

Abstract
The focus of this investigation was to prepare the cocrystal of carbamazepine (CBZ) using nicotinamide as a coformer and to compare its preformulation properties and stability profile with CBZ. The cocrystal was prepared by solution cooling crystallization, solvent evaporation, and melting and cryomilling methods. They were characterized for solubility, intrinsic dissolution rate, chemical identification by Fourier transform infrared spectroscopy, crystallinity by differential scanning calorimetry, powder X-ray diffraction, and morphology by scanning electron microscopy. Additionally, mechanical properties were evaluated by tensile strength and Heckel analysis of compacts. The cocrystal and CBZ were stored at 40°C/94% RH, 40°C/75% RH, 25°C/60% RH, and 60°C to determine their stability behavior. The cocrystals were fluffy, with a needle-shaped crystal, and were less dense than CBZ. The solubility profiles of the cocrystals were similar to CBZ, but its intrinsic dissolution rate was lower due to the high tensile strength of its compacts. Unlike CBZ, the cocrystals were resistant to hydrate transformation, as revealed by the stability studies. Plastic deformation started at a higher compression pressure in the cocrystals than CBZ, as indicated by the high yield pressure. In conclusion, the preformulation profile of the cocrystals was similar to CBZ, except that it had an advantageous resistance to hydrate transformation.
Key words: carbamazepine, cocrystal, mechanical property, nicotinamide, stability
INTRODUCTION
Carbamazepine (CBZ) was first approved for the US market by the FDA in 1968 under the brand name Tegretol® and currently approved as an anticonvulsant for selected epileptic seizures and for the treatment of pain associated with trigeminal neuralgia (1). CBZ is an iminostilbene derivative, which are structurally related to tricyclic antidepressants, with anticonvulsive and pain reduction due to a decreased potentiation of synaptic transmission in the affected areas (2).
CBZ is classified as a class 2 drug under the biopharmaceutics classification system (3). Class 2 drug substances are characterized as having low solubility in an aqueous media, but a high permeability across the human intestinal membrane or an appropriately predictive in vitro model (4). Therefore, formulation strategies are aimed at improving the solubility and dissolution rates in order to enhance the bioavailability of this highly permeable drug. The existence of a dihydrate and four anhydrous polymorphic forms of CBZ further complicates its formulation (5,6). CBZ polymorphs have been shown to yield different dissolution profiles and a significant difference in pharmacokinetic profiles when studied in dogs (7).
Techniques for physical and chemical alteration for enhanced drug dissolution profiles include complexation of poorly soluble drugs with β-cyclodextrans (8), jet milling to create microparticles (9), high-pressure homogenization to form nanoparticles (10), co-precipitation (11), and co-crystallization (12). Cocrystals are a multicomponent system in which two or more components crystallize into the same crystal lattice with distinctive physicochemical properties from the individual parent compounds. Cocrystals are formed through hydrogen bond interactions between the drug and coformers. Acids and amides have been widely studied as coformers for CBZ cocrystals due to their available hydrogen bonding sites (12,13).
Nicotinamide is a generally recognized as safe class 1 chemical and is often utilized in much larger doses to treat high cholesterol than seen in cocrystal formation (14). Nicotinamide has been utilized as a coformer for the co-crystallization of celecoxib (15), theophylline (16), ibuprofen (17), CBZ (18,19), and lamotrigine (20). Reported methods of cocrystal preparation are solution cooling crystallization (21), ambient co-milling (18), cryogenic co-grinding (22), melting (23), and solvent evaporation (24).
Although CBZ and nicotinamide (NCT) cocrystals have been reported in the literature (18,19), there is a dearth of published information regarding their physicochemical, mechanical, and stability properties. Thus, it is important to evaluate these properties prior to product development in order to select an appropriate dosage form. The primary aim of this study was to evaluate the effect of preparation methods on the quality of the cocrystals. The secondary objective was to evaluate its physicochemical and mechanical properties. Finally, an investigation of the cocrystal’s stability versus CBZ at 40°C/94%, 40°C/75% RH, 25°C/60% RH, and 60°C was performed.
MATERIALS AND METHODS
Materials
CBZ and NCT were purchased from Hangzhou Starshine Pharmaceuticals Co. Ltd. (HongZhou, China) and Acros Organics USA (Morris Plains, NJ, USA), respectively. Methanol, ethanol, acetonitrile, trifluoroacetic acid, beeswax, potassium hydrogen phosphate, potassium chloride, and sodium hydroxide were obtained from Fisher Scientific Co. (Norcross, GA, USA). Hydroxypropyl cellulose-L (HPC-L) and Avicel PH 200 were obtained from Shin-Etsu Chemical Co. Ltd, Tokyo, Japan, and FMC Biopolymer, Newark, DE, USA, respectively. All other chemicals and reagents were of analytical grade or better.
Preparation of Cocrystal
Solution Cooling Crystallization Method
The cocrystal was manufactured by alteration of the reported method (12). Briefly, equimolar weights of CBZ (10.635 g, 0.045 mol) and NCT (5.497 g, 0.045 mol) were added to 500 ml of a round-bottom flask attached to a condenser containing 200 ml of 70:30% (v/v) ethanol/methanol mixture. Solids were dissolved in the solvent by heating at 65°C and refluxed for 1 h while stirring. Stirring was continued and the solution was cooled to room temperature. The cocrystals of CBZ and NCT started to appear in the reaction vessel during the cooling period. The cocrystals were harvested by filtration, washed twice with 20 ml of ethanol, and vacuum oven-dried at 30°C for 48 h. Dried cocrystals were crushed and passed through a sieve 60 ASTM before the characterization studies. These cocrystals were used in solubility and dissolution profile, mechanical, and stability studies.
