycytidine of Primary Cell Cultures Restores Expression of Hypermethylated Genes in Benign and Malignant Parathyroid Tumors
In order to examine whether transcriptional repression to the CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4 genes was due to reversible epigenetic alterations, primary cell cultures from parathyroid adenomas (n=18) and a parathyroid carcinoma (n=1) were utilized. Cell cultures were treated with 5-aza-2′-deoxycytidine, an inhibitor of methyltransferase activity causing demethylation, and cells were harvested at 24h and 48h after treatment. Total RNA was prepared and gene expression of the previously demonstrated highly methylated genes CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4 were examined using quantitative RT-PCR. Treatment with 5-aza-2′-deoxycytidine restored measurable gene expression of the hypermethylated genes in cell cultures from parathyroid adenomas (Fig. 3) and the carcinoma (Supplementary Fig. 2). Thereby, indicating that for these 6 genes analyzed, all specimens (18 adenomas and 1 carcinoma) demonstrated evidence of promoter hypermethylation at the CpG island investigated.
Figure 3.
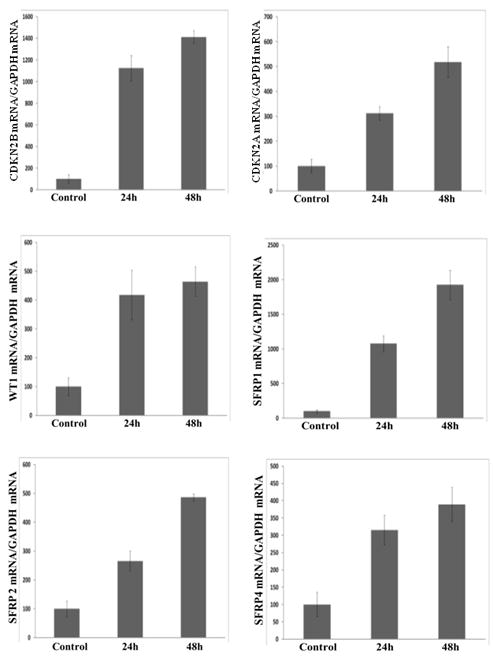
In vitro effect of 5-aza-2′-deoxycytidine treatment of primary cultures of parathyroid adenoma cells (n=18) showing restoration of gene expression of CDKN2A, CDKN2B, WT1, SFRP1, SFRP2 and SFRP4 after treatment at 24 and 48 hours compared to untreated cells (control). Target gene/GAPDH mRNA ratio was determined by quantitative RT-PCR analysis, normalized values are presented. Gene expression restoration was significant in all experiments (P ≤ 0.05)
DISCUSSION
Among epigenetic alterations, promoter hypermethylation of CpG islands, short regions 0.5–4.0 kilobases rich in CpG contents, has emerged as the best described epigenetic changes in tumors. Two key hypotheses have been formulated in understanding epigenetic changes in cancer development. Promoter hypermethylation can silence tumor suppressor genes and aberrant events occur across the genome resulting in specific patterns of gene inactivation contributing to the cancer state. Recently, the appreciation that DNA methylation alterations in tumors are genome-wide events has lead to the development of technologies to study the global patterns of methylation (Noushmehr et al., 2010).
Using the Illumina HumanMethylation27 platform, we characterized the DNA methylome of benign and malignant parathyroid tumors. We found it to be a sensitive, robust and reproducible technique for mapping DNA methylation patterns in parathyroid tumor genomes. Normal parathyroid tissue, adenomas and carcinomas suggested a distinct DNA methylation profile. Genes involved in regulation of apoptosis and cell cycle control as well as ion channels were frequently altered in benign and malignant parathyroid tumors. Overall, there were a higher number of genes that showed a significantly different β-value in parathyroid adenomas vs. normal parathyroid tissue (n=367) than parathyroid carcinomas vs. normal parathyroid tissue (n=175). However, when analyzing the unsupervised top 100 differentially methylated CpG islands it was evident that parathyroid carcinomas displayed a greater degree of hypermethylation as compared to parathyroid adenomas. This discrepancy is most likely due to the fact that a fewer number of the very rare parathyroid carcinomas (n=7) than adenomas (n=14) were available for analysis. It is conceivable that more genes would have reached statistically significant differences in parathyroid carcinomas than adenomas had the number of available cases been equal. Nonetheless, our goal was to identify genes that were highly and frequently altered and the used approach was able to identify such genes. Among the top 50 most hypermethylated genes in both benign and malignant parathyroid tumors were those previously reported to be frequently hypermethylated in parathyroid tumors such as RIZ1/PRDM2, CDKN2A, RASSF1A, and APC. Furthermore, we selected six genes (CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4) based on the array data, their known protein function and previous implication in oncogenesis. Frequent hypermethylation of these genes were validated by semi-quantitative MSP.
