

SUMMARY
Leptin acts in the brain to prevent obesity. The underlying neurocircuitry responsible for this is poorly understood, in part due to incomplete knowledge regarding first order, leptin-responsive neurons. To address this, we and others have been removing leptin receptors from candidate first order neurons. While functionally relevant neurons have been identified, the observed effects have been small suggesting that most first order neurons remain unidentified. Here we take an alternative approach and test whether first order neurons are inhibitory (GABAergic, VGAT+) or excitatory (glutamatergic, VGLUT2+). Remarkably, the vast majority of leptin’s anti-obesity effects are mediated by GABAergic neurons; glutamatergic neurons play only a minor role. Leptin, working directly on presynaptic GABAergic neurons, many of which appear not to express AgRP, reduces inhibitory tone to postsynaptic POMC neurons. As POMC neurons prevent obesity, their disinhibition by leptin action on presynaptic GABAergic neurons likely mediates, at least in part, leptin’s anti-obesity effects.
INTRODUCTION
Leptin is secreted by adipocytes in proportion to fat stores providing feedback on the status of lipid reserves (Friedman, 2009). Leptin circulates and binds its receptor (LEPR) in the brain where it decreases food intake and promotes energy expenditure (Myers et al., 2010). When fat stores are reduced, such as by fasting, dieting, lipodystrophy or uncontrolled type 1 diabetes, blood leptin levels fall and this stimulates hunger while decreasing energy expenditure - physiologic adaptations aimed at returning fat stores to normal. Genetic deficiency of leptin or its receptor removes this adipostat signal, “misinforming” the organism about its state of energy balance and abundant fat stores. Consequently, extreme hyperphagia, reduced energy expenditure and massive obesity result. Thus, circulating leptin, by restraining food intake and maintaining energy expenditure, prevents obesity.
The neurobiological mechanisms underlying these “anti-obesity” effects are unknown. Nevertheless, key components are likely to reside in the arcuate nucleus as suggested by the convergence of numerous lines of compelling research. First, the neuropeptides, αMSH (Smart et al., 2006; Yaswen et al., 1999) and AgRP (Ollmann et al., 1997; Shutter et al., 1997), and the neurons that express them (POMC and AgRP neurons which are located primarily in the arcuate) (Bewick et al., 2005; Gropp et al., 2005; Luquet et al., 2005; Xu et al., 2005), play key roles in regulating body weight. Second, POMC neurons and AgRP neurons project to brain regions likely to be important in regulating body weight (important examples include the paraventricular nucleus and the lateral parabrachial nucleus (Bagnol et al., 1999; Elias et al., 1998; Haskell-Luevano et al., 1999; Wu et al., 2009)). Third, αMSH and AgRP agonize and antagonize, respectively, melanocortin-4 receptors (MC4Rs) (Mountjoy et al., 1992; Ollmann et al., 1997), and importantly, MC4Rs mediate marked anti-obesity effects (Balthasar et al., 2005; Huszar et al., 1997). Since POMC and AgRP neurons are the sole sources of MC4R ligands (and since MC4Rs play a critical role in regulating energy balance), POMC and AgRP neurons must be playing a similarly important role. Fourth, LEPRs are expressed by most AgRP neurons and many POMC neurons (Baskin et al., 1999a; Elias et al., 1999; Williams et al., 2010; Wilson et al., 1999), and leptin, which promotes negative energy balance, inhibits AgRP neurons and excites POMC neurons (Cowley et al., 2001; Elias et al., 1999; Takahashi and Cone, 2005; van den Top et al., 2004). In addition, leptin decreases and increases, respectively, expression of the neuropeptide genes, Agrp and Pomc (Baskin et al., 1999a; Baskin et al., 1999b; Mizuno et al., 1998; Wilson et al., 1999). These effects of leptin on neuronal activity and neuropeptide gene expression are consistent with the catabolic effects of leptin, as well as the anabolic and catabolic nature, respectively, of AgRP and POMC neurons (Bewick et al., 2005; Gropp et al., 2005; Luquet et al., 2005; Xu et al., 2005) and their neuropeptides (Ollmann et al., 1997; Smart et al., 2006; Yaswen et al., 1999). Fifth, AgRP neurons, which also release NPY and GABA, send collaterals to POMC neurons, providing an additional means by which leptin can stimulate (via disinhibition) POMC neurons (Cowley et al., 2001; Horvath et al., 1992). The above findings suggest the following model: leptin binds directly to LEPRs on AgRP and POMC neurons, inhibiting AgRP neurons and activating POMC neurons, and this accounts for its anti-obesity actions.
If this model is correct, and if its the sole mechanism by which leptin regulates energy balance, then deletion of LEPRs on AgRP and POMC neurons should result in massive obesity – similar to that seen in mice with total lack of leptin action (i.e. Lepob/ob mice and Leprdb/db mice). To investigate this, we generated mice that lack LEPRs on POMC neurons (i.e. Pomc-Cre, Leprlox/lox mice), on AgRP neurons (i.e. Agrp-Cre, Leprlox/lox mice), and on both POMC and AgRP neurons (i.e. Pomc-Cre, Agrp-Cre, Leprlox/lox mice) (Balthasar et al., 2004; Hill et al., 2010; van de Wall et al., 2008). Of note, mice lacking LEPRs on either POMC neurons or on AgRP neurons developed very mild obesity (increase in body weight of ~ 5 g at 2–3 months of age) (Balthasar et al., 2004; van de Wall et al., 2008). This effect was much smaller than expected, especially when one compares this with the 26 g increase in body weight in 10 wk old mice with global deficiency of LEPRs (van de Wall et al., 2008)). One possibility for the smaller-than-expected effect is that deletion of LEPRs in one class of neurons (for example, the POMC neurons) might possibly be compensated by increased leptin action on the other class of neurons (for example, the AgRP neurons), or visa versa. However, this was not the case because an additive, and still much smaller-than-expected effect was observed in mice lacking LEPRs on both POMC and AgRP neurons (van de Wall et al., 2008). In total, the above findings suggest that direct leptin action on POMC and AgRP neurons plays a small role in controlling energy balance, and that there are likely to be other first-order, leptin responding neurons that contribute importantly to leptin’s anti-obesity actions.