Solvent Evaporation Method
An equimolar amount of CBZ (2.363 g, 0.01 mol) and NCT (1.221 g, 0.01 mol) were dissolved in 20 ml of ethanol by heating at 50°C while stirring. The stirring and heating were continued until all the solvent evaporated and left the solid residue of cocrystals. Solvent residue was further removed by vacuum oven at 30°C for 48 h. The obtained cocrystals were crushed in a mortar and pestle and passed through a sieve 60 ASTM before characterization.
Melting Method
A modified melting method was utilized based on previously reported technique (23). Equimolar amounts of CBZ (4.726 g, 0.02 mol) and NCT (2.442 g, 0.02 mol) were melted by heating on a liquid paraffin oil bath at 140°C for 10 min. The solid cocrystals were obtained by cooling the melted solution of the components to room temperature. The cocrystals were powdered by a mortar and pestle and passed through 60 mesh screen prior to characterization.
Cryomilling Method
An equimolar ratio of CBZ (1.181 g, 0.005 mol) and NCT (0.611 g, 0.005 mol) was co-ground in the cryomill (SPEX SamplePrep 6770 Freezer/Mill®, SPEX CertiPrep, Metuchen, NJ, USA) with a polycarbonate vial and stainless steel rod, which acted as an impactor for grinding (18). Liquid nitrogen was used as coolant for the mill. The sample was precooled for 2 min before the milling operation. The cryomill was operated for three cycles at 10 rpm/min with 10-min grinding time for each cycle and 2-min cooling period between the cycles. The vial was transferred to a desiccator after cryogrinding to prevent moisture condensation on the sample due to extremely low temperature. The sample was characterized daily by scanning electron microscopy (SEM), differential scanning calorimetry (DSC), X-ray diffraction, and Fourier transform infrared spectroscopy (FTIR) and continued for 7 days.
Solubility
The solubility of CBZ and the cocrystals were determined in water, HCl (pH 1.2), acetate (pH 4.5), and potassium phosphate (pH 6.8) buffers (0.2 M) at 25°C and 37°C, respectively. Excess amounts of CBZ and cocrystals were added to the vials containing the media, sonicated, and vortexed for 20 s. The vials were placed in a horizontal shaker maintained at 100 rpm for 72 h. The solubilized drug was estimated from the samples by filtration through a 0.45-μm membrane filter and diluted with mobile phase and injected into the HPLC system. The HPLC method reported by Bethune et al. (25) was modified to estimate CBZ and the cocrystals. An HP 1050 (Agilent Technologies, CA, USA) HPLC was fitted with quaternary pumps, autosampler, and UV detector set at a wavelength of 254 nm and column temperature was maintained at 26°C. The HPLC stationary phase was composed of a reverse phase Luna C18 (2), 4.6 × 254 mm (5-μm packing) column, and a C18, 4.6 × 12.5 mm (5-μm packing) Luna C18 (2) guard column (Phenomenex Torrance, CA, USA). The composition of the mobile phase was methanol/water containing 0.1% trifluoroacetic acid (55:45) and pumped isocratically at a flow rate of 1.25 ml/min. The HPLC method was validated as per ICH method validation guidance (26).
Intrinsic Dissolution
Intrinsic dissolution was carried by a static disk method (27). Non-disintegrating compact was prepared by a tablet press (Mini Press-1, Globe Pharma Inc., New Brunswick, NJ, USA) using a 8-mm die and flat-faced punches (Natoli Engineering Company Inc., Saint Charles, MO, USA). The die was manually filled with 200 mg of CBZ and cocrystals (303.4 mg) equivalent to 200 mg of CBZ and compressed at 2 ton of force for 30-s dwell time. The die was removed and the lower side opening was covered with beeswax to limit the drug release to the top side of the compact during the study. Each covered die was placed in USP apparatus 2 vessels and oriented so that the covered side of die should be touching the bottom of the vessel. The distance maintained between the paddle and the surface of compact was 2.54 cm. The intrinsic dissolution rate was determined in four media, namely, water, HCl (pH 1.2), acetate (pH 4.5), and potassium phosphate (pH 6.8) buffers (0.2 M) at 37°C and 100-rpm paddle speed. Aliquots of 2 ml were withdrawn at 15, 30, 45, 60, 90, 120, 150, 180, 210, and 240 min and the media not replaced but accounted for in the calculations. Sample was filtered by a 0.45-μm Millipore nylon filter and injected into the HPLC to determine the amount of drug dissolved in the medium.
Characterization Studies
Shape and surface morphology were studied by SEM (JSM-6390 LV, JEOL, Tokyo, Japan) and measured at the working distance of 20 mm and an accelerated voltage of 5 kV. Samples were gold coated with sputter coater (Desk V, Denton Vacuum, NJ, USA) before SEM observation under high vacuum 70 mTorr and high voltage of 30 mV. The cocrystals were chemically identified by FTIR. The spectra were collected by attenuated total reflectance-FTIR (Thermo Nicolet Nexus 670 FTIR, GMI Inc., Ramsey, Minnesota, USA) in absorbance mode with 50 scan and 4.0 resolutions. OMNIC ESP software (version 5.1) was used to capture and analyze the spectra. The cocrystals were also characterized for crystallinity and polymorphicity by powder X-ray diffraction. Diffractograms were obtained on an X-ray diffractometer (MD-10 mini diffractometer, MTI Corporation, Richmond, CA, USA) using Cu K2α rays (λ = 1.54056 Å) with a voltage of 25 kV and a current of 30 mA, in flat plate θ/2θ geometry, over the 2θ range 15–75°, and diffraction pattern was recorded for 20 min. Thermal properties were also investigated by DSC and thermal gravimetric analysis (TGA). DSC was measured with a SDT 2960 Simultaneous DSC/TGA (TA Instruments Co., New Castle, DE, USA). The temperature scanning rate was 10°C/min and scanned up to 250°C. This temperature range covered the melting point of both cocrystal components. Nitrogen gas was flowing at a pressure of 20 psi to provide inert atmosphere during the measurement to prevent oxidation reaction. TGA of the samples were collected at a heating rate of 10°C/min and scanned up to 300°C.
