

Abstract
Berberine chloride (BBR) is a natural isoquinoline alkaloid extracted from medicinal herbs. It has been reported that the intestinal absorption of BBR is very low. In this study, the absolute bioavailability of BBR was studied, and the enhancing effects of d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) on intestinal absorption were investigated in rats. BBR injection was administrated via the femoral vein at a dose of 1.0 mg kg−1 in intravenous group, and BBR oral formulations were administrated by oral gavage at a dose of 100 mg kg−1 in BBR control (control) group and BBR-TPGS (test) group, respectively. The result showed that BBR had a very low absolute bioavailability of 0.68%, and TPGS could enhance intestinal absorption of BBR significantly. TPGS at a concentration of 2.5% could improve peak concentration (Cmax) and area under the curve (AUC0–36) of BBR by 2.9 and 1.9 times, respectively. The absorption enhancing ability of TPGS may be due to its ability to affect the biological activity of P-glycoprotein and thereby reduce the excretion of absorbed BBR into the intestinal lumen. This study indicated that absolute bioavailability of BBR was 0.68% in rats, and TPGS was a good absorption enhancer capable of enhancing intestinal absorption of BBR significantly.
Key words: absorption enhancer, berberine chloride, bioavailability, P-glycoprotein, TPGS
INTRODUCTION
Berberine chloride (BBR) is a natural isoquinoline alkaloid isolated from medicinal herbs such as Coptis chinensis and Hydrastis canadensis, and it has been used for centuries in traditional Eastern medicines. It has been mainly used to treat diarrhea and gastroenteritis. Recent research has shown that BBR has many other biological activities, such as hypolipidemic, hypoglycemic, antiarrhythmic, antiproliferative, and antineoplastic activities (1–5). As a hypolipidemic agent, the blood lipid-reducing mechanism of BBR is distinct from that of statins. Statins have a hypolipidemic effect by increasing the transcription of LDLR mRNA. BBR lowers cholesterol by stabilizing LDLR mRNA, and thus, enhances the expression of LDL receptors (2). BBR has recently gained great interest because of its variety of bioactivities, low toxicity, and low cost properties. However, the wide application of BBR is restricted greatly by its poor intestinal absorption.
It has been reported that BBR is a substrate compound of P-glycoprotein (P-gp), and the action of P-gp plays an important role in the absorption of BBR (6). P-gp of epithelial is a membrane protein located in the apical membrane and functions as a multidrug efflux pump. It has wide substrate specificity and can lower intracellular drug concentration thereby reducing the cytotoxic activity of drugs (7,8). It has been reported that P-gp could limit the absorption of many drugs such as digoxin, etoposide, and paclitaxel as an absorption barrier (9–11). And the oral absorption of these drugs can be significantly improved by limiting the activity of P-gp effectively.
d-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS) is a water-soluble form of vitamin E and is comprised of a hydrophilic polar head and a lipophilic alkyl tail resulting in amphiphilic properties. It has a relatively low critical micelle concentration of 0.02 wt.% and has a hydrophile–lipophile balance value of 13.2 (12). Recently, TPGS has been described to be an effective oral absorption enhancer for improving bioavailability of several poor absorbed drugs. It has been reported that TPGS increased the oral absorption of cyclosporine through micelle formulation (13). And it is later reported that TPGS may interact with P-gp (14). Varma et al. also reported that the oral absorption of paclitaxel was enhanced significantly by coadministration of TPGS at a dose of 50 mg kg−1 in rats (15). Collnot et al. investigated the influence of vitamin E TPGS poly chain length on apical efflux transporters in Caco-2 cell monolayers and found that TPGS with a chain length of 1,000 was found to be the most potent analogs in the series (16).
In the present study, the absorption enhancing ability of TPGS on intestinal absorption of BBR was studied in rats. As few papers have reported the absolute bioavailability of BBR, the absolute bioavailability of BBR was also investigated in this study. Histopathological evaluation after using TPGS was performed in order to investigate the local toxicity of TPGS in the intestinal tract.
