

Abstract
Hydroxypropyl-sulfobutyl-β-cyclodextrin (HP-SBE-β-CD) inclusion complex was developed and used as a drug delivery system for DTX (DTX/HP-SBE-β-CD). The objective of the present study was to evaluate and compare the biological properties of DTX/HP-SBE-Β-CD with Taxotere®. The pharmacokinetics, biodistribution, antitumor efficacy in vivo and in vitro, and safety evaluation of DTX/HP-SBE-β-CD were studied. The most significant finding was that it was possible to prepare a Polysorbate-80-free inclusion complex for DTX. Studies based on pharmacokinetics, biodistribution, and antitumor efficacy indicated that DTX/HP-SBE-β-CD had similar pharmacokinetic properties and antitumor efficacy both in vitro and in vivo as Taxotere®. Fortunately, this new drug delivery system attenuated the side effects when used in vivo. As a consequence, DTX/HP-SBE-β-CD may be a promising alternative to Taxotere® for cancer chemotherapy treatment with reduced side effects. The therapeutic potential against a variety of human tumors and low toxicity demonstrated in a stringent study clearly warrant clinical investigation of DTX/HP-SBE-β-CD for possible use against human tumors.
Key words: antitumor efficacy, biodistribution, DTX/HP-SBE-β-CD, pharmacokinetics, safety evaluation
INTRODUCTION
Docetaxel (DTX, Taxotere®, Fig.1), an analog of paclitaxel, is a novel anticancer agent of the taxoid family. The drug was found to promote tubulin assembly in microtubules and to inhibit their depolymerization which leads to mitotic arrest in the G2M phase of the cell cycle (1–4). Over the past two decades, DTX has been shown to be a strong therapeutic which is active in clinical use against a wide range of tumors, including ovarian carcinoma, advanced breast cancer, non-small cell lung cancer, and head/neck cancer (5–7). Due to being highly lipophilic and practically insoluble in water (2.903 μg/mL), DTX is currently dissolved for clinical use in Polysorbate 80 and ethanol (50:50, v/v) as Taxotere®. However, some adverse effects occurred in the majority of patients treated with Taxotere®, for example, myelosuppression, severe hypersensitivity reaction, hemolysis, and fluid retention (8–10). These adverse effects caused by DTX are a limitation to its clinical use.
Fig. 1.

Chemical structures of DTX
Because of its broad efficacy and certain disadvantages, extensive investigations into improving its water solubility and lower adverse effects have been undertaken. Some approaches have been used to develop new drug delivery systems, for instance, liposomes (11), nanospheres (12), emulsions (13), polymeric micelles (14), cyclodextrin (15), folic acid conjugated nanoparticles (16), and water-soluble prodrugs (17). A bioequivalence study of DTX for injectable emulsion (ANX-514) without Polysorbate 80 in patients with advanced cancer has been completed in USA (http://clinicaltrials.gov/ct2/show/NCT00664170).
Cyclodextrin is a cyclic oligosaccharide with six to eight glucose units bonded by a α-1, 4-linkage. Cyclodextrins (CDs) can form supramolecular aggregates with hydrophobic drugs (18). They have been known to increase the water solubility of some medicinal agents through noncovalent inclusion complexation with a hydrophilic outer surface and a lipophilic central cavity which can encapsulate hydrophobic drug molecules or parts of these molecules. CDs can also enhance the stability and bioavailability of drug molecules (19). However, natural CDs (α-cyclodextrin, α-CD; β-cyclodextrin, β-CD; and γ-cyclodextrin, γ-CD) have low solubility both in aqueous and organic solvents and high hemolysis which limits their use in pharmaceutical formulation. As a result, a multitude of cyclodextrin derivatives have been prepared to extend the physicochemical properties and inclusion capacity of natural CDs as novel drug delivery systems including oral drug delivery, parenteral drug delivery, and ocular delivery among others (20–22). To obtain cyclodextrin derivatives, natural CDs are modified by selective substitution of aliphatic chains of different structures (2–18 C, linear or branched, linked with ester, ether, amide, thio, fluoro bonds) to different positions (primary face, secondary face, or both faces) (23, 24). Among them, some derivatives, such as 2-hydroxypropyl-β-cyclodextrin (2-HP-β-CD), sulfobutyl-β-cyclodextrin (SBE-β-CD), and dimethyl-β-cyclodextrin, have attracted growing interest because of their improved complexing ability, great water solubility, and low toxicity.
However, both natural cyclodextrins and derivatized cyclodextrins still result in renal toxicity and hemolysis depending on the mode of administration. CDs have been reported to interact with cell membrane constituents such as cholesterol, phospholipids, and phosphatidylinositols, resulting in the induction of hemolysis of RBC (25–28). The magnitude of the hemolytic activity of the parent CDs is reported to increase in the order of γ-CD < α-CD < β-CD (25). With the aim of reducing toxicity, we developed a new family of CDs based on hydroxypropyl-sulfobutyl-β-cyclodextrin (HP-SBE-β-CD) as a carrier for delivery of DTX. HP-SBE-β-CD is a β-cyclodextrin derivative which is substituted by a hydroxypropyl group and sulfobutyl group: n-(2,3,6-O-2-hydroxypropyl)-m-(2,3,6-O-sulfobutyl)-β-cyclodextrin. Previous investigations indicated that HP-SBE-β-CD had higher water solubility, stronger inclusion capability, lower hemolysis, and lower toxicity than β-CD, HP-β-CD, and SBE-β-CD. The preparation method, analytical method, and pharmaceutical application of HP-SBE-β-CD have been studied in our laboratory (http://www.freepatentsonline.com/20090012042.pdf). On that basis, we prepared the DTX inclusion complex with HP-SBE-β-CD (DTX/HP-SBE-β-CD) as a novel drug delivery system for DTX (http://www.freepatentsonline.com/20100048685.pdf).
