

Abstract
Yellow fever virus is the causative agent of Yellow fever. The genome of the virus contains three structural and seven non-structural proteins. Of these seven nonstructural proteins, NS2B-NS3 protein complex has protease activity required for viral replication. Predicting the 3D structure of this complex and studying the interaction of residues at the recognized catalytic triad of the complex is an integral part to understand the virus replication mechanism. In the present study, the structure was determined for NS2B-NS3 complex by Homology modeling and modeled structure was validated for its stability. Mutation studies at the residues His94, Asp118 and Ser176 revealed that Asp118-His94 bond played an important role in the structural stability of NS2B-NS3 complex. This indicates site-directed mutagenesis, controlling YFV replication, as one mechanism to design vaccine strains. Docking studies of the bioactive compounds at the active site of NS2B-NS3 complex also indicated 4-hydroxypanduratin A as potential lead compound for drug development. The theoretical models will further pave way to experimentally verify our mutation and docking studies, thus taking a lead in pharmacogenomics and drug development.
Abbreviations
YFV - Yellow Fever Virus, WNV - West Nile Virus, H-bonds - hydrogen bonds, SNP - Single nucleotide polymorphism.
Keywords: site-directed mutagenesis, docking, NS2B-NS3 complex
Background
Yellow fever is endemic in South America and in Sub-Saharan Africa. It was reported that 40,000 deaths occur in the endemic regions annually [1, 2]. The Yellow Fever Virus (YFV), causative agent of the Yellow fever disease is transmitted to vertebrates by mosquitoes during feeding. The RNA genome of YFV contains 10,862 nucleotides and encodes a large polyprotein precursor of 3411 amino acid residues which consist of three structural proteins (C, prM and E) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) [3]. Optimal activity of the NS3 serine protease is required for the maturation of the virus and the presence of the NS2B co-factor is a prerequisite for the optimal catalytic activity of NS3 [4]. Studies revealed that NS3, contains a serine proteinase catalytic triad within amino terminal regions of 181 amino acid residues and also it requires the 40 amino acid residues of NS2B for protease activity [5]. We focus on insilico modeling and mutation of YFV NS2B/NS3 serine protease complex. Three-dimensional models of the NS2B-NS3 complex would be extremely useful in design of pharmaceutical compounds.
Methodology
Homology model building
The Modeller software package (mod9v7) was used to build the NS2B/NS3 complex model [6]. The homology modelling of NS2B/NS3 complex of YFV was performed with WNV NS2B/NS3 in complex with Bz-nKRR-H (PDB ID: 2FP7) as a template [7]. The model generated was then submitted to the SAVES server for evaluation of its quality using PROCHECK [8].
Mutation and interaction studies
In NS2B-NS3 complex, residues His 94, Asp 118 and Ser 176 were mutated using Bioedit [9] and again modeled. To know the stability of complex, hydrogen bonds formed between the NS2B/NS3 complex was studied using CCP4 packages [10].
Docking studies
The docking of three competitive bioactive molecules, 4-hydroxypanduratin A, panduratin A and ester, onto the catalytic triad of the serine protease were performed using Autodock 4.0.1 (http://autodock.scripps.edu/). Polar hydrogen atoms were added to the model of YFV NS2B/NS3 complex and its non-polar hydrogen atoms were merged. Kollman charges were added to this molecule. For the ligands, non-polar hydrogen atoms were merged with Gasteiger charges assigned. All rotatable bonds of ligands were set to be rotatable. Docking was performed using genetic algorithm and lamarckian genetic algorithm methods. A population size of 150 and 10 millions energy evaluations were used for 50 times searches, with a 60 × 60 × 60 dimension of grid box size and 0.375 Ǻ grid spacing around the catalytic triad. Clustering histogram analyses were performed after the docking searches.
Results and Discussion
In this study, the crystal structure of WNV NS2B/NS3 in complex with BznKRR- H was chosen as template to generate the YFV NS2B/NS3 model. After modeling, mutations were done at the catalytic triad to study the stability of the modeled complex protein before and after mutation. Binding studies between inhibitors and YFV serine protease complex were studied by performing protein-ligand docking using Autodock 4.0.1. This program utilizes lamarckian genetic algorithm for configuration search and evaluates the energy using gridbased molecular affinity potential.