Areas beyond the arcuate could mediate important actions of leptin. Of note, LEPRs are present in many sites outside the arcuate. Within the hypothalamus, LEPRs are found in the ventromedial hypothalamus (VMH), the dorsomedial hypothalamus (DMH), the lateral hypothalamus (LH), and the ventral premammillary nucleus (PMv) (in addition to the arcuate); within the midbrain, in the ventral tegmental area and raphe; and within the brainstem, in the parabrachial nucleus, periaquiductal gray, and dorsal vagal complex (Elias et al., 2000; Figlewicz et al., 2003; Fulton et al., 2006; Grill et al., 2002; Hommel et al., 2006; Leinninger et al., 2009; Leshan et al., 2009; Mercer et al., 1996; Mercer et al., 1998; Munzberg, 2008; Scott et al., 2009). Strong arguments have been made that neurons outside the arcuate are well positioned to play important roles in regulating appetite (Berthoud, 2002; Grill and Kaplan, 2002). Indeed, injection of leptin into brain sites distant from the arcuate inhibit food intake (examples include injections into the 4th ventricle (Grill et al., 2002) and the lateral hypothalamus (Leinninger et al., 2009)).
The VMH is a site of interest given that gene knockout of SF1 causes abnormal VMH development and obesity (Majdic et al., 2002; Zhao et al., 2004). To investigate SF1 neurons, we generated Sf1-Cre, Leprlox/lox mice (Dhillon et al., 2006). These animals developed a small increase in body fat and body weight (~ 5 g increase in body weight at 2–3 months of age). Thus, as with LEPRs on POMC and AgRP neurons, the role of LEPRs on SF1 neurons is small. Another group has obtained qualitatively similar results regarding the role of LEPRs on SF1 neurons (Bingham et al., 2008).
Based on the above, the list of genetically verified, body weight-regulating, first order, leptin-responsive neurons includes POMC (Balthasar et al., 2004), AgRP (van de Wall et al., 2008) and SF1 neurons (Bingham et al., 2008; Dhillon et al., 2006). Given this, and the realization that these neurons account for only a portion of leptin’s effects (thus other neurons must also be involved), it has been proposed that leptin action is mediated by a distributed network of leptin-responsive neurons (Leinninger and Myers, 2008; Myers et al., 2009; Scott et al., 2009). With such a distributed model in mind, it is of interest to determine if any deeper logic underlies first-order, leptin-responsive neurons and/or their mode of communication with energy balance-regulating neurocircuits.
Since the obvious, “first order” candidates have already been directly tested, a new approach is needed for narrowing in on these “unidentified” first order neurons. In the present study we evaluate if leptin’s effects are mediated primarily by excitatory (glutamatergic, VGLUT2+) or inhibitory (GABAergic, VGAT+) neurons. This approach has two important features. First, it casts a wider net and provides insight into the neurotransmitter identity of the neurons mediating leptin’s anti-obesity effects. Second, it provides information regarding the function of the leptin-responsive neurons (excitatory versus inhibitory). With this goal in mind, we have generated mice that express Cre-recombinase in either glutamatergic (Vglut2-ires-Cre knockin mice) or GABAergic neurons (Vgat-ires-Cre knockin mice). VGLUT2 is one of three synaptic vesicle glutamate transporters (Takamori, 2006). VGLUT1 is expressed primarily by neurons in the cortex while VGLUT3 is expressed by isolated, select groups of neurons, none of which are in the hypothalamus. Consequently, VGLUT2 is the transporter utilized by glutamatergic neurons in the hypothalamus, thalamus, midbrain, and hindbrain – thus it is relevant to our investigation of leptin-responsive neurons. VGAT, on the other hand, is the only transporter capable of importing GABA into synaptic vesicles; hence, VGAT is expressed by all GABAergic neurons (Wojcik et al., 2006). Through use of these Cre-expressing mice, we show that the majority of leptin’s anti-obesity effects are mediated by inhibitory (GABAergic) neurons. We then build on this finding, and examine means by which leptin-responsive GABAergic neurons engage obesity-preventing POMC neurons.
RESULTS
Generation of Vgat-ires-Cre and Vglut2-ires-Cre Mice
To ensure eutopic expression of Cre recombinase by VGAT- and VGLUT2-expressing neurons, we inserted an ires-Cre cassette, by gene targeting, just downstream of the endogenous Vgat and Vglut2 stop codons, respectively (Figure 1A). The alleles are used in the heterozygous state (i.e. Vglut2ires-Cre/+, Vgatires-Cre/+), and do not have detectable effects on phenotype.
Figure 1. Generation of Vgat-ires-Cre and Vglut2-ires-Cre Mice.

(A) Mice expressing Cre recombinase under the control of the endogenous Vgat and Vglut2 genes were generated by inserting an ires-Cre cassette after the Vgat and Vglut2 stop codons, respectively.