The objective of the present study was to evaluate and compare the pharmacokinetics, biodistribution, antitumor efficacy, and toxicity of DTX/HP-SBE-β-CD with Taxotere® using in vitro and animal studies.
MATERIALS AND METHODS
Materials
DTX was obtained from Shanghai Jinhe Bio-Technology Co., Ltd. (Shanghai, China). HP-SBE-β-CD was prepared by Jiangsu Key Laboratory for Supramolecular Medicinal Materials and Applications (Nanjing, China). Taxotere® was purchased from Jiangsu Hengrui Medicine Co., Ltd. (Jiangsu, China). The reference standards, DTX (99.0%) and paclitaxel (99.5%), were obtained from Sigma-Aldrich (USA). High-performance liquid chromatography (HPLC)/spectra-grade reagents were used as the mobile phase in HPLC analysis, and all other reagents were of analytical grade and used without further purification. Distilled and deionized water was produced by a Millipore (Bedford, Massachusetts, USA) Milli-Q water system and used in all experiments.
Animals
Albino rabbits (1.8–2.3 kg), Kunming mice (18–22 g), and Wistar rats (180–200 g) were obtained from the Laboratory Animal Center of Southeast University (Nanjing, China). Animals were housed on a 12-h light/dark cycle with food and water provided ad libitum in a barrier facility which is fully accredited. The study and all procedures concerning animals were carried out according to the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication 86–23, revised 1986), conformed to the International Guidelines on the Ethical Use of Animals, and were approved by the Animal Care and Use Committee of Nanjing Normal University and Animal Ethics Committee of Nanjing Normal University.
Preparation of DTX/HP-SBE-β-CD
The DTX/HP-SBE-β-CD inclusion complex was prepared by freeze-drying method. DTX and HP-SBE-β-CD were weighted precisely in the 1:17 mass ratio. Specifically, the solution of DTX in absolute ethanol was slowly added into the distilled water solution of HP-SBE-β-CD. The mixture was magnetically stirred at room temperature for 2 h and passed through 0.4 and 0.2 μm pore size membranes. Then the filtrate was heated at 55°C for 30 min to thoroughly remove ethanol under reduced pressure and lyophilized for 48 h to obtain the dried powder of DTX/HP-SBE-β-CD.
Pharmacokinetic Studies
Albino rabbits (1.8–2.3 kg) were used in the pharmacokinetic study. Rabbits were randomly divided into the following two groups (n = 6, three of each sex): (1) DTX/HP-SBE-β-CD (7.5 mg/kg) and (2) Taxotere® (7.5 mg/kg). Drugs were administered as a 25-min continuous intravenous infusion through a marginal ear-vein. Blood samples of 1.5 mL each were collected in heparinized centrifuge tubes via another marginal ear-vein at various times (5, 10, 20, 30, 45, and 60 min and 1.5, 2, 3, 5, 8, 12, 16, and 24 h from the beginning of the infusion). The plasma was obtained by centrifugation at 3,000 rpm for 10 min and the DTX concentrations were determined by the HPLC method described below.
Pharmacokinetic parameters were determined by the 3p97 software provided by the Chinese Pharmacological Society. Distribution and elimination data were represented by the following parameters: area under the curve (AUC), mean residence time (MRT), total body clearance (CL), the apparent volume of distribution (Vd), and plasma half-life for the distribution and elimination phase (t1/2α, t1/2β).
Biodistribution
Kunming mice (18–22 g) were randomly divided into two groups (n = 18, nine of each sex), DTX/HP-SBE-β-CD and Taxotere®. DTX/HP-SBE-β-CD and Taxotere® were intravenously administrated via the tail vein at a dose of 20 mg/kg. After administration, mice were sacrificed at various times (5 min, 30 min, and 5 h) and various tissues (brain, heart, liver, spleen, lung, kidney, stomach, intestine, genitals, muscle, and fat) were rapidly removed. Tissue samples were removed, weighed, and homogenized with saline to a concentration of 10 mg/mL. The DTX concentrations were determined by the HPLC method described below.
Measurement of DTX Levels in Plasma and Tissue Samples by HPLC
The concentrations of DTX (both free and protein-bound formulation) in plasma and tissue were determined by RP-HPLC. Plasma samples and tissue samples were extracted by the liquid–liquid extraction technique. To 500 μL of plasma sample or tissue sample, 200 μL of sodium bicarbonate (1 mol/L) and 50 μL of internal standard solution containing paclitaxel (10 μg/mL) were added and mixed for 5 min by vortex. Then, the mixture was extracted with 4 mL of anhydrous diethyl ether. The total organic layer was separated by centrifugation at 3,000 rpm for 15 min, transferred to a clean tube, and evaporated to dryness at 40°C under a stream of nitrogen. The drug residue was finally reconstituted in an acetonitrile-ammonium acetate buffer (pH 5.0; 0.035 M)-tetrahydrofuran (47:48:5, v/v/v) solution followed by centrifugation at 4,000 rpm for 10 min before analysis, of which 20 μL were injected into the HPLC system.