Homology model of NS2B/NS3complex
The overall sequence identity between YFV NS2B/NS3 complex and WNV NS2B/NS3 was 69%, and was successfully modeled (Figure 1). Validation through PROCHECK shows that model after minimization contains 80.4% of the amino acid residues in the most favoured region, 14.1% in allowed regions, 3.8Å in generously regions and 1.6% in disallowed region in Ramachandran plot. The RMSD value of the modeled NS2B-NS3 complex was found to be 4.702 Å . The modeled structure was well within the described Ramachandran limits, and thus was considered to be the putative structure for NS2B-NS3 complex for further studies.
Figure 1.
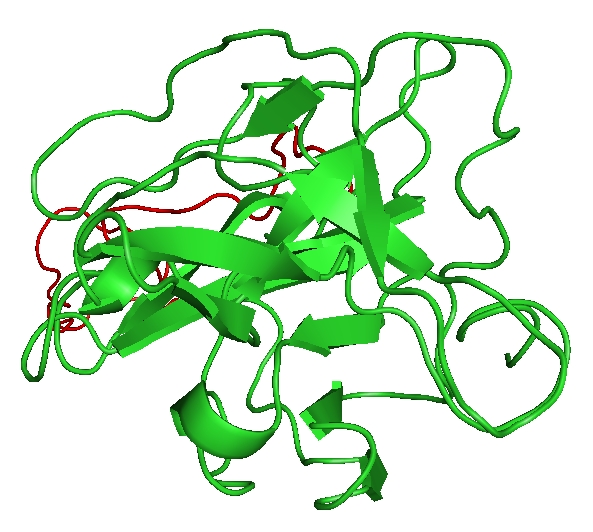
Three dimensional structure of YFV NS2B-NS3 complex modeled using Modeller9v7. The 2FP7 from WNV was used as template. NS2B is shown in red color and NS3 is shown in green color.
Mutation at the catalytic triad
NS2B-NS3 complex has a catalytic triad comprising of His94, Asp118 and Ser176 which has the proteolytic activity [4], important for viral replication. Thus abolishing or dramatically reducing protease activity has the ability to control the viral replication and thus stop spreading yellow fever [4, 11, 12]. Taking this factor into account, an insilico mutation study was done and comparison was drawn between the wild and the mutant models. The sequence retrieved from swiss-prot was mutated at 3 residues (His94, Asp118 and Ser 176) with 4 different combinations by using BioEdit software as tabularized below in Table 1(see Supplementary material). After mutation the sequence was modeled and validated.
Based on the three-dimensional structure of NS2B-NS3 complex, the hydrogen bonds in the active centre of NS2B-NS3 complex are discussed. As shown in (see Table 2Supplementary material), there are five hydrogen bonds in the NS2B-NS3 complex. These hydrogen bonds are important because they are formed between the catalytic triad and also catalytic triad residues with other residues in the complex. Studies by Lau et al. (1999) [13], suggests that the breaking of Asp118-His94 hydrogen bond of the catalytic triad has drastic effect on the structure and dynamics of the protein. The instability induced in the structure may also be reasoned to substantiate the reduced catalytic activity of the protein. Hence H-bonds formed between the residues were taken as deciding factor for judging the level of activity of the catalytic triad in various mutated models.
The effect of mutation was studied by considering the H-bonds formed within the catalytic triad. After mutation all bonds are formed, except one functionally important bond between His94 and Asp118 (See Table 3a in Supplementary material). Asp118-His94 bond was prominent in the wild model but this bond was non-existent in all of the mutant models. The RMSD values of the mutated models are given in Table 3b (See Table 3b in Supplementary material). From the values it is evident that the mutation of any of these residues disrupts the structure. Thus applying this study at molecular level may prove an important strategy for controlling the replication of Yellow Fever Virus.
Docking
The discovery of potential ligands as drug candidates was an important aspect for controlling diseases like dengue, swine flu based on homology models [14]. Inhibitors namely 4-hydroxypanduratin A, Panduratin A and Ester were used as ligands for binding interaction studies with the active site of the DEN2 serine protease [15]. Since the active residues of catalytic triad are found to be same in serine proteases of all flavi virus, the same three inhibitors 4- hydroxypanduratin A, Panduratin A and Ester are used in our docking studies. Docking analysis of the ligands 4-hydroxypanduratin A, Panduratin A and Ester with the NS2B-NS3 complex showed interesting pattern of interactions and binding mode to the active site. To have a better understanding of the interaction of ligands with the complex, binding energies as well as the hydrogen bond interactions, are tabulated in (see Table 4 in Supplementary material). Among the three ligands screened, 4-hydroxypanduratin A was found to have best docking in terms of interaction and forming hydrogen bond with active site residue, Asp118 (Figure 2). The minimum binding energy indicated the strong affinity of 4-hydroxypanduratin A with the NS2B-NS3 complex which had been stabilized by strong hydrogen bond interactions in the binding pocket.