Mechanical Property Characterization
Compaction and compression characteristics of the CBZ and cocrystals were studied by measuring the tensile strength and Heckel analysis of the compacts. The cocrystal compact laminated immediately upon ejection from die. This problem was solved by adding 10% Avicel PH 200 and 5% HPC-L to both the CBZ and cocrystals. The compact was prepared at nine different compression pressures (500–3,000 lb/cm2). Six compacts were prepared at each compression pressure by manually filling 200 mg of powder into 8-mm die and flat punches. Diameter, weight, thickness, and hardness (VK 200 Tablet hardness tester Varian Inc., Palo Alto, CA, USA) of the compacts were determined. The following equation was used to determine tensile strength, Q (MPa) (28):
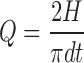 |
where H is the compact hardness (N), d the diameter, and t the thickness of the compact.
Plastic deformation of the compact is described by the Heckel equation (29):
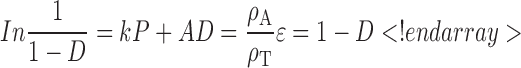 |
D is the relative density at pressure P, ρT and ρA are the true density and density at pressure P, respectively, ε is porosity, and k and A are the Heckel constants. k is related to the plasticity of compressed material and A related to die filling and particle rearrangement before deformation and bonding of discrete particles (30).
The true powder density of the CBZ and cocrystals with 10% (w/w) Avicel and 5% (w/w) HPC-L was determined by helium pycnometer (Ultrapycnometer 1000, Quantachrome Instruments, FL, USA). Standard steel spheres were used to calibrate the instrument. The sample was accurately weighed and transferred into a sample cell and the volume of sample measured by filling the sample chamber with ultrapure helium gas followed by discharge of the gas into a second empty chamber. The measurements were repeated four times per sample.
Stability
The stability of the CBZ and cocrystals was assessed by storing them at stressed, accelerated, and long-term conditions of ICH. The stressed condition were 60°C for 2 weeks and 40°C/94% RH for 1 month; accelerated and long-term conditions were 40°C/75% RH and 25°C/60% RH for 2 months, respectively. A saturated solution of potassium dihydrogen phosphate was used to maintain 94% RH to study the tendency of the CBZ and cocrystal conversion to their dihydrate forms. About 350 mg of the CBZ and cocrystals was placed as a thin layer in 20-ml open glass vials and placed at various conditions for stability studies. At the end of the studies, the samples were characterized for potency, crystallinity by powder X-ray diffraction (PXRD) and DSC, and dihydrate formation by TGA, DSC, and FTIR. Literature method (31) was modified to prepare the standard dihydrate form of CBZ. A 500-ml solution of carbamazepine (0.4 mg/ml) was prepared by heating at 70°C. The hot solution was filtered, refrigerated to form the hydrate of CBZ, and was characterized by FTIR, PXRD, DSC, and TGA.
RESULTS AND DISCUSSION
Solubility
The results of solubility of the CBZ and cocrystals are shown in Table I. There was no significant (p > 0.05) difference between the solubility of the parent drug and cocrystals. Essentially, the cocrystal is a single molecular entity in the solid state and its components are associated through hydrogen bonding, which dissociate into individual components upon contact with water and/or solvents. Nicotinamide has no effect on the solubility of the cocrystals in spite of being highly soluble in water (1 g/ml) (20). Thus, the solubility of the cocrystal is the intrinsic solubility of individual components in this case. A similar trend was observed on the solubility of the cocrystal of lamotrigine with nicotinamide as a coformer (20).
Table I.
Results of Solubility of CBZ and Cocrystal in Different Media
| Medium | CBZ (μg/ml) | Cocrystal (μg/ml) | ||
|---|---|---|---|---|
| 25°C | 37°C | 25°C | 37°C | |
| HCl buffer, pH 1.2 | 141.95 ± 1.52 | 243.09 ± 25.74 | 146.81 ± 2.36 | 235.74 ± 8.08 |
| Acetate buffer, pH 4.5 | 148.89 ± 6.25 | 243.49 ± 14.27 | 148.56 ± 1.53 | 237.29 ± 1.90 |
| Phosphate buffer, pH 6.8 | 136.80 ± 3.49 | 238.09 ± 23.11 | 139.74 ± 4.64 | 234.11 ± 10.95 |
| Water | 143.56 ± 5.14 | 238.70 ± 18.50 | 141.43 ± 1.05 | 241.25 ± 9.26 |
Values shown are the mean ± SD
Intrinsic Dissolution
Figure 1 shows the dissolution profile of CBZ and cocrystal, and their respective intrinsic dissolution rate (IDR) were 0.0357 ± 0.0006, 0.0421 ± 0.0017, 0.0350 ± 0.0002, and 0.0347 ± 0.0047 mg min−1 cm−2 and 0.0238 ± 0.0031, 0.0191 ± 0.0009, 0.0207 ± 0.0003, and 0.0184 ± 0.0002 mg min−1 cm−2 in water, HCl (pH 1.2), acetate (pH 4.5), and phosphate (pH 6.8) buffers, respectively. The statistically insignificant (p > 0.05) effect of media on the IDR of CBZ and cocrystals was observed. The higher IDR of CBZ and cocrystals at pH 1.2 and 4.5, respectively, might be explained by the variation in the compression force applied during the preparation of the compacts. Surprisingly, the cocrystals showed a lower intrinsic dissolution rate in all the dissolution media when compared with CBZ. It was expected that IDR of the cocrystal would be similar to the parent drug because it has the same pH solubility profile. This difference in the IDR might be related to the effect of the crystal habit on the mechanical properties of the cocrystals and CBZ.