MATERIALS AND METHODS
Materials
BBR was purchased from Northeast Pharmaceutical Group Co., Ltd. (Shenyang, People’s Republic of China). The internal standard, tetrahydropalmatine (IS), was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, People’s Republic of China). TPGS was purchased from the Wuhan Yuancheng Technology Development Co., Ltd. (Wuhan, People’s Republic of China). LC grade acetonitrile, methanol, and formic acid were purchased from Dikma (NY, USA). Purified water, obtained using a Barnstead EASYpure® II RF/UV ultra pure water system (Dubuque, Iowa, USA) and passed through a 0.22-μm filter, was used throughout the study. Male Wistar rats used in the study were provided by the Laboratory Animal Center of Shenyang Pharmaceutical University.
Preparation of Formulations
The formulation for injection was prepared by dissolving weighed quantity of BBR in dextrose solution, and then the solution was passed through a 0.22-μm filter. The BBR injection was transferred to vials and sealed under the nitrogen protection, and then autoclaved in a water steam bath at 121°C for 10 min. TPGS solution with concentration of 2.5% was prepared by dissolving TPGS in water and agitated for 5 h. The BBR-TPGS formulation for oral administration was prepared by adding weighed quantity of BBR in TPGS solution and mixed well. And the control formulation for oral administration was prepared by adding quantitative BBR in 0.5% sodium carboxymethyl cellulose solution. The analysis of content of BBR formulations was performed using a modified version of a HPLC method reported before (17). The content of BBR for injection and oral administration with or without TPGS was 0.22, 5.10, and 5.02 mg mL−1, respectively. The TPGS concentration in the test formulation was 2.5%, and the ratio of TPGS to BBR was 5.0.
Animal Experiments
All animal experiments were carried out according to the University Ethics Committee guidelines for experimental animal care (certificate number: scxk 2009–0004).
Male Wistar rats weighing 220–240 g were fasted overnight for at least 12 h, with free access to water, and randomly divided into three groups for intravenous and oral administration. BBR formulation for injection was administered via the femoral vein at a dosage of 1.0 mg kg−1, and the oral formulations were administered by oral gavage at a dosage of 100 mg kg−1. Blood samples (0.3 mL) were obtained from the retroorbital sinus before dosing and subsequently at 0.083, 0.167, 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 24, and 36 h following administration for intravenous group and at 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 24, and 36 h for oral groups. Blood samples were centrifuged in heparinized Eppendorf tubes at 4,000 rpm for 10 min. The supernatant plasma was transferred to clean tubes and frozen at −20°C until analysis. Five animals were used per sampling time point.
Preparation of Standard Solutions
Stock solutions of BBR (1.0 mg mL−1) and IS (1.0 mg mL−1) were prepared in methanol and stored at 4°C. Standard solutions of berberine at concentrations of 0.5, 1.0, 2.5, 5.0, 10, 25, 50, 100, 250, 500, and 1,000 ng mL−1 were prepared by serial dilution of the stock solution with methanol. Standard solutions for quality control (QC) samples were made at concentrations of 1.0, 25, and 1,000 ng mL−1. An IS solution of 10 ng mL−1 was also prepared by diluting the stock solution of IS with methanol. All solutions were stored at 4°C.
Preparation of Calibration Curves and QC Samples
The samples for standard calibration curves and QC samples were prepared by spiking the 100 μL blank plasma with 20 μL appropriate standard solutions. The sample concentrations for standard calibration curves were 0.1, 0.2, 0.5, 1.0, 2.0, 5.0, 10, 20, 50, 100, and 200 ng mL−1. The QCs were prepared at low, medium, and high concentrations of 0.2, 5.0, and 200 ng mL−1.