Figure 2.
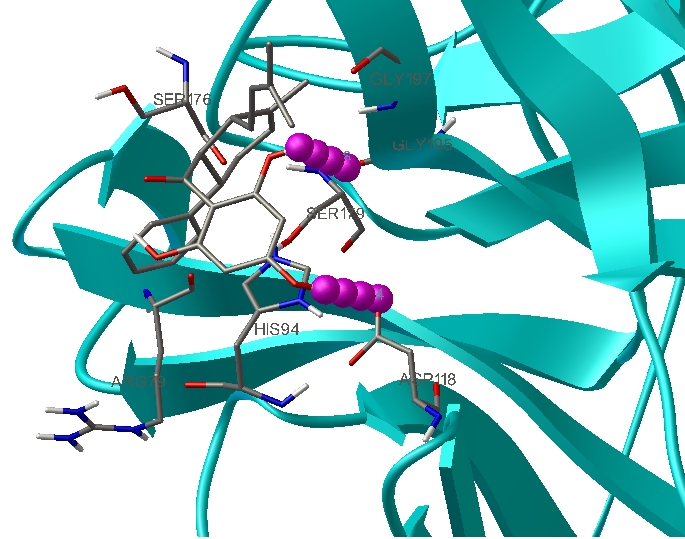
NS2B-NS3 complex docked with 4-hydroxypanduratin forming hydrogen bonds with Asp118 and Gly 195.
Conclusion
Working at the catalytic triad of YFV is one important way of studying viral replication and can provide an insight into the way to prevent its replication. Current work can be considered as a preliminary approach in determining the 3D structure of NS2B-NS3 complex. Docking studies of the 4- hydroxypanduratin A compound also yielded satisfactory result and thus can be used as potential lead compounds for drug trials against YFV. Thus structure validation, mutation and docking are approaches used to control YFV that needs to be potentially harnessed in lab and if successful, will prove a break in medical genetics. The SNP modification if carried out at molecular level will decrease viral activity and thus control viral replication leading to vaccine development. The X-ray crystallographic studies on our theoretically modeled NS2B-NS3 complex structure will lead to determination of 3D structure that may further help in the study of its mechanism. Mutation studies will help us determine the structure modification undergone due to instability of the structure.
Supplementary material
Footnotes
Citation:Kannappan & Narayanan, Bioinformation 6(8): 303-306 (2011)
References
- 1.TP Monath. Lance. 1999;353:1541. doi: 10.1016/S0140-6736(99)00155-5. [DOI] [PubMed] [Google Scholar]
- 2.SE Robertson, et al. JAMA. 1996;276:1157. [Google Scholar]
- 3.BD Lindenbach, CM Rice. Adv Virus Res. 2003;59:23. doi: 10.1016/s0065-3527(03)59002-9. [DOI] [PubMed] [Google Scholar]
- 4.TJ Chambers, et al. J Virol. 1993;67:6797. doi: 10.1128/jvi.67.11.6797-6807.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DA Droll, et al. Virology. 2003;275:335. [Google Scholar]
- 6.A Sali. Mol Med Today. 1995;1:270. doi: 10.1016/s1357-4310(95)91170-7. [DOI] [PubMed] [Google Scholar]
- 7.P Erbel, et al. Nat Struct Mol Biol. 2006;13:372. doi: 10.1038/nsmb1073. [DOI] [PubMed] [Google Scholar]
- 8.AL Morris, et al. Proteins. 1992;12:345. [Google Scholar]
- 9.TA Hall, et al. Nucl Acids Symp. 1999;41:95. [Google Scholar]
- 10.Collaborative Computational Project. Acta Crystallogr D Biol Crystallogr. 1994;D50:760. doi: 10.1107/S1399004714018070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.TJ Chambers, et al. J Virol. 1991;65:6042. doi: 10.1128/jvi.65.11.6042-6050.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.TJ Chambers, et al. Proc Nati Acad Sci U S A. 1990;87:8898. doi: 10.1073/pnas.87.22.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.EY Lau, TC Bruice. Biophys J. 1999;77:85. [Google Scholar]
- 14.N Frimayanti. Int J Mol Sci. 2011;12:1089. [Google Scholar]
- 15.LY Kee, et al. Asia Pacific Journal of Molecular Biology and Biotechnology. 2007;15:53. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.