The HPLC system consisted of a Shimadzu LC10A instrument equipped with an SPD-10AV (UV–VIS) detector, along with a LC solution Chromopac data processor. Sample separation was performed on a reversed-phase column (platisil C18, 150 × 4.6 mm, 5 μm; Dikma, USA). The mobile phase consisting of acetonitrile–ammonium acetate buffer (pH 5.0; 0.035 M)–tetrahydrofuran (47:48:5, v/v/v) was used for chromatographic separations. Column temperature was 30°C and the flow rate was 1 mL/min, respectively. The detector was set at the wave length 227 nm UV.
Antitumor Efficacy
In Vitro Test
Human tumor cell lines (A549 lung adenocarcinoma, HCT-116 colon adenocarcinoma, SKOV-3 ovarian adenocarcinoma, and Hep G2 hepatoma) were obtained from America Type Culture Collection (ATCC, Manassas, Virginia, USA). Cells were cultured in suspension in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Biochrom, Berlin) and antibiotics (100 mg/mL of streptomycin and 100 unit/mL of penicillin, Sigma) at 37°C in a balanced air humidified incubator with an atmosphere of 5% CO2. Cells were maintained in exponential growth phase by periodic dilutions with fresh medium.
Briefly, 5 × 103 cells/well were seeded in 100 μL of growth medium in 96-well plates and allowed to attach overnight. A total of 24 h later, the monolayers were exposed to DTX/HP-SBE-β-CD (48 h) or Taxotere® (48 h). After drug exposure, 10 μL of MTT (3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium bromide) solution was added to cells cultured for the designed time. The plates were incubated for 4 h and absorbance was measured at 570 nm using a 96-well plate reader. Each assay was carried out in triplicate. The drug concentration which inhibited the cell growth by 50% (IC50) was determined from semilogarithmic dose–response plots.
In Vivo Test
The antitumor activities of DTX/HP-SBE-β-CD were further investigated by in vivo tests against the mice sarcoma tumor S180 and mice hepatocellular carcinoma H22 according to the method of Keiko et al (29) and Zhang et al (30) with minor modifications. S180 Sarcoma and H22 hepatocarcinoma cells were cultured and used for assessing the in vivo antitumor efficacy of DTX/HP-SBE-β-CD. Kunming mice (18–22 g) received subcutaneous injections into the hind flank on day 0 with 0.2 mL of cell suspension containing S180 or H22 (5 × 106 cells). The day of injection was defined as day 0. After 24 h, animals were weighed and randomly divided into five different groups (n = 10, five of each sex): (1) negative control group (HP-SBE-β-CD-based vehicle); (2) DTX/HP-SBE-β-CD 2.5 mg/kg; (3) DTX/HP-SBE-β-CD 5 mg/kg; (4) DTX/HP-SBE-β-CD 10 mg/kg; and (5) positive control group (Taxotere®, 10 mg/kg). Drugs were administered via the tail vein every other day for the subsequent 13 days. Twenty-four hours later after the last drug administration, animals were sacrificed, and then their tumors were resected and weighed. Antitumor efficacy calculations were expressed as the percentage of inhibition ratio which was calculated by the following formula: inhibition ratio (%) = [(A − B)/A] × 100, where A is the average tumor weight of the negative control and B is that of the treatment group.
Safety Evaluation
Hemolysis Test
Hemolysis studies were carried out on DTX/HP-SBE-β-CD and Taxotere®. Blood samples were drawn from rabbit. In brief, 15 mL of rabbit blood was obtained from the carotid artery and the fibrinogen was removed. To obtain a pure suspension of erythrocytes, freshly collected rabbit blood was washed more than three times with saline by centrifugation at 1,500 rpm for 10 min. Finally, after repeated washing and centrifugation, the erythrocyte suspension was obtained and diluted with saline up to a concentration of 2% for the hemolysis assay. DTX/HP-SBE-β-CD and Taxotere® were respectively dissolved in saline at a concentration of 4 mg/mL, and various volumes of DTX/HP-SBE-β-CD and Taxotere® (0.1–0.5 mL) were each added into 2.5 mL 2% of erythrocyte suspension with the final concentrations ranging from 0.08 to 0.4 mg/mL (all diluted through saline); 2.5 mL of distilled water and 2.5 mL of saline were respectively added into 2.5 mL of 2% erythrocyte suspension as the positive control (100% hemolysis) and negative control (0% hemolysis). After incubation at 37°C for 3 h, all samples were centrifuged at 1,500 rpm for 10 min, and the supernatant was analyzed for hemoglobin release by spectrophotometric determinations at 545 nm. Hemolysis (percent) was calculated by the following equation:
Hemolysis 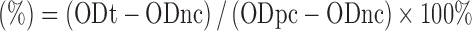 , where ODt, ODnc, and ODpc are the absorbances of the test samples, negative control (0% hemolysis), and positive control (100% hemolysis).
, where ODt, ODnc, and ODpc are the absorbances of the test samples, negative control (0% hemolysis), and positive control (100% hemolysis).