CDKN2B and CDKN2A are tumor suppressor genes located on 9p21, and encode a cyclin-dependent kinase inhibitors. They regulate two critical cell cycle regulatory pathways, the TP53 pathway and the retinoblastoma (RB1) pathway. They are frequently deleted in cancer and hypermethylation of the genes occurs in a number of both solid and hematological malignancies (Yu et al., 2008). Studies have failed to identify mutational aberrations of CDKN2B and CDKN2A in parathyroid tumors, although 9p21 is among the 5 most frequently deleted loci in parathyroid tumors (Tahara et al., 1996). The WT1 (Wilm’s tumor 1) gene encodes a transcription factor and plays an important role in cell growth and differentiation and is a well characterized tumor suppressor gene in leukemia and various types of solid tumors. Hypermethylation of WT1 has been demonstrated in cancer, including in breast, ovarian, hepatocellular carcinoma as well as myelodysplastic syndrome ( Huang et al., 1997; Kaneuchi et al., 2005; Yu et al., 2003; Hopfer et al., 2009). The secreted frizzled-related proteins (SFRPs) comprise a family of five secreted glycoproteins that antagonize Wnt signaling and are putative tumor suppressors. Epigenetic inactivation of SFRP genes causing constitutive Wnt signaling was initially demonstrated in colorectal cancer (Suzuki et al., 2004), and has now been substantiated in a number of solid tumors (Dahl et al., 2007). Hypermethylation of CDKN2B, CDKN2A, WT1, SFRP1, SFRP2 and SFRP4 was related to lack/or reduced expression of these genes in both benign and malignant parathyroid tumors. Furthermore, treatment with a 5 μM solution of the demethylating agent 5-aza-2′-deoxycytidine restored gene expression of the hypermethylated genes in primary cell cultures from parathyroid adenomas and a parathyroid carcinoma (only one fresh specimen could be obtained due to the rarity of this specific pathology) suggesting that the epigenetic alterations are reversible.
Our studies substantiate the importance of alterations of cell cycle regulation and transcriptional control in parathyroid tumors via epigenetic inactivation of pathways controlled by CDKN2B and CDKN2A and RB1 and RIZ/PRDM2. The APC tumor suppressor gene has been shown previously to be hypermethylated in parathyroid adenomas and carcinomas (Svedlund et al., 2010), which is in agreement with the results presented here. In the examined parathyroid carcinomas versus normal parathyroid tissue, hypermethylation at 5 different CpG sites of the APC gene was noted with a differential β-value of 0.44 at the most differentially methylated sites. Furthermore, the frequent hypermethylation of SFRP genes likely resulting in constitutive Wnt signaling further implicates an altered Wnt/β-catenin signaling pathway in parathyroid tumorigenesis. This study provides the framework for understanding epigenetic mechanisms contributing to parathyroid tumor development. In addition, it may aid in the notoriously difficult histopathological distinction between benign and malignant parathyroid tumors. Finally, it identifies genes and pathways showing frequent epigenetic alterations, which may be pharmaceutically targeted in metastatic parathyroid carcinoma.
Supplementary Material
Acknowledgments
We thank Sheila Umlauf and Shrikant Mane and the Yale Center for Genome Analysis for assistance in the development of critical methodologies.
Supported by: Ohse Research Award, the Yale Clinical and Translational Science Award UL1 RR024139 from NIH, and Ake Wiberg Foundation. R.P.L. is an Investigator of the Howard Hughes Medical Institute. P.B. is a Swedish Society for Medical Research fellow. T.C. is a Doris Duke-Damon Runyon Clinical Investigator supported in part by the Damon Runyon Cancer Research Foundation and the Doris Duke Charitable Foundation.