UPLC-MS/MS Analysis of Plasma Samples
Many papers have reported LC-MS methods for BBR determination in biological samples (18,19). In this study, a UPLC-MS/MS method was used for the analysis of BBR in rat plasma. The 100 μL plasma samples were transferred to polyethylene tubes after the addition of 20 μL IS solution and vortexed for 1 min. Then 3 mL ether–dichloromethane (3:2, v/v) was added, and the mixture was mixed for 10 min. After centrifugation and separation, the supernatant was transferred to a clean tube and evaporated to dryness at 40°C in a centrifugal concentrator (Labconco Corp., Missouri, USA). The residue was reconstituted in 120 μL methanol, and an aliquot of 5 μL was injected into the UPLC-ESI-MS/MS system. The UPLC-ESI-MS/MS system consisted of a Waters ACQUITY™ TQD triple-quadrupole tandem mass spectrometer (Waters Corp., Manchester, UK) equipped with an electrospray ionization interface, an ACQUITY™ UPLC system (Waters Corp., Milford, MA, USA) with a conditioned autosampler at 4°C and an ACQUITY UPLC™ BEH C18 column at 35°C (50 × 2.1 mm i.d., 1.7 μm; Waters Corp., Milford, MA, USA). The mobile phase for linear gradient elution was comprised of (A) water (containing 0.1% formic acid) and (B) acetonitrile. The gradient conditions started with solvent B increasing from 20% to 90% within 1.0 min and maintained at this ratio for 1.0 min. Then, solvent A was increased to 80% within the next 0.5 min and maintained at this level from 2.5 to 3.0 min for column equilibration. The flow rate was 0.2 mL min−1. The total run time was 3.0 min, and the partial loop mode was used for sample injection. The ionization was carried out in the positive ionization mode with the capillary voltage set at 0.6 kV. The cone voltage was 50 V, and the extractor and RF lens voltages were set at 3.0 and 0.2 V, respectively. The source and desolvation temperatures were 100°C and 450°C, respectively. Nitrogen was used as the desolvation gas (450 L h−1) and cone gas (50 L h−1) for nebulization. For collision-induced dissociation, argon was used as the collision gas at a flow rate of 0.20 mL min−1. Quantification was carried out using multiple reaction monitoring mode. The m/z transitions were 336 → 320 for BBR and 356 → 192 for IS. All data collected in centroid mode were obtained using Masslynx™ NT4.1 software (Waters Corp., Milford, MA, USA). Post-acquisition quantitative analyses were carried out using a QuanLynx™ program (Waters Corp., Milford, MA, USA). The method was validated over the concentration range of 0.1–200 ng mL−1. The lower limit of quantification was 0.1 ng mL−1.
Pharmacokinetics Analysis
Pharmacokinetic parameters were calculated by non-compartmental analysis using the drug and statistics version 2.0 software (Mathematical Pharmacology Professional Committee of China, Shanghai, China). Values for area under the BBR concentration–time curve (AUC0–36 h) were calculated by the trapezoidal method from the beginning of administration to the final sampling time. The time to reach the maximum BBR concentration, Tmax, and the maximum plasma BBR concentration, Cmax, was determined directly from the plasma concentration–time profiles. The absolute bioavailability of BBR was calculated from the following equation:
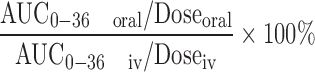 |
1 |
All values are expressed as the mean ± SD. Differences in pharmacokinetics were analyzed by the Student’s t test. A value of p < 0.05 was considered statistically significant.
Histopathological Evaluation of Local Toxicity
The histopathological evaluation was carried out by an experienced veterinary histopathologist. The ileums of the control group and the test group were removed at 5 h after oral gavage administration of drugs and washed using saline. Segments were removed and immersed in a 10% aqueous solution of formalin. A vertical section was prepared, stained using hematoxylin–eosin, and examined by light microscopy.
RESULTS
Method Validation
Selectivity
Assay selectivity was evaluated by analyzing six different lots of blank rat plasma. The retention time of BBR and IS was 1.45 and 1.36 min, respectively. The results showed that drug-free plasma samples had no interference at the retention time of BBR and the IS.