Anaphylaxis Test
Wistar rats (180–200 g) were randomly assigned to three different groups (n = 8, male): (1) negative control group; (2) DTX/HP-SBE-β-CD group; and (3) positive control group. The sensitization and challenge protocols were performed as described below. In 7 days, each animal was intraperitoneally injected every other day with 1 mL of 5% glucose injection solution for the negative control group, 1 mL of phosphate-buffered saline (PBS) with 1.0 mg chicken ovalbumin (OVA; Grade V, Sigma), and 10 mg aluminum hydroxide (Al(OH)3) for the positive control group, and 1 mL of DTX/HP-SBE-β-CD solution (2.0 mg/mL in 5% glucose injection solution) for the DTX/HP-SBE-β-CD group. After day 7, each group was divided into two sub-groups (n = 4). Animals in sub-group one were intravenously injected after 14 days of the first injection and animals in sub-group two were intravenously injected after 21 days of the first injection.
Airway responsiveness (AHR) of the rats for each group was detected using the animal lung function analysis system (AniRes2005, Beijing BestLab Co., China). Rats were anesthetized intraperitoneally with pentobarbital sodium, and then respirated via a tracheal cannula connected to a computer-controlled small-animal ventilator. The respiratory rate was preset at 75/min and the time ratio of expiration/inspiration at 1.5:1. An injector needle was then inserted into the jugular vein. AHR was assessed by AniRes2005 software with indexes of respiratory resistance R-area (the area between the peak value and baseline), including expiratory resistance (Re), inspiratory resistance (Ri), and the peak value of dynamic pulmonary compliance (Cldyn). After a steady basal line, 1 mL of 5% glucose injection solution for the negative control group, 1 mL of PBS with 1.0 mg OVA, and 10 mg Al(OH)3 for the positive control group, and 1 mL of DTX/HP-SBE-β-CD solution (2.0 mg/mL in 5% glucose injection solution) for the DTX/HP-SBE-β-CD group were respectively intravenously administrated via a tail vein. After the administration, data were collected continuously from 5 s to 30 min and the average values of Re, Ri, and Cldyn were taken to express changes in airway function.
After assessment of airway reactivity, the rats were sacrificed and bronchoalveolar lavage fluid (BALF) was collected for assay of histamine. The BALF was centrifuged; the supernatant was saved and frozen at −80°C for subsequent detecting histamine by enzyme-linked immunosorbent assay (Histamine EIA Kit, SPI-Bio, USA).
Statistical Analysis
The results were expressed as mean ± SD and the statistical analysis was done by analysis of variance. A probability level of p < 0.05 was considered statistically significant.
RESULTS
Pharmacokinetic Experiments
The concentration of DTX was detected by HPLC analysis and the method was validated. Endogenous components produced no interference in the chromatograms. After intravenous infusion of DTX/HP-SBE-β-CD and Taxotere® in rabbits, the plasma concentration–time profiles of DTX are illustrated in Fig. 2 and the main pharmacokinetic parameters are summarized in Table I. A two-compartmental model was used to describe the pharmacokinetics of both formulations. In addition, the plasma concentration was detectable up to 24 h in rabbits using the analysis method established in this study. As shown in Fig. 2, the concentration levels showed a rapid decline in the distribution phase for the first 0.2 h after dosing with two formulations, which was consistent with Marchettini’s study (27). The plasma maximum concentration (Cmax) for DTX/HP-SBE-β-CD and Taxotere® were 3.974 and 4.026 mg/L. The values of the area under the plasma concentration–time curve (AUC0-∞) for DTX/HP-SBE-β-CD and Taxotere® were 3.885 and 3.907 mg/L/h, respectively. Both formulations showed a similar distribution phase (t1/2α = 0.215 h for DTX/HP-SBE-β-CD and 0.283 h for Taxotere®) and a similar elimination phase (t1/2β = 5.182 h for DTX/HP-SBE-β-CD and 5.989 h for Taxotere®). Furthermore, the total body clearance for DTX/HP-SBE-β-CD and Taxotere® were 2.006 and 2.124 L/h/kg, respectively.
Fig. 2.

Mean plasma concentration–time profiles of DTX after intravenous infusion administration of a single 7.5-mg/kg dose of DTX/HP-SBE-β-CD and Taxotere® to rabbits (each point represents the mean ± SD, n = 6)
Table I.
Pharmacokinetic Parameters of DTX After Intravenous Infusion Administration of a Single 7.5-mg/kg Dose of DTX/HP-SBE-β-CD and Taxotere® to Rabbits (n = 6)
| Drug (mg/kg) | DTX/HP-SBE-β-CD | Taxotere® |
|---|---|---|
| V d (L/kg) | 0.836 ± 0.173 | 1.080 ± 0.295 |
| t 1/2α (h) | 0.215 ± 0.026 | 0.283 ± 0.056 |
| t 1/2β (h) | 5.182 ± 1.485 | 5.989 ± 1.394 |
| AUC0–24h (mg/L/h) | 3.740 ± 0.754 | 3.530 ± 0.326 |
| AUC0-∞ (mg/L/h) | 3.885 ± 0.752 | 3.907 ± 0.331 |
| CL (L/h/kg) | 2.006 ± 0.420 | 2.124 ± 0.211 |
| MRT (h) | 2.750 ± 0.356 | 2.469 ± 0.713 |
All data are presented as mean ± SD (n = 6)
V d apparent volume of distribution, t 1/2α plasma half-life for the distribution phase, t 1/2β plasma half-life for the elimination phase, AUC 0-24h area under the plasma concentration–time curve from 0 to 24 h, AUC 0-∞ area under the plasma concentration–time curve from time zero to infinity, CL total body clearance, MRT mean residence time, DTX docetaxel, HP-SBE-β-CD hydroxypropyl-sulfobutyl-β-cyclodextrin
Biodistribution Experiments
The biodistribution profile of DTX after intravenous administration of DTX/HP-SBE-β-CD in mice was compared with Taxotere® as a control. The mean tissue concentrations of DTX after intravenous administration of the DTX/ HP-SBE-β-CD and Taxotere® at a single dose of 20 mg/kg at different sample times are shown in Fig. 3. Because both the ovaries and uterus are too small to determine singly, they were placed together for the analysis. As shown in Fig. 3, the results demonstrated that DTX was absorbed rapidly and distributed widely into most tissues after intravenous administration of DTX/HP-SBE-β-CD, and the drug concentration was the highest in liver, followed by kidney, heart, and lung. After 30 min, the concentrations in these tissues for DTX/HP-SBE-β-CD in a descending order was lung > kidney > spleen > stomach. Finally after 5 h, the corresponding order for DTX/HP-SBE-β-CD was lung > kidney > spleen > stomach.