References
- Arnold A, Kim HG. Clonal loss of one chromosome 11 in a parathyroid adenoma. J Clin Endocrinol Metab. 1989;69:496–499. doi: 10.1210/jcem-69-3-496. [DOI] [PubMed] [Google Scholar]
- Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, Vollmer E, Goldmann T, Seifart C, Jiang W, Barker DL, Chee MS, Floros J, Fan JB. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–393. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund P, Akerstrom G, Westin G. An LRP5 receptor with internal deletion in hyperparathyroid tumors with implications for deregulated WNT/beta-catenin signaling. PLoS Med. 2007;4:e328. doi: 10.1371/journal.pmed.0040328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling T, Åkerström G, Rastad J, Westin G. Vitamin D receptor (VDR) and parathyroid hormone mRNA levels correspond to polymorphic VDR alleles in human parathyroid tumors. J Clin Endocrinol Metab. 1998;83:2255–2259. doi: 10.1210/jcem.83.7.4862. [DOI] [PubMed] [Google Scholar]
- Carling T, Du Y, Fang W, Correa P, Huang S. Intragenic allelic loss and promoter hypermethylation of the RIZ1 tumor suppressor gene in parathyroid tumors and pheochromocytomas. Surgery. 2003;134(6):932–939. doi: 10.1016/s0039-6060(03)00422-7. discussion 939–940. [DOI] [PubMed] [Google Scholar]
- Carling T, Rastad J, Ridefelt P, Gobl A, Hellman P, Oberg K, Rask L, Larsson C, Juhlin C, Akerstrom G, Skogseid B. Hyperparathyroidism of multiple endocrine neoplasia type 1: candidate gene and parathyroid calcium sensing protein expression. Surgery. 1995;118:924–930. doi: 10.1016/s0039-6060(05)80095-9. [DOI] [PubMed] [Google Scholar]
- Carling T, Szabo E, Bai M, Ridefelt P, Westin G, Gustavsson P, Trivedi S, Hellman P, Brown EM, Dahl N, Rastad J. Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J Clin Endocrinol Metab. 2000;85:2042–2047. doi: 10.1210/jcem.85.5.6477. [DOI] [PubMed] [Google Scholar]
- Dahl E, Wiesmann F, Woenckhaus M, Stoehr R, Wild PJ, Veeck J, Knuchel R, Klopocki E, Sauter G, Simon R, Wieland WF, Walter B, Denzinger S, Hartmann A, Hammerschmied CG. Frequent loss of SFRP1 expression in multiple human solid tumours: association with aberrant promoter methylation in renal cell carcinoma. Oncogene. 2007;26:5680–5691. doi: 10.1038/sj.onc.1210345. [DOI] [PubMed] [Google Scholar]
- Fraker D. Adrenal Tumors. In: DeVita V Jr, Lawrence T, Rosenberg S, editors. Cancer: Principles & Practice of Oncology. 8. Philadelphia: Lippincott-Raven; 2008. pp. 1690–1702. [Google Scholar]
- Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC, Manickam P, Olufemi S-E, Skarulis MC, Doppman JL, Alexander RH, Kim YS, Saggar SK, Lubensky IA, Zhuang Z, Liotta LA, Chandrasekharappa SC, Collins FS, Speigel AM, Burns AL, Mark SJ. Somatic mutations of the MEN1 gene in parathyroid tumours. Nature Genet. 1997;16:375–378. doi: 10.1038/ng0897-375. [DOI] [PubMed] [Google Scholar]
- Hopfer O, Komor M, Koehler IS, Freitag C, Schulze M, Hoelzer D, Thiel E, Hofmann WK. Aberrant promotor methylation in MDS hematopoietic cells during in vitro lineage specific differentiation is differently associated with DNMT isoforms. Leuk Res. 2009;33:434–442. doi: 10.1016/j.leukres.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Hsi E, Zukerberg L, Yang W-I, Arnold A. Cyclin D1/PRAD1 expression in parathyroid adenomas: An immunohistochemical study. J Clin Endocrinol Metab. 1996;81:1736–1739. doi: 10.1210/jcem.81.5.8626826. [DOI] [PubMed] [Google Scholar]