Fig. 1.

Intrinsic dissolution profile of CBZ (a) and cocrystals (b) in different media
Fourier Transform Infrared Spectroscopy
Figure 2 shows the FTIR spectrum of CBZ, NCT, their physical mixture, and cocrystal prepared by different methods. The stretching of the primary amide group in CBZ was represented by absorption bands at 3,465 and 3,157 cm−1, respectively, which could be assigned to the free anti-NH (asymmetrical) and hydrogen-bonded syn-NH (symmetrical) vibration (5,32). The absorption band at 1,677 cm−1 was due to carbonyl (−C=O) stretching. Peaks due to C=C– and –C=O vibration and −NH deformation have appeared at 1,605 and 1,593 cm−1 (5,6). It indicated that the CBZ used in the studies was polymorph III (5). Its dihydrate form showed the carbonyl (−C=O) and NH stretching absorption bands at 1,677 and 3,430 cm−1. The shift of NH stretching to a lower wavenumber is due to hydrogen bonding between free anti-NH (asymmetrical) of CBZ and oxygen atom of water (22). Similarly, NCT showed asymmetrical and symmetrical stretching vibrations due to the −NH2 group at 3,353 and 3,143 cm−1, respectively. Absorption bands due to carbonyl (−C=O) and −CN stretching vibrations appeared at 1,672 and 1,392 cm−1, respectively (33).
Fig. 2.

FTIR spectra of CBZ, NCT, and cocrystals
The cocrystals prepared by the solution cooling crystallization method showed changes in the peak positions of carbonyl and amide groups, which suggested interactions between the CBZ and NCT and a new phase formation. The shift in the carbonyl stretching of NCT and CBZ was observed at 1,655 and 1,681 cm−1 in the cocrystals. Additionally, it also showed a shift in the N–H vibration band at 3,386 and 3,446 cm−1. The cocrystals prepared by the solvent evaporation and melting method also showed characteristic peaks of the cocrystals at 1,655, 1,681, 3,386, and 3,446 cm−1, which indicated hydrogen bond formation between CBZ and NCT. Interestingly, a peak appeared at 3,465 cm−1, which corresponded to an absorption band of asymmetrical NH stretching of CBZ. This indicated the presence of unreacted CBZ which had not transformed into the cocrystals.
The FTIR of the cryomilled sample was taken immediately after milling and showed a spectrum similar to their equimolar physical mixture (Fig. 2), which was an additive spectrum encompassing the peaks of both the CBZ and NCT. After 1 day, the cryomilled sample stored at room temperature started to show the characteristic peaks of the cocrystal at 1,655, 1,681, 3,386, and 3,446 cm−1. Additionally, peaks of the asymmetrical NH stretching vibration band of the CBZ appeared at 3,465 cm−1. These data indicated an incomplete transformation of CBZ and NCT into its cocrystal form. The intensity of this peak diminished and the intensity of the cocrystal peaks increased in the 7-day cryomilled sample. This conversion of the cryomilled sample into cocrystals can be explained by an amorphous form generation phenomenon. It is generally accepted that the amorphous form of a substance has a higher molecular mobility than its crystalline counterpart (34). Increased mobility caused more interaction between the CBZ and NCT molecules through hydrogen bonding. The mobility of a molecule in amorphous materials and its transformation to the crystalline form is directly proportional to their storage temperature and duration. Cryomilling did not generate the cocrystals in spite of producing their amorphous forms due to the extremely low temperature of the process which restricts the molecular mobility and their interaction to produce the cocrystals (22).
Three polymorphs of the cocrystal of CBZ and NCT were reported in the literature, namely, CBZ-NCT-I, CBZ-NCT-II, and PN-CBZ-NCT. The cocrystals prepared by different methods showed N–H vibration bands at 3,386 and 3,446 cm−1. This is in agreement with the literature report that the polymorph cocrystals are the CBZ-NCT-I form (19).
Powder X-Ray Diffraction
Diffractograms of the CBZ, NCT, and cocrystals are shown in Fig. 3. The diffractogram of CBZ and its dihydrate forms showed sharp peaks at 2θ values of 20.01°, 21.40°, 23°, 24.14°, 26.50°, 28.42°, 31.15°, and 45.21° and 18.39°, 18.85°, 21.41°, 24.20°, 30.29°, 31.61°, and 36.51°, respectively. Similarly, the diffractogram of NCT showed peaks at 2θ values of 21.67°, 22.63°, 24.81°, 26.43°, 29.91°, 31.57°, 33.55°, 35.88°, 37.52°, and 40.28°. Their physical mixture encompassed the peaks of CBZ and NCT. The cocrystal prepared by solubility, solvent evaporation, and melting methods showed a distinct peak of the cocrystals at 25.77° which was absent either in the CBZ or NCT. This indicated the formation of a new crystalline phase. An initial diffractogram of the cryomilled sample showed diffuse and low-intensity peaks of NCT and CBZ, which were similar to their physical mixture. This diffuse diffractogram pointed to the amorphous form of the materials. The 7-day cryomilled sample stored at room temperature started to show characteristic peaks of the cocrystal at 2θ values of 20.05° and 25.77°, and peaks corresponding to CBZ and NCT diminished with storage time. This could be explained by the amorphous material-mediated transformation of the CBZ and NCT into their cocrystals.