Fig. 3.

Distribution in tissues in mice after intravenous administration of DTX/HP-SBE-β-CD and Taxotere® at a dose of 20 mg/kg at different sample times a 5 min, b 30 min, c 5 h. Each column represents the mean ± SD (n = 6). *p < 0.05, **p < 0.01, compared with Taxotere®
Antitumor Efficacy
In Vitro
To investigate the in vitro antitumor efficacy of DTX/HP-SBE-β-CD and Taxotere®, we analyzed cell growth by a MTT cell proliferation assay kit. The mean IC50 values obtained in the MTT assay of DTX/HP-SBE-β-CD and Taxotere® are shown in Table II. Our data showed that DTX/HP-SBE-β-CD and Taxotere® displayed similar inhibition of cell viability for A549, HCT-116, SKOV-3, and Hep G2 cancer cells.
Table II.
In Vitro Antitumor Efficacy (IC50) of DTX/HP-SBE-β-CD and Taxotere®
| Cell line | IC50 values (nM) | |
|---|---|---|
| DTX/HP-SBE-β-CD | Taxotere® | |
| A549 | 37.5 ± 5.1 | 35.1 ± 4.3 |
| HCT-116 | 11.2 ± 3.2 | 14.4 ± 1.2 |
| SKOV-3 | 41.8 ± 7.3 | 43.0 ± 5.2 |
| Hep G2 | 1.5 ± 0.2 | 1.7 ± 0.3 |
Data represent mean value ± SD, n = 3
DTX docetaxel, HP-SBE-β-CD hydroxypropyl-sulfobutyl-β-cyclodextrin
In Vivo
To determine whether DTX/HP-SBE-β-CD had similar potent activity to Taxotere®, we examined the response to DTX/HP-SBE-β-CD and Taxotere® treatment of different tumors established in mice. The ability of DTX/HP-SBE-β-CD and Taxotere® to inhibit the growth of tumors was evaluated in mice bearing established S180 or H22 tumor models. Results are summarized in Fig. 4a and b. The high-dose of DTX used in this study was 10 mg/kg because significant toxic death of mice occurred above this dose. Administration of DTX/HP-SBE-β-CD and Taxotere® effectively inhibited the growth of S180 and H22 tumors both in male and female mice. DTX/HP-SBE-β-CD significantly inhibited the growth of S180 and H22 at dose of 5 mg/kg and 10 mg/kg (p < 0.01).
Fig. 4.

Antitumor effect of intravenous administration of DTX/HP-SBE-β-CD and Taxotere® on tumor weight in mice after sacrifice. a S180-bearing mice and b H22-bearing mice. Each column represents the mean ± SD (n = 10). *p < 0.05, **p < 0.01, compared with negative control
Safety Evaluation
Hemolysis Test
Hemolytic properties of DTX/HP-SBE-β-CD was assessed and compared with Taxotere®. The positive control group presented total hemolysis, but no hemolysis was observed in the DTX/HP-SBE-β-CD solution at concentrations ranging from 0.08 to 0.4 mg/mL and the negative control group after 3 h (data not shown). Compared with DTX/HP-SBE-β-CD, Taxotere® caused hemolysis at concentrations of 0.32 and 0.4 mg/mL after 3 h (data not shown). The results indicated that the DTX/HP-SBE-β-CD solution at a high concentration of 0.4 mg/mL did not cause hemolysis and was less toxic than Taxotere®.
Anaphylaxis Test
The airway reactivity measurement and levels of histamine in BALF were determined to evaluate injection anaphylaxis in Wistar rats. As shown in Table III, the values of Re and Ri in the positive control group were significantly increased compared with the negative control group and the DTX/HP-SBE-β-CD group both on days 14 and 21 (p < 0.01). The values of Cldyn in the positive control group were significantly decreased compared with negative control group and DTX/HP-SBE-β-CD group both on days 14 and 21(p < 0.01). However, no significant changes of Re, Ri, and Cldyn were observed between the negative control group and DTX/HP-SBE-β-CD group both on days 14 and 21. All the results demonstrated that DTX/HP-SBE-β-CD at a dose of 10 mg/kg did not change AHR in rats. Moreover, after antigen challenge, the level of histamine in the positive control group increased significantly compared with the negative control group and DTX/HP-SBE-β-CD group (p < 0.01). However, the levels of histamine were similar between the negative control group and DTX/HP-SBE-β-CD group. These results obtained indicated that the DTX/HP-SBE-β-CD at a dose of 10 mg/kg did not obviously induce histamine release in rats.