- Huang TH, Laux DE, Hamlin BC, Tran P, Tran H, Lubahn DB. Identification of DNA methylation markers for human breast carcinomas using the methylation-sensitive restriction fingerprinting technique. Cancer Res. 1997;57:1030–1034. [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhlin CC, Kiss NB, Villablanca A, Haglund F, Nordenstrom J, Hoog A, Larsson C. Frequent promoter hypermethylation of the APC and RASSF1A tumour suppressors in parathyroid tumours. PLoS One. 2010;5:e9472. doi: 10.1371/journal.pone.0009472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneuchi M, Sasaki M, Tanaka Y, Shiina H, Yamada H, Yamamoto R, Sakuragi N, Enokida H, Verma M, Dahiya R. WT1 and WT1-AS genes are inactivated by promoter methylation in ovarian clear cell adenocarcinoma. Cancer. 2005;104:1924–1930. doi: 10.1002/cncr.21397. [DOI] [PubMed] [Google Scholar]
- Killian JK, Bilke S, Davis S, Walker RL, Killian MS, Jaeger EB, Chen Y, Hipp J, Pittaluga S, Raffeld M, Cornelison R, Smith WI, Jr, Bibikova M, Fan JB, Emmert-Buck MR, Jaffe ES, Meltzer PS. Large-scale profiling of archival lymph nodes reveals pervasive remodeling of the follicular lymphoma methylome. Cancer Res. 2009;69:758–764. doi: 10.1158/0008-5472.CAN-08-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- Lundgren E, Rastad J, Åkerström G, Ljunghall S. Population-based health screening for primary hyperparathyroidism with serum calcium and parathyroid hormone values in menopausal women. Surgery. 1997;121:287–294. doi: 10.1016/s0039-6060(97)90357-3. [DOI] [PubMed] [Google Scholar]
- Mosimann C, Hausmann G, Basler K. Parafibromin/Hyrax activates Wnt/Wg target gene transcription by direct association with beta-catenin/Armadillo. Cell. 2006;125:327–341. doi: 10.1016/j.cell.2006.01.053. [DOI] [PubMed] [Google Scholar]
- Motokura T, Bloom T, Kim HG, Jüppner H, Ruderman JV, Kronenberg HM, Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350:512–515. doi: 10.1038/350512a0. [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, Copeland TD, Guszczynski T, Resau JH, Meyerson M. The parafibromin tumor suppressor protein is part of a human Paf1 complex. Mol Cell Biol. 2005;25:612–620. doi: 10.1128/MCB.25.2.612-620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siilin H, Lundgren E, Mallmin H, Mellstrom D, Ohlsson C, Karlsson M, Orwoll E, Ljunggren O. Prevalence of primary hyperparathyroidism and impact on bone mineral density in elderly men: MrOs Sweden. World J Surg. 2011 doi: 10.1007/s00268-011-1062-2. (Epub ahead of Print) [DOI] [PubMed] [Google Scholar]
- Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, Toyota M, Tokino T, Hinoda Y, Imai K, Herman JG, Baylin SB. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- Svedlund J, Auren M, Sundstrom M, Dralle H, Akerstrom G, Bjorklund P, Westin G. Aberrant WNT/beta-catenin signaling in parathyroid carcinoma. Mol Cancer. 2010;9:294. doi: 10.1186/1476-4598-9-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara H, Smith A, Gaz R, Arnold A. Loss of chromosome arm 9p DNA and analysis of the p16 and p15 cyclin-dependent kinase inhibitor genes in human parathyroid adenomas. J Clin Endocr Metab. 1996;81:3663–3667. doi: 10.1210/jcem.81.10.8855819. [DOI] [PubMed] [Google Scholar]
- Thirlwell C, Eymard M, Feber A, Teschendorff A, Pearce K, Lechner M, Widschwendter M, Beck S. Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the Illumina Infinium HumanMethylation27 BeadChip. Methods. 2010;52:248–254. doi: 10.1016/j.ymeth.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Yi Y, Nowak NJ, Pacchia AL, Morrison C. Chromosome 11 genomic changes in parathyroid adenoma and hyperplasia: array CGH, FISH, and tissue microarrays. Genes Chromosomes Cancer. 2008;47:639–648. doi: 10.1002/gcc.20565. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang HY, Ma ZZ, Lu W, Wang YF, Zhu JD. Methylation profiling of twenty four genes and the concordant methylation behaviours of nineteen genes that may contribute to hepatocellular carcinogenesis. Cell Res. 2003;13:319–333. doi: 10.1038/sj.cr.7290177. [DOI] [PubMed] [Google Scholar]
- Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–206. doi: 10.1038/nature06468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.