Fig. 3.

PXRD diffractograms of CBZ, NCT, and cocrystals
Thermal Property
DSC thermograms of pure components and the cocrystals prepared by various methods are shown in Fig. 4a. The thermogram of CBZ showed endotherm at 165.4°C followed immediately by a weak exotherm and another endotherm at 191°C. These thermal events could be explained by the polymorphic transformation of form III to form I. The first endotherm was attributed to the melting of form III, following exotherm explained by crystallization to form I, and the subsequent endotherm was due to its melting (5). This further proved that the starting CBZ was form III. Similarly, NCT showed sharp melting endothermic peaks at 130.6°C. Equimolar physical mixture of CBZ and NCT showed an endothermic peak at 127.4°C and 159°C, which corresponds to the melting point of NCT and the cocrystal that were formed by melt fusion phenomenon, respectively. The dihydrate form of CBZ showed shallow broad twin peaks at 92.8°C and 105.85°C that could be ascribed to the loss of water molecules from its crystal lattice and later on the evaporation of water. It also showed a melting peak of the CBZ at 191°C. The cocrystals prepared by the solution cooling crystallization and solvent evaporation methods showed sharp melting peaks at 160°C and 160.3°C, respectively. These peaks were different from either the melting point of the NCT or CBZ which indicated the formation of a new crystalline phase. The melting method gives a broad exothermic and a sharp endothermic peak at 96.38°C and 159.2°C which could be suggestive of the crystallization and melting of the cocrystals. This indicated that the melting method of cocrystal formation yields a mixture of amorphous and crystalline forms during the cooling step of the melt. This could be explained by the rapid solidification of the melt which prevented proper fitting of the molecules into the crystalline lattice of the cocrystals. This was further supported by their enthalpy of fusion which was 87.08, 150.6, and 150.4 J/g for the melting, solvent evaporation, and solution cooling crystallization methods of the cocrystal formation, respectively. The crystalline form of material has a higher enthalpy of fusion than its amorphous or mixture of amorphous and crystalline forms (35). The DSC thermogram of the cryomilled sample immediately after milling showed a broad exothermic peak and two endothermic peaks. An exothermic peak appeared at 101.2°C, which could be assigned to the crystallization peak of the cocrystals. One broad and small endothermic peak showed up at 123.6°C and another sharp peak at 159.7°C that corresponded to the melting point of the NCT and cocrystals. Furthermore, the presence of the NCT peak at a lower melting point indicated the presence of either a very small quantity of unreacted amorphous or crystalline and/or a mixture of both forms of NCT in the sample. The presence of the cocrystal peak could be explained by an amorphous form reversion to the crystalline form upon heating. Cryomilling produced the amorphous form of the CBZ and NCT. The amorphous form has a higher molecular mobility, and this mobility of molecules increases with temperature and causes an interaction between the amorphous NCT and CBZ to produce the cocrystals. The 7-day cryomilled sample showed only two endothermic peaks at 120.8°C and 159.2°C. The absence of the crystallization peak indicated that the amorphous cocrystal had transformed into its crystalline form during storage. Additionally, the increase in the enthalpy of fusion from 122.5 to 145.7 J/g of the initial and 7-day cryomilled sample, respectively, supports this transformation.
Fig. 4.

DSC (a) and TGA (b) thermograms of CBZ, NCT, and cocrystals
TGA thermograms of the CBZ, NCT, and cocrystals prepared by various methods are shown in Fig. 4b. Pure components and the cocrystals started losing their weight immediately after passing their melting points. The weight loss profiles of all the cocrystals were similar irrespective of their methods of preparation. The dihydrate form of CBZ was similar to its anhydrous form, except that it lost about 13.20% from 60°C to 85°C, which corresponded to the loss of two water molecules from its crystalline lattice.
Scanning Electron Microscopy
The morphology of CBZ, NCT, and the cocrystal prepared by various methods are shown in Fig. 5. The CBZ showed broken crystals, while NCT showed pebble-shaped crystals. The cocrystals prepared by the solution cooling crystallization and solvent evaporation methods showed a needle-shaped structure, which is in agreement with the reported shape of CBZ and NCT cocrystals (19). The cocrystal prepared by the melting method showed irregular-shaped crystals which are probably due to the milling process. The initial cryomilled sample showed an amorphous-looking lump (not shown), and the 7-day sample showed needle-shaped crystals similar to the solution cooling crystallization and solvent evaporation methods.
Fig. 5.

SEM photomicrographs of CBZ (a), NCT (b), solution cooling crystallization method (c), solvent evaporation (d), cryomilling (e), and melting method (f) cocrystals
Compressibility and Compaction Characteristics
The cocrystals were fluffy and bulky powders as revealed by a lower true density value (1.337 g/cm3) when compared to the CBZ powder (1.381 g/cm3). The impact of compression pressure was more prominent on the cocrystal tensile strength than CBZ (Fig. 6a). It was evident from the high tensile strength of the cocrystal compact subjected to the same compression pressure as the CBZ compact. Thus, the cocrystal powder is more suitable for tabletting than the CBZ. Furthermore, good correlation were identified in the compacts between the tensile strength and compression pressure, as indicated by the high R2 value of 0.984 and 0.992 for CBZ and the cocrystals, respectively. The high tensile strength of cocrystal would probably explain its lower intrinsic dissolution rate than CBZ. It is generally accepted that the tensile strength of a compact and dissolution rate holds an inverse relationship, and this has been reported by investigators for both hydrophilic and hydrophobic drugs (35–37).
Fig. 6.