Calibration Curve Linearity and Lower Limit of Quantification
The calibration curve was prepared by plotting the peak area ratios of berberine to IS versus the nominal concentrations of berberine. Calibration curves showed good linearity over the range of 0.1–200 ng mL−1 by linear least squares linear regression using 1/x2 as a weight factor. Typical regression equation for three consecutive days was:
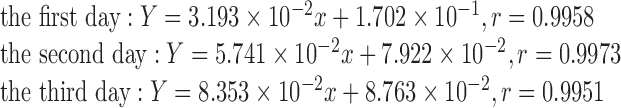 |
The LLOQ was established at 0.1 ng mL−1. The assay precision was calculated using the relative standard deviation (RSD%), and the accuracy was obtained from the relative deviation expressed as a percentage (RE%). The RSD and RE of the LLOQ are shown in Table I.
Table I.
Accuracy and Precision of the LLOQ Analysis (n = 6)
| Concentration added (ng mL−1) | Concentration found (ng mL−1) | Mean (ng mL−1) | RSD (%) | RE (%) |
|---|---|---|---|---|
| 0.100 | 0.09 | 0.098 | 11.8 | −1.67 |
| 0.11 | ||||
| 0.09 | ||||
| 0.09 | ||||
| 0.09 | ||||
| 0.11 |
Precision and Accuracy
The precision and accuracy of the assay were determined by analyzing QCs at three concentration levels (n = 6) at 0.2, 5.0, and 200 ng mL−1 on three consecutive days. The intra- and inter-day precision and accuracy are summarized in Table II. The results demonstrated that the precision and accuracy of this assay were acceptable.
Table II.
Accuracy and Precision of the Analysis of Berberine in Rat Plasma (Based on Assay of Six Replicates per Day on Three Consecutive Days)
| Concentration added (ng mL−1) | Concentration found (ng mL−1) | Intra-day RSD (%) | Inter-day RSD (%) | RE (%) |
|---|---|---|---|---|
| 0.2 | 0.20 | 12.5 | 9.3 | 0.75 |
| 5 | 4.87 | 8.4 | 8.7 | −2.59 |
| 200 | 197 | 10.0 | 5.2 | −1.70 |
Extraction Recovery and Stability
The extraction recovery was estimated by analyzing six replicates of berberine at 0.2, 5.0, and 200 ng mL−1. The extraction recovery was evaluated by comparing the peak areas of berberine extracted from plasma samples with those of six spike-after-extraction samples that represent 100% recovery. The extraction recovery of IS was calculated in the same manner as the QC samples of 5.0 ng mL−1. The recovery values of berberine at three concentrations of 0.2, 5.0, and 200 ng mL−1 from rat plasma with ether–dichloromethane (3:2, v/v) were 85.6 ± 10.2%, 84.1 ± 5.5%, and 84.3 ± 7.0% (n = 6), respectively. Also, the recovery of IS at a concentration of 5.0 ng mL−1 was 87.9 ± 8.3%. The recovery of berberine and the IS met the requirements for the analysis of biological samples.
The stability of berberine in plasma was studied by analyzing three replicates of both low (0.2 ng mL−1) and high (200 ng mL−1) concentrations under various storage and process conditions. Processed samples were stable after three cycles of freezing and thawing with a variation of no more than ±15%. After storage at 4°C for 12 h in the autosampler and at room temperature for 6 h, berberine remained stable. In the long-term stability test in which berberine in plasma was kept at −20°C for 2 weeks, no significant difference in concentration between the test samples and the standards was found. The stability results of berberine in rat plasma under various storage conditions are shown in Table III.
Table III.
Stability Results of Berberine in Rat Plasma Under Various Storage Conditions (n = 3)
| Conditions | Concentration added (ng mL−1) | Concentration found (ng mL−1) | RSD (%) | RE (%) |
|---|---|---|---|---|
| Room temperature (6 h) | 0.2 | 0.205 | 6.2 | 2.7 |
| 200 | 194 | 9.7 | −3.2 | |
| Three freeze-thaw cycles | 0.2 | 0.201 | 13.2 | 0.7 |
| 200 | 195 | 3.6 | −2.7 | |
| Autosampler (4°C for 12 h) | 0.2 | 0.204 | 10.1 | 2.0 |
| 200 | 197 | 5.8 | −1.7 | |
| 2 weeks at −20°C | 0.2 | 0.197 | 6.2 | −1.5 |
| 200 | 197 | 9.6 | −1.6 |
Matrix Effect
The matrix effect was investigated at three concentration levels of 0.2, 5.0, and 200 ng mL−1 using six replicates by comparing the peak areas of the analyte standards dissolved in the blank plasma extraction solvent with those dissolved in blank solution. The blank plasma used in the present study was obtained from six different lots of blank rat plasma. The results were in the range of 85–115%, indicating that there was no matrix effect in this study.