Table III.
Airway Reactivity Measurement and Histamine Levels in BALF at Last Challenge in Experimental Rats
| Group | Time of last challenge | Airway reactivity measurement | Histamine in BALF (nM/mL) | ||
|---|---|---|---|---|---|
| Re (cmH2O/mL/s) | Ri (cmH2O/mL/s) | Cldyn (mL/cmH2O) | |||
| Negative control | D14 | 0.797 ± 0.106 | 0.571 ± 0.053 | 0.087 ± 0.004 | 2.35 ± 0.843 |
| D21 | 0.863 ± 0.093 | 0.611 ± 0.060 | 0.085 ± 0.004 | 2.43 ± 0.806 | |
| Positive control | D14 | 1.347 ± 0.150** | 1.028 ± 0.125** | 0.053 ± 0.007** | 18.4 ± 4.057** |
| D21 | 1.380 ± 0.136** | 1.093 ± 0.107** | 0.051 ± 0.006** | 15.4 ± 4.473** | |
| DTX/HP-SBE-β-CD | D14 | 0.836 ± 0.049 | 0.611 ± 0.034 | 0.086 ± 0.007 | 3.83 ± 1.357 |
| D21 | 0.830 ± 0.035 | 0.666 ± 0.161 | 0.089 ± 0.003 | 3.15 ± 0.850 | |
Data represent mean value ± SD, n = 4
*p < 0.05; **p < 0.01 (compared with negative control)
Re expiratory resistance, Ri inspiratory resistance, Cldyn dynamic pulmonary compliance, DTX docetaxel, HP-SBE-β-CD hydroxypropyl-sulfobutyl-β-cyclodextrin, BALF bronchoalveolar lavage fluid
DISCUSSION
Due to its poor water solubility, DTX is currently dissolved in Polysorbate 80 and ethanol (50:50, v/v) for clinical use as Taxotere®. However, some adverse effects occurred in the majority of patients with Taxotere®, such as severe hypersensitivity reaction and fluid retention (8–10). In order to avoid these adverse reactions of DTX, we prepared the DTX inclusion complex with HP-SBE-β-CD without Polysorbate 80.
DTX is administered by intravenous infusion in clinical cancer therapy. As a consequence, in our pharmacokinetic study, intravenous infusion administration was used in rabbits, which was different to the intravenous administration presented in other articles. There was no significant difference of pharmacokinetic parameters between DTX/HP-SBE-β-CD and Taxotere® which showed that HP-SBE-β-CD did not influence the pharmacokinetic behavior of DTX.
In our tissue distribution experiments, the results of DTX/HP-SBE-β-CD were similar to that of Taxotere®. DTX was distributed widely into most tissues in DTX/HP-SBE-β-CD, which was in accordance with the results reported by Bissery et al (2). Biodistribution studies also showed high concentrations of DTX were observed in heart, liver, spleen, lung, and kidney, organs which have high blood perfusion. Both in DTX/HP-SBE-β-CD and Taxotere®, drug retention in lung occurred. However, the time of drug retention in DTX/HP-SBE-β-CD was longer than in Taxotere® (not significantly). Based on this, it was thought that DTX/HP-SBE-β-CD could be used to treat non-small cell lung in a clinical setting. High concentrations were observed in the liver at 5 min after intravenous administration. In contrast, concentrations became low in the liver at 30 min and 5 h after intravenous administration. In addition, no DTX was detected in mouse brain both in DTX/HP-SBE-β-CD and Taxotere®. This may due to the fact that DTX could not cross the blood–brain barrier because of its large molecular size. Levels of DTX in reproductive organs were higher in females than males in both DTX/HP-SBE-β-CD and Taxotere®, which was also consistent with the results reported by Bissery et al (2). It was demonstrated that DTX/HP-SBE-β-CD had similar antitumor efficacy to Taxotere® both in vitro. Compared with Taxotere®, DTX/HP-SBE-β-CD at the dose of 10 mg/kg showed similar antitumor activity to S180 or H22 tumor animal models in vivo (Fig. 4a, b). In additional, no obvious necrosis at the injection sites was observed in all of animal tested. The results could have important clinical implications.
A hemolysis test was conducted to evaluate the toxicity of the formulation. In this work, the highest concentration of HP-SBE-β-CD in the DTX/HP-SBE-β-CD solution was 20 mg/mL and in our previous unpublished work, we found that β-CD and 2-HP-β-CD caused 50% hemolysis in rabbit erythrocyte suspension at concentrations of 1.14 and 12.61 mg/mL, respectively. This implied that HP-SBE-β-CD did not cause hemolysis at a concentration of 20 mg/mL and was less toxic to red blood cells than β-CD and 2-HP-β-CD.