Tensile strength plot (a) and Heckel plots (b) of CBZ and cocrystal
Both CBZ and its cocrystal showed typical Heckel plots, which plot the compression pressure and logarithm of reciprocal porosity In(1/1 − D), as indicated by the initial curvature followed by a linear curve (Fig. 6b). The curvature region in the plot is due to the initial movement and rearrangement of particles in the die. The Heckel plot became linear after 385 and 910 lb/cm2 compression pressure for CBZ and the cocrystals, respectively (Table II). Additionally, the slope of the plot was greater in the case of CBZ than the cocrystals, resulting in greater yield pressure, which is the minimum pressure to cause plastic deformation. Thus, the cocrystals have a higher yield pressure than CBZ, which suggests that plastic deformation started at higher pressure.
Table II.
Results of Heckel and Tensile Strength Equation Analysis of CBZ and Cocrystal
| Drug | Heckel equation constant | Tensile strength, σ 0 (MPa) | b | |||
|---|---|---|---|---|---|---|
| k (1/lb/cm2) | Yield pressure (lb/cm2) | A | R 2 | |||
| CBZ | 1.16 × 10−3 | 859.22 | 0.579 | 0.960 | 9.85 | 6.85 |
| Cocrystal | 1.48 × 10−3 | 676.69 | 0.477 | 0.985 | 27.89 | 6.86 |
Porosity reduction was measured to indicate the compressibility of the powders. The compressibility of CBZ was higher than the cocrystals, as indicated by the lower porosity value for CBZ at any compression pressure when compared to the cocrystals (Fig. 7a). This phenomenon could be explained by the difference in the densities of CBZ and cocrystal (true, bulk, and tapped densities of CBZ and cocrystal were 1.381, 1.337, 0.452, 0.174, 0.782, and 0.292 g/cm3, respectively).
Fig. 7.

Porosity (a) and compaction plots (b) of CBZ and cocrystal
The porosity decreased exponentially with increased tensile strength. The relation between tensile strength and porosity is given by Rayshkewitch (38):
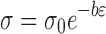 |
where σ is the tensile strength, σ0 is the tensile strength at zero porosity, and ε is the porosity. b is a constant related to the pore distribution in the compact, and its high value is indicative of higher yield stress or anisotropic deformation (39). A good correlation was obtained between Inσ and porosity for both the cocrystal (0.971) and CBZ (0.979; Fig. 7b). Tensile strength at zero porosity was higher for the cocrystal than CBZ. Similarly, the value of b was higher for the cocrystals (Table II), which indicated a non-homogenous pore distribution in the cocrystal compact. This might be due to a better crystal lattice and anisotropic deformation behavior of the cocrystals.
Stability
The potency results (Table III) indicate that the chemical stability of the cocrystals was similar to carbamazepine. DSC, TGA, FTIR, and PXRD thermograms and spectra of the cocrystals stored at 60°C, 25°C/60%, 40°C/75% RH, and 40°C/94% RH showed no change in their thermograms and spectra when compared with the initial sample. These results suggested that cocrystals are resistant to hydrate transformation when exposed to extreme conditions of storage. Likewise, CBZ showed the same behavior of stability as that of the cocrystal at all the conditions of stability testing, except at 40°C/94% RH.
Table III.
Potency Results of CBZ and Cocrystal
| Drug | Initial | 60°C (2 weeks) | 25°C/60% RH (2 months) | 40°C/75% RH (2 months) |
|---|---|---|---|---|
| CBZ | 97.68 ± 0.93 | 97.75 ± 0.84 | 98.66 ± 0.13 | 97.71 ± 0.75 |
| Cocrystal | 99.21 ± 1.28 | 96.24 ± 1.90 | 97.64 ± 0.31 | 97.55 ± 0.44 |
Values shown are the mean ± SD
The FTIR spectrum of CBZ stored at 40°C/94% RH showed absorption bands at 3,430 and 3,465 cm−1, which could be assigned to the NH stretch of the anhydrous and the dihydrate forms of CBZ (Fig. 8a). This indicated that the sample was a mixture of anhydrous and dihydrate forms of the CBZ and that only a partial conversion took place during storage at high humidity. This was further supported by the PXRD diffractogram of CBZ stored at high humidity. Its diffractogram encompassed the peaks of anhydrous and the dihydrate forms of CBZ, as indicated by the major peaks of anhydrous CBZ at 24.27° and 26.43° and dihydrate CBZ at 36.47° (Fig. 8b). This finding was further supported by thermal studies such as DSC and TGA. The TGA thermogram of CBZ stored at high humidity lost about 5.1% of its weight from 60°C to 85°C, which indicated water loss from the CBZ sample (Fig. 3b). It was assumed that it is a crystalline water of the dihydrate form of CBZ and calculated to represent 38.58% transformation of the anhydrous CBZ to its dihydrate form. This assumption was further proven by DSC studies. It showed two small and broad endothermic peaks shown by the dihydrate forms of CBZ at 85.81°C and 97.32°C which correspond to the dehydration of the water form of the crystal lattice of CBZ and subsequent evaporation (Fig. 9). Physically adsorbed water would show a single peak at lower temperature, and these findings indicated that CBZ stored at 40°C/94% RH has partially transformed to its dihydrate form.
Fig. 8.

FTIR spectra (a) and PXRD diffractograms (b) of CBZ, CBZ dihydrate, and cocrystal before and after storage at 40°C/94% RH
Fig. 9.

DSC thermograms of CBZ, CBZ dihydrate, and cocrystal before and after storage at 40°C/94% RH
CONCLUSION
The cocrystal of CBZ and NCT showed physicochemical properties similar to the parent CBZ compound and a superior chemical stability to resist conversion to its dihydrate form. This transformation has quality and performance implications as the dihydrate form of CBZ has approximately half the dissolution rate as the anhydrous form (31). Thus, the cocrystal form of CBZ provides another approach to product development, with greater consistency of physicochemical quality attributes resulting in stable products when exposed to severe processing conditions and environmental changes.