(B–D) Immunohistochemistry for eGFP expression in Vgat-ires-Cre, lox-GFP mice; brown-staining represents Cre activity. (D) Enlargement of boxed region in (C).
(E–G) Immunohistochemistry for eGFP expression in Vglut2-ires-Cre, lox-GFP mice; brown staining represents Cre activity. (G) Enlargement of boxed region in (F).
Arc=arcuate, BLA=basolateral amygdala, CeA=central amygdala, CPu=caudate putamen, DMH=dorsomedial hypothalamus, LOT=nucleus of the lateral olfactory tract, PVN=paraventricular nucleus, SCh=suprachiasmatic nucleus, TH=thalamus, VMH=ventromedial hypothalamus, ZI=zona incerta.
To confirm that Cre is eutopically expressed, Vgat-ires-Cre and Vglut2-ires-Cre mice were crossed with lox-GFP reporter mice (Novak et al., 2000), and brains were processed for immunohistochemical detection of GFP. As is evident from Figures 1B-G and S1A, Cre activity is detected in sites where it is expected (i.e. known to be composed primarily of GABAergic or glutamatergic VGLUT2+ neurons) and is not seen in sites where it is unexpected. Brain areas known to be composed primarily of GABAergic cell bodies (see Figure S1B for Vgat mRNA in situ hybridization and supplemental text for detailed discussion and supporting references), which are depicted in Figure 1, include the caudate putamen (CPu), suprachiasmatic nucleus (SCh), central amygdaloid nucleus (CeA), and zona incerta (ZI). GABAergic areas depicted in Figure S1A include, in addition to those previously mentioned, the nucleus accumbens (ACB), lateral (LS) and medial septum (MS), reticular nucleus of the thalamus (Rt), substantia nigra pars reticulata (SNr), and Purkinje cell layer of the cerebellum. Brain areas known to be composed primarily of glutamatergic (VGLUT2+) cell bodies (see Figure S1B for Vglut2 mRNA in situ hybridization and supplemental text for detailed discussion and supporting references), which are depicted in Figure 1, include the thalamus (TH), paraventricular nucleus (PVN), nucleus of the lateral olfactory tract (LOT), basolateral nucleus of the amygdala (BLA), and ventromedial hypothalamus (VMH). Glutamatergic (VGLUT2+) areas depicted in Figure S1A include, in addition to those previously mentioned, the piriform cortex (PIR), posterior hypothalamus (PH), ventral premammillary nucleus (PMv), subthalamic nucleus (STh), medial geniculate nucleus (MG), reticulotegmental nucleus (RTg), pontine grey (PG), external cuneate nucleus (ECu) and lateral reticular nucleus (LRt). Of note, the arcuate nucleus (ARC), dorsomedial nucleus of the hypothalamus (DMH) and lateral hypothalamus contain both glutamatergic and GABAergic neurons, with GABAergic neurons predominating. A striking feature of Figures 1 and S1A, in addition to what has previously been mentioned, is the lack of Cre activity where it should not be found – i.e. in areas where cell bodies of the opposing neurotransmitter predominate. For example, in sections from Vglut2-ires-Cre mice, there is an absence of Cre activity in known GABAergic sites (in Figure 1: CPu, SCh, CeA, and ZI; in Figure S1A: ACB, LS, MS, Rt and SNr) and in sections from Vgat-ires-Cre mice, an absence of Cre activity in known glutamatergic sites (in Figure 1: TH, PVN, LOT, BLA and VMH; in Figure S1A: PIR, PH, PMv, STh, MG, RTg, PG, ECu and LRt). In accordance with this, sections from Vglut2-ires-Cre mice versus Vgat-ires-Cre mice appear as “negative images” of each other. These results are consistent with Cre being expressed in all VGLUT2+ or VGAT+ neurons, and demonstrate that Vglut2-ires-Cre and Vgat-ires-Cre mice subdivide the brain into neurons that are either excitatory (glutamatergic, VGLUT2+), inhibitory (GABAergic), or neither.
Energy Balance in Vgat-ires-Cre, Leprlox/lox and Vglut2-ires-Cre, Leprlox/lox mice
To generate study subjects, Leprlox/lox mice were mated with either Vgatires-Cre/+, Leprlox/lox mice or Vglut2ires-Cre/+, Leprlox/lox mice. From such matings, ~ 50% of all offspring are controls (i.e. Leprlox/lox mice) and ~ 50% have deletion of LEPRs in either GABAergic (Vgatires-Cre/+, Leprlox/lox mice) or glutamatergic (i.e. Vglut2ires-Cre/+, Leprlox/lox mice) neurons. Remarkably, deletion of LEPRs in GABAergic neurons of both male and female mice resulted in a massive increase in body weight (Figure 2A) and fat mass (Figure 2B), which was associated with marked hyperphagia (Figure 2C). Deletion of LEPRs in glutamatergic neurons, on the other hand, produced minimal effects (Figures 2A-2C). These latter, small effects are likely to be due to deletion of LEPRs in the VMH as neurons in this site are glutamatergic (Figure 1 and (Tong et al., 2007)) and the magnitude of effect is similar to that seen in Sf1-Cre, Leprlox/lox mice (Dhillon et al., 2006). As an additional comparison group, we generated mice that are global knockouts for a germline-deleted lox-Lepr allele (i.e. LepΔ/Δ mice) (Figure S2). Of note, the weight gain seen in Vgat-ires-Cre, Leprlox/lox mice is ~86% (in males) and ~83% (in females) of that seen in mice with total lack of LEPRs (LeprΔ/Δ mice). The weight gain seen in Vglut2-ires-Cre, Leprlox/lox mice, on the other hand, is only a small fraction of that seen in LeprΔ/Δ mice. These results demonstrate that LEPRs on GABAergic neurons mediate the vast majority of leptin’s anti-obesity effects; in comparison, LEPRs on glutamatergic (VGLUT2+) neurons play only a small role.