Animal Studies
The plasma BBR concentration–time profiles of the iv group, control group, and test group after intravenous or oral administration different BBR formulations to rats are shown in Fig. 1. The main pharmacokinetic parameters are shown in Table IV. The absolute bioavailability of BBR after intravenous and oral administration of BBR formulation without TPGS was calculated from Eq. 1. The result showed that BBR has an absolute bioavailability of 0.68%.
Fig. 1.

Plasma BBR concentration–time profile after intravenous (iv) and oral administration of BBR along (control) and in combination with TPGS (test) in rats. Data are shown as mean concentration, and error bars represent SD (n = 5). The iv BBR dose was 1.0 mg kg−1, and the oral dose was 100 mg kg−1. The concentration of TPGS in the formulation was 2.5%
Table IV.
The Mean (± SD) Main Pharmacokinetic Parameters of BBR After Intravenous and Oral Administration to Rats at Dosage of 1.0 mg kg−1 and 100 mg kg−1, Respectively (n = 5)
| Group | C max (ng mL-1) | T max (h) | AUC0–36 h (ng h mL−1) |
|---|---|---|---|
| iv | 44.9 ± 6.64 | 0.083 ± 0 | 68.7 ± 10.5 |
| control | 9.48 ± 3.40 | 2.60 ± 1.14 | 46.5 ± 12.8 |
| test | 27.9 ± 14.2a | 0.40 ± 0.14a | 87.8 ± 13.4a |
BBR was intravenously administered at the dose of 1.0 mg kg−1 and orally administered at the dose of 100 mg kg−1 with or without TPGS (500 mg kg−1). Control refers to the oral administration of BBR without TPGS. Test refers to the oral administration of BBR with TPGS
BBR berberine chloride, TPGS d-α-tocopheryl polyethylene glycol 1000 succinate, AUC 0–36 h area under the BBR concentration–time curve calculated by the trapezoidal method from 0 to36h, C max the maximum plasma BBR concentration, T max the time to reach maximum BBR concentration, iv intravenous administration of BBR
aSignificantly different form control group, p < 0.05
As shown in Table IV, there was very little BBR absorbed from the gastrointestinal tract in the control group without any absorption enhancers after oral administration of BBR at a dose of 100 mg kg−1 in rats, and the Cmax and AUC0–36 h values were 9.48 ± 3.40 ng mL−1 and 46.5 ± 12.8 ng h mL−1, respectively. After adding 2.5% TPGS to BBR formulation, it resulted an increased absorption of BBR with a Cmax value of 27.9 ± 14.2 ng mL−1 and an AUC0–36 h value of 87.8 ± 13.4 ng h mL−1. With the addition of 2.5%TPGS to the BBR solutions, the Cmax and AUC0–36 h of BBR were increased by about 1.9 and 2.9 times compared with the control group, respectively. It indicated that TPGS produced a significant enhancement of BBR absorption (p < 0.05).
Histopathological Evaluation
Photomicrographs of the intestinal mucosa exposed to BBR and BBR combined with TPGS are shown in Fig. 2. The ileum cavity was chosen as the site of testing because it is very sensitive to absorption enhancer. As indicated in Fig. 2, the epithelium of each group was undamaged, and the villus structure was intact. There was no significant difference between BBR alone and TPGS. It is indicated that TPGS has no significant local toxicity in the intestinal tract.
Fig. 2.