Acknowledgment
The authors would like to thank the Oak Ridge Institute for Science and Education (ORISE) for supporting the post doctoral research program. The authors also thank Mr. Alan Carlin for helping with the FTIR spectrum.
Disclaimer
The views and opinions expressed in this paper are only those of the authors and do not necessarily reflect the views or policies of the FDA.
References
- 1.Food and Drug Administration. Drugs@FDA. Label and Approval History of Tegretol. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Label_ApprovalHistory. Accessed 02 September 2010.
- 2.Granger P, Biton B, Faure C, Vige X, Depoortere H, Graham D, Langer SZ, Scatton B, Avenet P. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol Pharmacol. 2005;47:1189–1196. [PubMed] [Google Scholar]
- 3.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernäs H, Hussain AS, Junginger HE, Stavchansky SA, Midha KK, Shah VP, Amidon GL. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm. 2003;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 4.Food and Drug Administration. Guidance for Industry, Waiver of in vitro bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. 2000.
- 5.Grzesiak AL, Lang M, Kim K, Matzger AJ. Comparison of the four anhydrous polymorphs of carbamazepine and the crystal structure of form I. J Pharm Sci. 2003;92:2260–2271. doi: 10.1002/jps.10455. [DOI] [PubMed] [Google Scholar]
- 6.Rustichelli C, Gamberini G, Ferioli V, Gamberinig MC, Ficarra R, Tomasini S. Solid-state study of polymorphic drugs: carbamazepine. J Pharm Biomed Anal. 2000;23:41–54. doi: 10.1016/S0731-7085(00)00262-4. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi Y, Ito S, Itai S, Yamamoto K. Physicochemical properties and bioavailability of carbamazepine polymorphs and dihydrate. Int J Pharm. 2000;193:137–146. doi: 10.1016/S0378-5173(99)00315-4. [DOI] [PubMed] [Google Scholar]
- 8.Rahman Z, Zidan AS, Khan MA. Risperidone solid dispersion for orally disintegrating tablet: its formulation design and non-destructive methods of evaluation. Int J Pharm. 2010;400:49–58. doi: 10.1016/j.ijpharm.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 9.Hoyer H, Schlocker W, Krum K, Bernkop-Schnnrch A. Preparation and evaluation of microparticles from thiolated polymers via air jet milling. Eur J Pharm Biopharm. 2008;69:476–485. doi: 10.1016/j.ejpb.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Hecq J, Deleers M, Fanara D, Vranckx H, Boulanger P, Le Lamer S, Amighi K. Preparation and in vitro/in vivo evaluation of nano-sized crystals for dissolution rate enhancement of ucb-35440-3, a highly dosed poorly water-soluble weak base. Eur J Pharm Biopharm. 2006;64:360–368. doi: 10.1016/j.ejpb.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Khan MA, Karnachi AA, Agarwal V, Vaithiyalingam SR, Nazzal S, Reddy IK. Stability characterization of controlled release coprecipitates and solid dispersions. J Control Release. 2000;63:1–6. doi: 10.1016/S0168-3659(99)00172-8. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez-Hornedo N, Nehm SJ, Seefeldt KF, Falkiewicz CF. Reaction crystallization of pharmaceutical molecular complexes. Mol Pharm. 2006;3:362–367. doi: 10.1021/mp050099m. [DOI] [PubMed] [Google Scholar]
- 13.Weyna DR, Shattock T, Vishweshwar P, Zaworotko MJ. Synthesis and structural characterization of cocrystals and pharmaceutical cocrystals: mechanochemistry vs slow evaporation from solution. Cryst Growth Des. 2009;9:1106–1123. doi: 10.1021/cg800936d. [DOI] [Google Scholar]
- 14.Food and Drug Administration. Database of select committee on GRAS substances (SCOGS) reviews: niacinamide (nicotinamide). Report No. 108. http://www.accessdata.fda.gov/scripts/fcn/fcnDetailNavigation.cfm?rpt=scogsListing&id=221. Accessed 02 September 2010.