The pathophysiology of DTX-induced SHR is unknown. Bronchospasm, laryngeal edema, gastrointestinal symptoms, and hypotension can occur during mast cell/basophil mediator release induced by IgE and non-IgE mechanisms. Polysorbate 80 has also been associated with these hypersensitivity reactions, sometimes with severe and irreversible sensory and motor neuropathies. Moreover, Polysorbate 80 can alter membrane fluidity, leading to cumulative fluid retention (31). This toxicity may be reduced by prophylactic corticosteroids or antihistaminic drugs. Therefore, during recent years some special efforts have been made to avoid these problems. AHR and levels of histamine in BALF were measured as useful indexes for evaluating anaphylaxis of rats in our study. Histamine is a component of the storage granules of myeloid cells including basophils and mast cells. The clinical importance of histamine in the pathogenesis of allergy and asthma has long been appreciated. Histamine contributes to airway constriction, edema, vascular congestion, and inflammatory cell recruitment seen in AHR (32). The measurement of histamine level is therefore of importance in evaluating system hypersensitivity reactions. Our experimental results demonstrated that DTX/HP-SBE-β-CD did not change AHR and levels of histamine in BALF of rats, which implied that DTX/HP-SBE-β-CD did not cause anaphylaxis in rats. However, additional studies will be required to fully characterize the nature of toxicity for DTX/HP-SBE-β-CD.
CONCLUSION
In conclusion, the most significant finding from our study is that it was possible to prepare a Polysorbate-80-free formulation for DTX. Studies based on pharmacokinetics, biodistribution, and antitumor efficacy indicated that DTX/HP-SBE-β-CD had similar pharmacokinetic properties and antitumor efficacy as Taxotere® both in vitro and in vivo. As a consequence, DTX/HP-SBE-β-CD seemed to be a promising alternative to Taxotere® for cancer chemotherapy with reduced side effects. The therapeutic potential against a variety of human tumors and low toxicity demonstrated in this stringent study clearly warrant clinical investigation of DTX/HP-SBE-β-CD for possible use against human tumors.
Acknowledgment
This work was supported by the Society Development Science Foundation of Jiangsu, China, no: BE2008676; and was supported by the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University.
References
- 1.Diaz JF, Andreu JM. Assembly of purified GDP-tubulin into microtubules induced by Taxol and Taxotere: reversibility, ligand stoichiometry and competition. Biochemistry. 1993;32:2747–2755. doi: 10.1021/bi00062a003. [DOI] [PubMed] [Google Scholar]
- 2.Bissery MC, Nohynek G, Sanderink GJ, Lavelle F. Docetaxel (Taxotere): a review of preclinical and clinical experience. Part I: preclinical experience. Anticancer Drugs. 1995;6(339–55):363–368. doi: 10.1097/00001813-199506000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Ajani JA. Chemotherapy for advanced gastric or gastroesophageal cancer: defining the contributions of docetaxel. Expert Opin Pharmacother. 2006;7:1627–1631. doi: 10.1517/14656566.7.12.1627. [DOI] [PubMed] [Google Scholar]
- 4.Bouvier E, Thirot S, Schmidt F, Monneret C. First enzymatically activated Taxotere prodrugs designed for ADEPT and PMT. Bioorg Med Chem. 2004;12:969–977. doi: 10.1016/j.bmc.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 5.Rowinsky EK. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu Rev Med. 1997;48:353–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- 6.Capri G, Tarenzi E, Fulfaro F, Gianni L. The role of taxanes in the treatment of breast cancer. Semin Oncol. 1996;23:68–75. [PubMed] [Google Scholar]
- 7.Kaye SB. Taxoids. Eur J Cancer. 1995;31A:824–826. doi: 10.1016/0959-8049(95)00104-Q. [DOI] [PubMed] [Google Scholar]
- 8.Valero V, Holmes FA, Walters RS, Theriault RL, Esparza L, Fraschini G. Phase II trial of docetaxel: a new, highly effective antineoplastic agent in the management of patients with anthracycline-resistant metastatic breast cancer. J Clin Oncol. 1995;13:2886–2894. doi: 10.1200/JCO.1995.13.12.2886. [DOI] [PubMed] [Google Scholar]
- 9.Ferraresi V, Milella M, Vaccaro A, D’Ottavio AM, Papaldo P, Nistico C. Toxicity and activity of docetaxel in anthracycline-pretreated breast cancer patients: a phase II study. Am J Clin Oncol. 2000;23:132–139. doi: 10.1097/00000421-200004000-00006. [DOI] [PubMed] [Google Scholar]
- 10.Esmaeli B, Valero V, Ahmadi MA, Booser D. Canalicular stenosis secondary to docetaxel (Taxotere)—a newly recognized side effect. Ophthalmology. 2001;108:994–995. doi: 10.1016/S0161-6420(00)00640-0. [DOI] [PubMed] [Google Scholar]