Histopathological comparison between test and control rats. Light micrograph samples were taken at 5 h after oral gavage administration of drugs (original magnification, ×100). I Control, oral gavage administration of BBR. II Test, oral gavage administration of BBR formulation containing 2.5% TPGS. The BBR doses were all 100 mg kg−1
DISCUSSION
BBR has an extremely low absolute bioavailability of 0.68%, which is comparable with the hypothesis that drugs containing the quaternary ammonium groups in their structures commonly have low bioavailability (20). Poor bioavailability of BBR is due to many factors, and one important reason is the physicochemical property. BBR is a hydrophilic compound with a temperature-dependent aqueous solubility which increases with an increase in temperature. It has been reported that BBR has a log p value of −1.5 and is considered as hydrophilic compound (21), which indicates that BBR is a biopharmaceutical classification system (BCS) class III drug. Drugs that belong to BCS class III such as BBR are mostly lipophobic with poor membrane permeability, and the absorption of the drugs is mostly limited to the paracellular pathway, and this limits the intestinal absorption and leads to the low bioavailability (22). Another important reason is that BBR is a substrate of P-gp, and the bioavailability of BBR is greatly limited by this ATP-dependent multidrug transporter whereby absorbed BBR is secreted into the intestinal lumen (6). An important method used to increase the absorption of drugs being the substrate compounds of P-gp such as BBR is to inhibit the activity of P-gp. It has been previously reported that BBR absorption is substantially enhanced by coadministration of P-gp inhibitors such as cyclosporine A, verapamil, and the monoclonal antibody C219 in in vivo and in vitro models (6). TPGS has been described to be capable of inhibiting the biological activity of P-gp and the cytotoxicity of many drugs such as doxorubicin, vinblastine, and paclitaxel in G185 cells was enhanced by TPGS (14). Therefore, the absorption enhancing ability of TPGS at a dose of 500 mg kg−1 as a P-gp inhibitor was investigated in this study.
It has been reported that TPGS enhances bioavailability of drugs by enhancing solubility and/or permeability (13,23). In this study, the solubility enhancing effect of TPGS on BBR was investigated. The equilibrium solubility of BBR in water was 2.08 ± 0.3 mg mL−1 (n = 3) after shaking for consecutive 3 days in a thermostatic shaking water bath (Zhicheng, People’s Republic of China). And the equilibrium solubility of BBR in 2.5% TPGS solution was 1.88 ± 0.06 mg mL−1 (n = 3). The results indicated that BBR is slightly soluble in water, and the 2.5% TPGS solution has no significant solubility enhancing ability on BBR (p > 0.05). As mentioned above, BBR is a substrate of P-gp and the absorption was significantly enhanced by P-gp inhibitors such as cyclosporine A, verapamil, and monoclonal antibody C219. Therefore, it is speculated that TPGS enhanced the gastrointestinal absorption of BBR mainly by enhancing permeability.
As shown in Fig. 1, the Tmax of control group and test group was greatly different. After oral administration of TPGS-BBR formulation, the Tmax of BBR shifted to 0.40 ± 0.14 h, which was significantly smaller than that of the control group. There was no paper reported this before. It is speculated that the shift of Tmax of BBR is due to the change of physiological activity of gastrointestinal tract in rats after administration of TPGS. However, the precise reason of obtaining a shifted Tmax with the addition 2.5% is not clear. It has been reported that surfactants affect the permeability of drugs in intestinal tract via disruption and fluidization of the cell membrane (24). And the surfactant-induced alteration in intestinal tract is associated with acute epithelial damage. The extent of damage is correlated with the type of surfactant, the quantity of surfactant, and the time of exposure; and the damage is rapidly reversible after removal of the surfactants by means of villus shortening and epithelial cell migration to cover the injured area (25,26). Prasad et al. suggested that surfactants such as TPGS alter the epithelium transport of drugs by changing the biophysical characteristics of the intestinal epithelial membrane (27). The interfacial tension between hydrophilic drugs and intestinal mucosa with lipid nature resulted in less contact between the formulation and the epithelium. By using surfactants, the interfacial tension was reduced and the contact between formulation and epithelium was improved. This change might lead to the absorption enhancement of low-permeability drugs such BBR, and it also might play a role in the shift of Tmax. However, further studies should be performed to verify the hypothesis.