- 15.Remenar JF, Peterson ML, Stephens PW, Zhang Z, Zimenkov Y, Hickey MB. Celecoxib:nicotinamide dissociation using excipients to capture the cocrystal’s potential. Mol Pharm. 2007;4:386–400. doi: 10.1021/mp0700108. [DOI] [PubMed] [Google Scholar]
- 16.Lu J, Rohani S. Preparation and characterization of theophylline–nicotinamide cocrystal. Org Pro Res Dev. 2009;13:1269–1275. doi: 10.1021/op900047r. [DOI] [Google Scholar]
- 17.Berry DJ, Seaton CC, Clegg W, Harrington RW, Coles SJ, Horton PN, Hursthouse MB, Storey R, Jones W, Friscic T, Blagden N. Applying hot-stage microscopy to co-crystal screening: a study of nicotinamide with seven active pharmaceutical ingredients. Cryst Growth Des. 2008;8:1697–1712. doi: 10.1021/cg800035w. [DOI] [Google Scholar]
- 18.Chieng N, Hubert M, Saville D, Rades T, Aaltonen J. Formation kinetics and stability of carbamazepine–nicotinamide cocrystals prepared by mechanical activation. Cryst Growth Des. 2009;9:2377–2386. doi: 10.1021/cg801253f. [DOI] [Google Scholar]
- 19.Porter W, Elie S, Matzger A. Polymorphism in carbamazepine cocrystals. Cryst Growth Des. 2008;8:14–16. doi: 10.1021/cg701022e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheney ML, Shan N, Healey ER, Hanna M, Wojtas L, Zaworotko MJ, Sava V, Song S, Sanchez-Ramos JR. Effects of crystal form on solubility and pharmacokinetics: a crystal engineering case study of lamotrigine. Cryst Growth Des. 2010;10:394–405. doi: 10.1021/cg901010v. [DOI] [Google Scholar]
- 21.Trask AV, Motherwell SWD, Jones W. Physical stability enhancement of theophylline via cocrystallization. Int J Pharm. 2006;320:114–123. doi: 10.1016/j.ijpharm.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 22.Jayasankar A, Somwangthanaroj A, Shao Z, Rodriguez-Hornedo N. Cocrystal formation during cogrinding and storage is mediated by amorphous phase. Pharm Res. 2006;23:2381–2392. doi: 10.1007/s11095-006-9110-6. [DOI] [PubMed] [Google Scholar]
- 23.Seefeldt K, Miller J, Alvarez-Nunez F, Rodriguez-Hornedo N. Crystallization pathways and kinetics of carbamazepine–nicotinamide cocrystals from the amorphous state by in situ thermomicroscopy, spectroscopy and calorimetry studies. J Pharm Sci. 2007;96:1147–1158. doi: 10.1002/jps.20945. [DOI] [PubMed] [Google Scholar]
- 24.Fleischman SG, Kuduyam SS, McMahon JA, Moulton B, Walsh RDB, Rodriguez-Hornedo N, Zaorotko MJ. Crystal engineering of the composition of pharmaceutical phases: multiple-component crystalline solids involving carbamazepine. Cryst Growth Des. 2003;3:909–919. doi: 10.1021/cg034035x. [DOI] [Google Scholar]
- 25.Bethune SJ, Huang N, Jayasankar A, Rodrıguez-Hornedo N. Understanding and predicting the effect of cocrystal components and pH on cocrystal solubility. Cryst Growth Des. 2009;9:3976–3988. doi: 10.1021/cg9001187. [DOI] [Google Scholar]
- 26.International Conference of Harmonization–Validation of analytical Procedures Q2(R1); 2005.
- 27.Ito S, Nishimura M, Kobayashi Y, Itai S, Yamamoto K. Characterization of polymorphs and hydrates of GK-128, a serotonin3 receptor antagonist. Int J Pharm. 1997;151:133–143. doi: 10.1016/S0378-5173(97)04867-9. [DOI] [Google Scholar]
- 28.Hairian I, Newton JM. Tensile strength of circular flat and convex faced Avicel PH102 tablets. DARU. 1999;7:36–40. [Google Scholar]
- 29.Heckel RW. An analysis of powder compaction phenomena. Trans Metall Soc AIME. 1961;221:1001–1008. [Google Scholar]
- 30.Sun C, Grant DJW. Influence of crystal shape on the tableting performance of l-lysine monohydrochloride dihydrate. J Pharm Sci. 2001;90:569–579. doi: 10.1002/1520-6017(200105)90:5<569::AID-JPS1013>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 31.Murphy D, Rodríguez-Cintrón F, Langevin B, Kelly RC, Rodríguez-Hornedo N. Solution-mediated phase transformation of anhydrous to dihydrate carbamazepine and the effect of lattice disorder. Int J Pharm. 2002;246:121–134. doi: 10.1016/S0378-5173(02)00358-7. [DOI] [PubMed] [Google Scholar]
- 32.Nair R, Nyamweya N, Gonen S, Martinez-Miranda LJ, Hoag SW. Influence of various drugs on the glass transition temperature of poly(vinylpyrrolidone): a thermodynamic and spectroscopic investigation. Int J Pharm. 2001;225:83–96. doi: 10.1016/S0378-5173(01)00767-0. [DOI] [PubMed] [Google Scholar]
- 33.Bayarıa S, Atac A, Yurdakul S. Coordination behaviour of nicotinamide: an infrared spectroscopic study. J Mol Struct. 2003;655:163–170. doi: 10.1016/S0022-2860(03)00256-4. [DOI] [Google Scholar]
- 34.Rahman Z, Zidan AS, Khan MA. Formulation and evaluation of a protein-loaded solid dispersions by non-destructive methods. AAPS J. 2010;12:158–70. doi: 10.1208/s12248-009-9171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adeyemi MO, Pilpel N. The effects of interacting variables on the tensile strength, disintegration and dissolution of oxytetracycline-lactose tablets. Int J Pharm. 1984;20:171–186. doi: 10.1016/0378-5173(84)90228-X. [DOI] [Google Scholar]
- 36.Hendriksen BA, Williams JD. Characterization of calcium fenoprofen 2. Dissolution from formulated tablets and compressed rotating discs. Int J Pharm. 1991;69:175–180. doi: 10.1016/0378-5173(91)90222-A. [DOI] [Google Scholar]
- 37.Jacob JT, Plein EM. Factors affecting the dissolution rate of medicaments from tablets II. Effect of binder concentration, tablet hardness, and storage conditions on the dissolution rate of phenobarbital from tablets. J Pharm Sci. 2006;57:802–805. doi: 10.1002/jps.2600570517. [DOI] [PubMed] [Google Scholar]
- 38.Ryshkewitch E. Compression strength of porous sintered alumina and zirconia. J Am Cer Soc. 1953;36:65–68. doi: 10.1111/j.1151-2916.1953.tb12837.x. [DOI] [Google Scholar]
- 39.Robert RJ, Rowe RC, York P. The relationship between the fracture properties, tensile strength, and critical stress intensity factor of organic solids and their molecular structure. Int J Pharm. 1995;125:157–162. doi: 10.1016/0378-5173(95)00157-E. [DOI] [Google Scholar]