Figure 2. Effect of Deleting LEPRs from VGAT+ and VGLUT2+ Neurons on Body Weight, Body Composition, and Food Intake.

(A) Body weight curves of male and female mice with LEPR deleted from VGAT+ neurons (open boxes), VGLUT2+ neurons (closed triangles) or controls (Leprlox/lox, closed circles). Data are presented as mean +/− SEM. Compared to the Leprlox/lox controls, all values are significantly different except in week 4 and 7 for the Vglut2-ires-Cre, Leprlox/lox females (assessed by one-way ANOVA with Dunnett’s test of all groups compared to Leprlox/lox mice ).
(B) Fat mass and lean mass of male and female Vgat-ires-Cre, Leprlox/lox mice (left) and Vglut2-ires-Cre, Leprlox/lox mice (right) at 10 weeks of age were analyzed by EchoMRI. Data are presented as mean +/− SEM.*, p< 0.05; ***, p<0.001; unpaired t-test compared to Leprlox/lox controls.
(C) Food intake of Vgat-ires-Cre, Leprlox/lox mice (left) and Vglut2-ires-Cre, Leprlox/lox mice (right). Data are presented as mean +/− SEM.***, p<0.001; unpaired t-test compared to Leprlox/lox controls.
To determine if deletion of LEPRs in GABAergic or glutamatergic (VGLUT2+) neurons had effects on glucose homeostasis, blood glucose and insulin levels were measured (Supplemental Table I). Vgat-ires-Cre, Leprlox/lox mice had significantly elevated fed and fasted blood glucose and serum insulin levels, which is consistent with the development of obesity-induced type 2 diabetes. In contrast, but consistent with the minimal increase in fat stores, fed and fasted blood glucose levels were unchanged, and serum insulin levels were only slightly increased (fed state only) in Vglut2-ires-Cre, Leprlox/lox mice.
Of the Known First Order Neurons, Only AgRP Neurons are GABAergic
Previously established, functionally relevant, first order leptin-responsive neurons include POMC and AgRP neurons in the arcuate nucleus and SF1 neurons in the VMH (Balthasar et al., 2004; Dhillon et al., 2006; van de Wall et al., 2008), and possibly serotonin neurons in the raphe (Yadav et al., 2009). An important question is which, if any, of these neurons are GABAergic, as determined by Cre activity in Vgat-ires-Cre mice, and consequently contribute to the obesity phenotype of Vgat-ires-Cre, Leprlox/lox mice.
As previously mentioned, SF1 neurons are glutamatergic (Figure 1 and (Tong et al., 2007)). In support of this, no GABAergic neurons were found in the VMH (Figure 1). To determine if POMC neurons are GABAergic or glutamatergic, and to confirm that AgRP neurons are GABAergic (Cowley et al., 2001; Tong et al., 2008), we used immunodetectable hrGFP, expressed from POMC-hrGFP (Parton et al., 2007) and NPY-hrGFP (van den Pol et al., 2009) BAC transgenes, to identify POMC and AgRP neurons, and colocalized this with Cre activity (tdTomato, as described below). Note that NPY and AgRP are coexpressed in the arcuate nucleus (van den Pol et al., 2009). GABAergic (VGAT+) and glutamatergic (VGLUT2+) neurons were identified by immunodetectable tdTomato in Vgat-ires-Cre, lox-tdTomato mice and Vglut2-ires-Cre, lox-tdTomato mice, respectively (lox-tdTomato, Ai9, (Madisen et al., 2010)). Of note, essentially no POMC neurons (< 1 %) were VGAT+ (Figure 3A) and ~10% of POMC neurons were VGLUT2+ (Figure 3B). AgRP neurons, as expected, were GABAergic (Figure 3C). It is important to note, however, that AgRP neurons represent only a subset of all GABAergic neurons in the arcuate (Figure 3C). Also of note, POMC neurons, which are not GABAergic, are situated in a dense background of GABAergic neurons (Figure 3A). Finally, serotonin neurons in the raphe (as identified by immunohistochemistry for TpH), do not express Cre activity in Vgat-ires-Cre mice (Figure S3). Thus, AgRP neurons, but not POMC or SF1 neurons, are GABAergic and therefore are the only previously established first order neurons that contribute, directly, to the obesity seen in Vgat-ires-Cre, Leprlox/lox mice. However, as previously discussed, the contribution of LEPRs on AgRP neurons to regulation of energy balance is small (van de Wall et al., 2008). Thus, the majority of leptin’s anti-obesity must be mediated by previously uncharacterized first order leptin-responsive GABAergic neurons.
Figure 3. GABAergic (VGAT+) and Glutamatergic (VGLUT2+) Nature of POMC and AgRP Neurons.