- 11.Zhao L, Wei YM, Zhong XD, Liang Y, Zhang XM, Li W. PK and tissue distribution of docetaxel in rabbits after i.v. administration of liposomal and injectable formulations. J Pharm Biomed Anal. 2009;49:989–996. doi: 10.1016/j.jpba.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 12.Musumeci T, Ventura CA, Giannone I, Ruozi B, Montenegro L, Pignatello R. PLA/PLGA nanoparticles for sustained release of docetaxel. Int J Pharm. 2006;325:172–179. doi: 10.1016/j.ijpharm.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 13.Gao K, Sun J, Liu K, Liu XH, He ZG. Preparation and characterization of a submicron lipid emulsion of docetaxel: submicron lipid emulsion of docetaxel. Drug Dev Ind Pharm. 2008;34:1227–1237. doi: 10.1080/03639040802005057. [DOI] [PubMed] [Google Scholar]
- 14.Yang M, Ding YT, Zhang LY, Qian XP, Jiang XQ, Liu BR. Novel thermosensitive polymeric micelles for docetaxel delivery. J Biomed Mater Res A. 2007;81:847–857. doi: 10.1002/jbm.a.31129. [DOI] [PubMed] [Google Scholar]
- 15.Quaglia F, Ostacolo L, Mazzaglia A, Villari V, Zaccaria D, Sciortino MT. The intracellular effects of non-ionic amphiphilic cyclodextrin nanoparticles in the delivery of anticancer drugs. Biomaterials. 2009;30:374–382. doi: 10.1016/j.biomaterials.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 16.Liu YT, Li K, Pan J, Liu B, Feng SS. Folic acid conjugated nanoparticles of mixed lipid monolayer shell and biodegradable polymer core for targeted delivery of Docetaxel. Biomaterials. 2010;31:330–338. doi: 10.1016/j.biomaterials.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 17.Du WT, Hong L, Yao TW, Yang XC, He QJ, Yang B. Synthesis and evaluation of water-soluble docetaxel prodrugs-docetaxel esters of malic acid. Bioorg Med Chem. 2007;15:6323–6330. doi: 10.1016/j.bmc.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Loftsson T, Magnusdottir A, Masson M, Sigurjonsdottir JF. Self-association and cyclodextrin solubilization of drugs. J Pharm Sci. 2002;91:2307–2316. doi: 10.1002/jps.10226. [DOI] [PubMed] [Google Scholar]
- 19.Loftsson T, Duchene D. Cyclodextrins and their pharmaceutical applications. Int J Pharm. 2007;329:1–11. doi: 10.1016/j.ijpharm.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 20.Szente L, Szejtli J. Highly soluble cyclodextrin derivatives: chemistry, properties, and trends in development. Adv Drug Deliv Rev. 1999;36:17–38. doi: 10.1016/S0169-409X(98)00092-1. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda H, Arima H. Cyclodextrins in transdermal and rectal delivery. Adv Drug Deliv Rev. 1999;36:81–99. doi: 10.1016/S0169-409X(98)00056-8. [DOI] [PubMed] [Google Scholar]
- 22.Challa R, Ahuja A, Ali J, Khar RK. Cyclodextrins in drug delivery: an updated review. AAPS PharmSciTech. 2005;6:E329–357. doi: 10.1208/pt060243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wouessidjewe D, Skiba M, Leroy LF, Lemos SE, Puisieux F, Duchene D. A new concept in drug delivery based on skirt-shaped cyclodextrin aggregates: present state and future prospects. S.T.P. Pharm Sci. 1996;6:21–28. [Google Scholar]
- 24.Sallas F, Darcy R. Amphiphilic cyclodextrins—advances in synthesis and supramolecular chemistry. Eur J Org Chem. 2008;6:957–969. doi: 10.1002/ejoc.200700933. [DOI] [Google Scholar]
- 25.Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Differential effects of α-, β- and γ-cyclodextrins on human erythrocytes. Eur J Biochem. 1989;186:17–22. doi: 10.1111/j.1432-1033.1989.tb15171.x. [DOI] [PubMed] [Google Scholar]
- 26.Fauvelle F, Debouzy JC, Crouzy S, Goschl M, Chapron Y. Mechanism of α-cyclodextrin-induced hemolysis. 1. The two-step extraction of phosphatidylinositol from the membrane. J Pharm Sci. 1997;86:935–943. doi: 10.1021/js9602453. [DOI] [PubMed] [Google Scholar]
- 27.Debouzy JC, Fauvelle F, Crouzy S, Girault L, Chapron Y, Goschl M. Mechanism of α-cyclodextrin-induced hemolysis. 2. A study of the factors controlling the association with serine-ethanolamine-, and choline-phospholipids. J Pharm Sci. 1998;87:59–66. doi: 10.1021/js970180j. [DOI] [PubMed] [Google Scholar]
- 28.Drori S, Eytan GD, Assaraf YG. Potentiation of anticancer drug cytotoxicity by multidrug-resistance chemosensitizers involves alterations in membrane fluidity leading to increased membrane permeability. Eur J Biochem. 1995;228:1020–1029. doi: 10.1111/j.1432-1033.1995.tb20352.x. [DOI] [PubMed] [Google Scholar]
- 29.Keiko I, Izumi H, Shohei Y, Masataka M. A potential role of bradykinin in angiogenesis and growth of S-180 mouse tumors. Jpn J Pharmacol. 2001;87:318–326. doi: 10.1254/jjp.87.318. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C, Qu GW, Sun YJ, Wu XL, Yao Z, Guo QL, Ding QL, Yuan ST, Shen ZL, Ping QN, Zhou HP. Pharmacokinetics, biodistribution, efficacy and safety of N-octyl-O-sulfate chitosan micelles loaded with paclitaxel. Biomaterials. 2008;29:1233–1241. doi: 10.1016/j.biomaterials.2007.11.029. [DOI] [PubMed] [Google Scholar]
- 31.Marchettini P, Stuart OA, Mohamed F, Yoo D, Sugarbaker PH. Docetaxel: pharmacokinetics and tissue levels after intraperitoneal and intravenous administration in a rat model. Cancer Chemother Pharmacol. 2002;49:499–503. doi: 10.1007/s00280-002-0439-1. [DOI] [PubMed] [Google Scholar]
- 32.Akdis CA, Simons FE. Histamine receptors are hot in immunopharmacology. Eur J Pharmacol. 2006;533:69–76. doi: 10.1016/j.ejphar.2005.12.044. [DOI] [PubMed] [Google Scholar]