CONCLUSION
The absolute bioavailability of BBR in rats was studied, and the results showed that BBR has an extreme low bioavailability of 0.68%. TPGS (2.5%) was studied as an absorption enhancer for BBR, and it showed a significant absorption enhancing effect. This absorption enhancing effect may be due to the P-gp inhibition ability. Histopathological evaluation performed after applying TPGS did not show any significant morphological changes. This study showed that TPGS is a good pharmaceutical excipient for use as an absorption enhancer to improve the absorption of BBR.
Acknowledgment
This work is supported by the Liaoning Provincial Science and Technology Department (2009ZX09301-012).
References
- 1.Lau CW, Yao XQ, Chen ZY, Ko WH, Huang Y. Cardiovascular actions of berberine. Cardiovasc Drug Rev. 2001;19(3):234–44. doi: 10.1111/j.1527-3466.2001.tb00068.x. [DOI] [PubMed] [Google Scholar]
- 2.Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10(12):1344–51. doi: 10.1038/nm1135. [DOI] [PubMed] [Google Scholar]
- 3.Leng SH, Lu FE, Xu LJ. Therapeutic effects of berberine in impaired glucose tolerance rats and its influence on insulin secretion. Acta Pharmacol Sin. 2004;25(4):496–502. [PubMed] [Google Scholar]
- 4.Piyanuch R, Sukhthankar M, Wandee G, Baek SJ. Berberine, a natural isoquinoline alkaloid, induces NAG-1 and ATF3 expression in human colorectal cancer cells. Cancer Lett. 2007;258(2):230–40. doi: 10.1016/j.canlet.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holy EW, Akhmedov A, Luscher TF, Tanner FC. Berberine, a natural lipid-lowering drug, exerts prothrombotic effects on vascular cells. J Mol Cell Cardiol. 2009;46(2):234–40. doi: 10.1016/j.yjmcc.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 6.Pan GY, Wang GJ, Liu XD, Fawcett JP, Xie YY. The involvement of P-glycoprotein in berberine absorption. Pharmacol Toxicol. 2002;91(4):193–7. doi: 10.1034/j.1600-0773.2002.t01-1-910403.x. [DOI] [PubMed] [Google Scholar]
- 7.Endicott JA, Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem. 1989;58:137–71. doi: 10.1146/annurev.bi.58.070189.001033. [DOI] [PubMed] [Google Scholar]
- 8.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 9.Leu BL, Huang JD. Inhibition of intestinal P-glycoprotein and effects on etoposide absorption. Cancer Chemother Pharmacol. 1995;35(5):432–6. doi: 10.1007/s002800050258. [DOI] [PubMed] [Google Scholar]
- 10.Su SF, Huang JD. Inhibition of the intestinal digoxin absorption and exsorption by quinidine. Drug Metab Dispos. 1996;24(2):142–7. [PubMed] [Google Scholar]
- 11.Varma MV, Sateesh K, Panchagnula R. Functional role of P-glycoprotein in limiting intestinal absorption of drugs: contribution of passive permeability to P-glycoprotein mediated efflux transport. Mol Pharm. 2005;2(1):12–21. doi: 10.1021/mp0499196. [DOI] [PubMed] [Google Scholar]
- 12.Wu SH, Hopkins WK. Characteristics of d-α-tocopheryl PEG 1000 succinate for applications as an absorption enhancer in drug delivery systems. Pharm Technol. 1999;23:52–68. [Google Scholar]
- 13.Sokol RJ, Johnson KE, Karrer FM, Narkewicz MR, Smith D, Kam I. Improvement of cyclosporin absorption in children after liver transplantation by means of water-soluble vitamin E. Lancet. 1991;338(8761):212–4. doi: 10.1016/0140-6736(91)90349-T. [DOI] [PubMed] [Google Scholar]
- 14.Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by d-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) Pharm Res. 1999;16(10):1550–6. doi: 10.1023/A:1015000503629. [DOI] [PubMed] [Google Scholar]