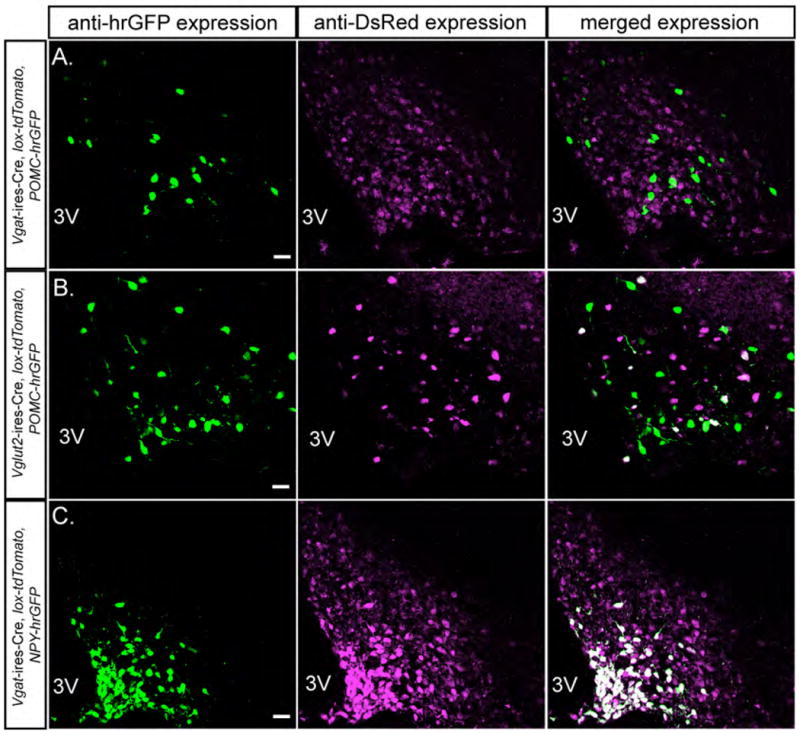
(A) Colocalization of hrGFP (POMC neurons) and DsRed (GABAergic neurons) in the arcuate in Vgat-ires-Cre, lox-tdTomato, POMC-hrGFP mice.
(B) Colocalization of the hrGFP (POMC neurons) and DsRed (glutamatergic VGLUT2+ neurons) in the arcuate in Vglut2-ires-Cre, lox-tdTomato, POMC-hrGFP mice.
(C) Colocalization of the hrGFP (AgRP/NPY neurons) and DsRed (GABAergic neurons) in the arcuate in Vgat-ires-Cre, lox-tdTomato, NPY-hrGFP mice.
Green=anti-hrGFP, Magenta=anti-DsRed, white=hrGFP+DsRed. Scale bar=20 um.
Colocalization of LEPRs with GABAergic and Glutamatergic Neurons
Given the above, a key question becomes the location of leptin-responisve GABAergic neurons. To address this, we colocalized LEPR activity, as assessed by leptin-inducible STAT3 phosphorylation, with Cre activity in Vgat-ires-Cre and Vglut2-ires-Cre, lox-GFP reporter mice, with or without neuron-specific deletion of LEPRs. Note, leptin-inducible P-STAT3 is a robust means of detecting LEPR activity (Munzberg et al., 2004). Leptin was injected (4 mg/kg BW i.p.) into mice that were fasted overnight, and 1 hour later brains were removed and assessed for P-STAT3 and GFP expression. With regards to GABAergic neurons, in control mice, P-STAT3 colocalized with GFP only in the arcuate (Figure 4A), the DMH (Figure 4B) and in the lateral hypothalamus (not shown). In the dorsal vagal complex (NTS/DMV), all P-STAT3 expression was detected in non-GABAergic neurons (Figure 4C). When LEPRs were deleted from GABAergic neurons, all colocalization disappeared; residual P-STAT3 was restricted to non-GABAergic neurons (Figures 4D, 4E and 4F). Thus, leptin-responsive GABAergic neurons are located in the arcuate, the DMH and the lateral hypothalamus.
Figure 4. Leptin-induced pSTAT3 Expression in GABAergic and Glutamatergic (VGLUT2+) Neurons With and Without LEPRs.

(A–F) Double immunohistochemical staining for pSTAT3 and eGFP in the arcuate, DMH, and NTS/DMV from leptin-injected Vgat-ires-Cre, Lepr+/+, lox-GFP mice (with LEPRs intact, A–C) and Vgat-ires-Cre, Leprlox/lox, lox-GFP mice (with LEPRs deleted from GABAergic neurons, D–F).
(G–N) Double immunohistochemical staining for pSTAT3 and eGFP in the arcuate, VMH, PMv, and NTS/DMV from leptin-injected Vglut2-ires-Cre, Lepr+/+, lox-GFP mice (with LEPRs intact, G-J) and Vglut2-ires-Cre, Leprlox/lox, lox-GFP mice (with LEPRs deleted from glutamatergic VGLUT2+ neurons, K-N).
Green=anti-eGFP, Magenta=anti-pSTAT3, white=eGFP+pSTAT3. Scale bar=20 um.
With regards to glutamatergic (VGLUT2+) neurons, in control mice, P-STAT3 colocalized with GFP only in the arcuate (small number of neurons, Figure 4G), the VMH (Figure 4H), the PMv (Figure 4I) and in the NTS/DMV (Figure 4J). When LEPRs were deleted from glutamatergic neurons, colocalization disappeared in the arcuate (Figure 4K), and in the VMH, PMv and NTS/DMV, all P-STAT3 signal was lost (Figures 4L, 4M and 4N). These findings indicate that leptin-responsive glutamatergic neurons are located primarily in the VMH, the PMv and the NTS/DMV (with a smaller number also found in the arcuate), and of note, in the VMH, PMv and NTS/DMV, 100% of LEPR-expressing neurons are glutamatergic.
Acute (In Vitro) Leptin Regulation of GABAergic Tone to POMC Neurons
POMC neurons play a critical role in preventing obesity as evidenced by massive weight gain in mice lacking αMSH (Smart et al., 2006; Yaswen et al., 1999), its receptor, MC4R (Balthasar et al., 2005; Huszar et al., 1997), and in mice with ablation of POMC neurons (Xu et al., 2005). Given this, we examined if POMC neurons are downstream of leptin-responsive GABAergic neurons. Specifically, we recorded inhibitory postsynaptic currents (IPSCs) in POMC neurons (visualized with the POMC-hrGFP BAC transgene) and assessed effects of leptin. Of interest, a prior study using 200-μm thick coronal slices found that leptin reduced IPSC frequency in POMC neurons by 25% in one third of POMC neurons, and this was attributed to AgRP/NPY GABAergic neurons (Cowley et al., 2001). In our studies, we prepared thicker slices (300-μm), positing that this might preserve more connections between the GABAergic and POMC neurons. In Figures 5, 6 and 7, we report effects on all neurons tested.
Figure 5. Effects of Leptin Addition on sIPSC Frequency in POMC Neurons.

(A) Left–Time course of effects of leptin (100 nM) (blue squares) or of no additions (pink circles) on sIPSC frequency in POMC neurons from control (Leprlox/lox) mice. Right - Example traces of sIPSCs recorded from a POMC neuron i) just before and ii) after 25–30 minutes of leptin addition.
(B) Effects of 25–30 minutues of leptin treatment on sIPSC frequency, expressed as percent of baseline (prior to leptin addition), in POMC neurons from Leprlox/lox (control) mice, Vgat-ires-Cre, Leprlox/lox mice, AgRP-ires-Cre, Leprlox/lox mice, and POMC-Cre, Leprlox/lox mice, and also from Vgatlox/lox (control) mice and AgRP-ires-Cre, Vgatlox/lox mice (lacking VGAT in AGRP neurons). Data are presented as mean +/− SEM. **, p<0.01; ***, p<0.001; ****, p<0.0001, paired t-test for effect of leptin versus baseline (see “Supplemental Procedures” for details on how percent baseline was determined).
Figure 6. Effects of Deleting LEPRs Globally (Lepr Δ/Δ mice), from GABAergic Neurons, from AgRP neurons or from POMC Neurons on Inhibitory Tone to POMC Neurons.

(A) Summary of sIPSC and mIPSC frequency (top left) and amplitude (bottom left) in POMC neurons from Leprlox/lox (control) mice, Vgat-ires-Cre, Leprlox/lox mice, LeprΔ/Δ mice, POMC-Cre, Leprlox/lox, and AgRP-ires-Cre, Leprlox/lox mice. Data are presented as mean +/− SEM. **, p<0.01; ***, p<0.001; ****, p<0.0001 one-way ANOVA with Dunnett’s test of all groups compared to Leprlox/lox mice. (Top right) Example traces of sIPSCs recorded from POMC neurons of a Leprlox/lox (control) mouse and a Vgat-ires-Cre, Leprlox/lox mouse. (Bottom right) Cumulative probability distribution of mIPSC amplitudes from POMC neurons showing a significant rightward shift in Vgat-ires-Cre, Leprlox/lox mice compared to Leprlox/lox (control) mice.
(B) Summary of membrane potential and firing rate of POMC neurons from Leprlox/lox (control) mice and Vgat-ires-Cre, Leprlox/lox mice before and after the addition of the GABAA receptor blocker, picrotoxin. Data are presented as mean +/− SEM.
Figure 7. Effects of Fasting on sIPSC Frequency and Amplitude in POMC Neurons.

(A) Summary of the effects of fasting on sIPSC frequency (left) and amplitude (right) in POMC neurons of fed Leprlox/lox mice, fasted Leprlox/lox mice, and fasted+saline- or fasted+leptin-injected Leprlox/lox mice. Data are presented as mean +/− SEM. ***, p<0.001 one-way ANOVA with Dunnett’s test of all groups compared to fed Leprlox/lox mice.
(B) Summary of the effects of fasting on sIPSC frequency (left) and amplitude (right) in POMC neurons of fed and fasted Vgat-ires-Cre, Leprlox/lox mice. Data are presented as mean +/− SEM. ***, p<0.001; ****, p<0.0001; t-test compared to fed Vgat-ires-Cre, Leprlox/lox mice.
Addition of leptin decreased spontaneous IPSC (sIPSC) frequency in POMC neurons by 40% (Figures 5A and 5B). This effect was not dependent upon action potentials because, in the presence of tetrodotoxin (TTX), leptin reduced miniature-IPSC (mIPSC) frequency to a similar extent (Figure S4A). We and others (Cowley et al., 2001; Pinto et al., 2004) have observed that frequency and amplitude of sIPSCs in POMC neurons are minimally affected by the addition of TTX, demonstrating that most sIPSCs in POMC neurons, in the context of brain slice preparations, originate from spontaneous vesicle fusion events in presynaptic GABAergic neurons. Unlike the situation in POMC neurons, leptin failed to reduce sIPSC frequency in AgRP neurons (visualized with the NPY-hrGFP BAC transgene) (Figure S4B), indicating that leptin-mediated suppression of IPSCs is not a generalized phenomenon. Of interest, leptin’s inhibitory effect on IPSCs in POMC neurons was absent in Vgat-ires-Cre, Leprlox/lox mice and persisted, unaffected, in Pomc-Cre, Leprlox/lox mice (Figure 5B), demonstrating that LEPRs on presynaptic GABAergic neurons, and not postsynaptic POMC neurons, mediate this response. We next evaluated the role of LEPRs on AgRP neurons, which as mentioned above, are GABAergic and are thought to be in synaptic contact with POMC neurons (Cowley et al., 2001). Of note, in mice lacking LEPRs on AgRP neurons, leptin reduced IPSC frequency in POMC neurons by 31%, representing a small attenuation of the inhibitory response (40%) noted above in control mice. This suggests that the majority of leptin’s effect is mediated by LEPRs on “non-AgRP” GABAergic neurons. To confirm a prominent role for non-AgRP GABAergic neurons, we utilized mice in which AgRP neurons are unable to release GABA (Agrp-ires-Cre, Vgatlox/lox mice (Tong et al., 2008)). In this case, all IPSCs originate exclusively from non-AgRP GABAergic neurons. Of note, leptin’s inhibitory effect on IPSC frequency was still present (Figure 5B), establishing an important role for non-AgRP GABAergic neurons. To summarize, addition of leptin to brain slices reduces IPSC frequency in POMC neurons and this effect is mediated by LEPRs on presynaptic GABAergic neurons, the majority of which appear to be non-AgRP GABAergic neurons.