- 15.Varma MV, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci. 2005;25(4–5):445–53. doi: 10.1016/j.ejps.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Collnot EM, Baldes C, Wempe MF, Hyatt J, Navarro L, Edgar KJ, et al. Influence of vitamin E TPGS poly(ethylene glycol) chain length on apical efflux transporters in Caco-2 cell monolayers. J Control Release. 2006;111(1–2):35–40. doi: 10.1016/j.jconrel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Misaki T, Sagara K, Ojima M, Kakizawa S, Oshima T, Yoshizawa H. Simultaneous determination of berberine, palmatine and coptisine in crude drugs and oriental pharmaceutical preparations by ion-pair high-performance liquid chromatography. Chem Pharm Bull (Tokyo). 1982;30(1):354–7. doi: 10.1248/cpb.30.354. [DOI] [PubMed] [Google Scholar]
- 18.Gupta PK, Hubbard M, Gurley B, Hendrickson HP. Validation of a liquid chromatography-tandem mass spectrometric assay for the quantitative determination of hydrastine and berberine in human serum. J Pharm Biomed Anal. 2009;49(4):1021–6. doi: 10.1016/j.jpba.2009.01.036. [DOI] [PubMed] [Google Scholar]
- 19.Lu T, Liang Y, Song J, Xie L, Wang GJ, Liu XD. Simultaneous determination of berberine and palmatine in rat plasma by HPLC-ESI-MS after oral administration of traditional Chinese medicinal preparation Huang-Lian-Jie-Du decoction and the pharmacokinetic application of the method. J Pharm Biomed Anal. 2006;40(5):1218–24. doi: 10.1016/j.jpba.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 20.Hua W, Ding L, Chen Y, Gong B, He J, Xu G. Determination of berberine in human plasma by liquid chromatography–electrospray ionization–mass spectrometry. J Pharm Biomed Anal. 2007;44(4):931–7. doi: 10.1016/j.jpba.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 21.Battu SK, Repka MA, Maddineni S, Chittiboyina AG, Avery MA, Majumdar S. Physicochemical characterization of berberine chloride: a perspective in the development of a solution dosage form for oral delivery. AAPS PharmSciTech. 2010;11(3):1466–75. doi: 10.1208/s12249-010-9520-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madara JL. Loosening tight junctions. Lessons from the intestine. J Clin Invest. 1989;83(4):1089–94. doi: 10.1172/JCI113987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu L, Bridgers A, Polli J, Vickers A, Long S, Roy A, et al. Vitamin E-TPGS increases absorption flux of an HIV protease inhibitor by enhancing its solubility and permeability. Pharm Res. 1999;16(12):1812–7. doi: 10.1023/A:1018939006780. [DOI] [PubMed] [Google Scholar]
- 24.Liu DZ, LeCluyse EL, Thakker DR. Dodecylphosphocholine-mediated enhancement of paracellular permeability and cytotoxicity in Caco-2 cell monolayers. J Pharm Sci. 1999;88(11):1161–8. doi: 10.1021/js990094e. [DOI] [PubMed] [Google Scholar]
- 25.Erickson RA. Effect of 16,16-dimethyl PGE2 and indomethacin on bile acid-induced intestinal injury and restitution in rats. J Lab Clin Med. 1988;112(6):735–44. [PubMed] [Google Scholar]
- 26.Nakanishi K, Masada M, Nadai T. Effect of pharmaceutical adjuvants on the rectal permeability of drugs. III. Effect of repeated administration and recovery of the permeability. Chem Pharm Bull (Tokyo). 1983;31(11):4161–6. doi: 10.1248/cpb.31.4161. [DOI] [PubMed] [Google Scholar]
- 27.Prasad YV, Puthli SP, Eaimtrakarn S, Ishida M, Yoshikawa Y, Shibata N, et al. Enhanced intestinal absorption of vancomycin with Labrasol and d-alpha-tocopheryl PEG 1000 succinate in rats. Int J Pharm. 2003;250(1):181–90. doi: 10.1016/S0378-5173(02)00544-6. [DOI] [PubMed] [Google Scholar]