Long-term (In Vivo) Leptin Regulation of GABAergic Tone to POMC Neurons
We next evaluated if manipulation of LEPRs, in vivo, altered GABAergic tone to POMC neurons. Of note, a previous study reported that mice lacking leptin (Lepob/ob mice) have a marked increase in IPSC frequency in POMC neurons (Pinto et al., 2004). In agreement with this, we observed that mice with global lack of LEPRs (LeprΔ/Δ mice) have a large increase in sIPSC and mIPSC frequency in POMC neurons (Figure 6A), and also in amplitude. This latter effect on amplitude appears to occur in Lepob/ob mice as well (Pinto et al., 2004). In addition, we observed an equally large increase in IPSC frequency and amplitude in POMC neurons from Vgat-ires-Cre, Leprlox/lox mice, but found no increase in POMC neurons from Pomc-Cre, Leprlox/lox mice (Figure 6A), demonstrating that deficient leptin signaling in presynaptic GABAergic neurons, but not postsynaptic POMC neurons, increases inhibitory tone in POMC neurons. This effect appears not to be caused by deletion of LEPRs from AgRP neurons since no increase in IPSC frequency or amplitude was observed in POMC neurons from AgRP-ires-Cre, Leprlox/lox mice (Figure 6A).
Deletion of LEPRs in VGAT+ neurons did not increase IPSC frequency or amplitude in AgRP neurons - while not statistically significant, IPSC frequency in AgRP neurons actually tended to decrease in Vgat-ires-Cre, Leprlox/lox mice (Figure S5A). Furthermore, deletion of LEPRs in glutamatergic (VGLUT2+) neurons did not affect excitatory postsynaptic currents (sEPSCs and mEPSCs) in POMC neurons (Figure S5B). In summary, deficiency of leptin signaling in presynaptic, non-AgRP GABAergic neurons, but not postsynaptic POMC neurons, selectively increases inhibitory tone in POMC neurons.
To determine if POMC neurons are affected by this increased GABAergic tone, we assessed their membrane potential and firing rate. POMC neurons from Vgat-ires-Cre, Leprlox/lox mice, compared with neurons from control mice, tended to be hyperpolarized (−62.1 +/−1.94 mV compared with −57.8 +/− 2.8 mV in control mice; Figure 6B, left panel) and, consistent with this, addition of the GABAA receptor blocker, picrotoxin (PTX) in Vgat-ires-Cre, Leprlox/lox mice produced a greater degree of depolarization. PTX addition increased membrane potential by 6.4 +/− 0.97 mV in Vgat-ires-Cre, Leprlox/lox mice compared with only 3.2 +/− 1.01 mV in control mice (p<0.05, t-test). In agreement with this, their firing rate was markedly reduced, 0.32 +/− 0.11 Hz in Vgat-ires-Cre, Leprlox/lox mice compared with 1.81 Hz +/− 0.37 in control mice (p=0.01, t-test; Figure 6B, right panel), and this reduction was markedly attenuated by PTX. PTX addition increased firing rate by 11.6 +/− 6.2 fold in Vgat-ires-Cre, Leprlox/lox mice and by only 1.2 +/− 0.1 fold in control mice (p=0.01, Mann-Whitney test). These findings support the view that deficiency of leptin signaling in presynaptic GABAergic neurons inhibits the activity of POMC neurons.
Physiologic (In Vivo) Leptin Regulation of GABAergic Tone to POMC Neurons
We next evaluated whether a physiologic reduction in circulating leptin, as occurs with fasting (Ahima et al., 1996), also increases inhibitory input to POMC neurons. This is a key question because the marked effects observed in Figure 6, while suggestive of important regulation, might be seen only with “unphysiologic”, total absence of leptin signaling. Our studies, described below, were motivated by a prior study in which fasting markedly increased the firing rate of AgRP neurons (which are GABAergic) - an effect that was prevented by leptin treatment 3 hours prior to sacrifice (Takahashi and Cone, 2005). Of interest, we found that fasting for 24 hours produced a marked increase in sIPSC frequency and amplitude in POMC neurons (Figure 7A). Importantly, these fasting-mediated effects were completely prevented by injection of leptin (4 mg/kg), but not saline, 3 hrs prior to sacrifice. Complete prevention of the fasting-stimulated increase in IPSCs by leptin treatment is consistent with the view that increased inhibitory tone caused by fasting is, indeed, due to the fasting-mediated fall in leptin. We then assessed the effects of fasting in mice lacking LEPRs on GABAergic neurons (Vgat-ires-Cre, Leprlox/lox mice). Of note, fasting in these mice failed to increase sIPSC frequency and amplitude (Figure 7B), which, as noted earlier, are increased in the fed state compared with control mice (Figure 6A). Amplitude not only failed to increase but actually decreased with fasting in Vgat-ires-Cre, Leprlox/lox mice, which is a paradoxical response presumably unmasked by the absence of leptin signaling. To summarize, a physiologic fall in circulating leptin (induced by fasting in this case) causes a marked increase in inhibitory tone to POMC neurons, and this effect is likely mediated by decreased leptin action on presynaptic GABAergic neurons.
DISCUSSION
Despite intensive investigation, the neuronal, subcellular and molecular mechanisms responsible for leptin’s anti-obesity effects are incompletely understood. While strong evidence suggests a role for AgRP and POMC neurons in the arcuate, it is unclear to what degree, and by what means (directly or indirectly), leptin engages these functionally important neurons. Uncertainty about the direct neuronal targets of leptin (i.e. the first order, leptin-responsive neurons) has been a key obstacle. The elusive nature of these first order neurons, combined with other findings, suggests the possibility that leptin action occurs through a distributed network of leptin-responsive neurons. If this is so, it then becomes important to establish if any deeper organizing principles underlie leptin-responsive first order neurons and their integration with body weight-regulating neurocircuitry.
As part of a search for higher order, we have determined the inhibitory/excitatory nature of leptin-responsive, body weight-regulating neurons. Specifically, we used Vgat-ires-Cre and Vglut2-ires-Cre knock-in mice to manipulate LEPR expression on GABAergic (VGAT+) and glutamatergic (VGLUT2+) neurons, and then tested for effects on energy balance. Remarkably, we find that leptin’s anti-obesity effects are mediated predominantly by GABAergic (VGAT+) neurons, and that glutamatergic (VGLUT2+) neurons play only a small role. Importantly, this raises the likely possibility that modulation of GABAergic output is a key aspect of leptin action. Consistent with this, we find, using multiple approaches, that leptin action on presynaptic GABAergic neurons markedly decreases inhibitory tone to postsynaptic POMC neurons. This regulation was observed using the following paradigms: a) addition of leptin in vitro (Figure 5), b) removal of leptin signaling selectively from presynaptic GABAergic neurons in vivo (Figure 6), and c) importantly, with physiologic reductions in circulating leptin brought about by fasting (Figure 7). Given the previously established role of POMC neurons in preventing obesity (Smart et al., 2006; Xu et al., 2005; Yaswen et al., 1999), these effects of leptin on presynaptic GABAergic neurons provide a basis for leptin’s anti-obesity effects. Of note, indirect regulation of POMC neurons by leptin reconciles the known important role of POMC neurons in regulating body weight with the relatively unimportant role played by direct leptin action on POMC neurons (Balthasar et al., 2004; Hill et al., 2010; van de Wall et al., 2008). While in our studies we have focused on POMC neurons as the downstream target of leptin-responsive GABAergic neurons, it’s certainly possible and perhaps likely that other anti-obesity neurons are similarly disinhibited by leptin action on presynaptic GABAergic neurons. This will need to be tested as other anti-obesity neurons are identified. Given the above-mentioned findings, GABAergic output appears to be an important, direct target of leptin action.
Identity and Location of Leptin-Responsive GABAergic Neurons
Using leptin-inducible P-STAT3 and GFP reporter expression (in Vgat-ires-Cre mice) to colocalize LEPRs and GABAergic neurons, we observed that LEPR-expressing GABAergic neurons are located in the arcuate, the DMH, and the lateral hypothalamus. Consequently, LEPR-expressing GABAergic neurons in one, two or all three of these sites mediate leptin’s anti-obesity effects. At present, our results do not allow us to rule in or out any one, or any combination of these sites. Nevertheless, for reasons listed below, we favor an important role for neurons in the arcuate. First, LEPR-expressing arcuate neurons have unparalleled access to circulating leptin (Faouzi et al., 2007). Second, the arcuate has many GABAergic neurons, a small fraction of which are AgRP neurons (Figure 3C and (Acuna-Goycolea et al., 2005; Horvath et al., 1997; Ovesjo et al., 2001)). Third, POMC neurons, which are key targets of leptin-responsive GABAergic neurons (Figures 5–7 and (Cowley et al., 2001)), are located within the arcuate, surrounded by a dense population of GABAergic neurons (Figure 3A). Fourth, neurons in the arcuate make many local connections (Matsumoto and Arai, 1978; van den Pol and Cassidy, 1982), providing the apparatus for local regulation of POMC neurons. One previously defined local circuit, likely to be physiologically important, is that between AgRP neuron collaterals and POMC neurons (Cowley et al., 2001; Horvath et al., 1992). As discussed below, we postulate that this is just one of many local leptin-responsive GABAergic inputs to POMC neurons.
Role of AgRP GABAergic Neurons versus Other GABAergic Neurons
An earlier study (Cowley et al., 2001) established that leptin reduces the frequency of IPSCs in POMC neurons (25% reduction in one third of POMC neurons). The source of the reduced GABAergic input was attributed to AgRP neurons (which also express NPY). In the present study, we confirm leptin’s inhibitory effect on IPSC frequency, but, of interest, note a larger effect (40% inhibition in all POMC neurons), perhaps due to our use of thicker brain slices (300 μm versus 200 μm). A key outcome of that prior study was the compelling proposal that leptin indirectly regulates POMC neurons via AgRP/NPY-GABAergic collaterals. The degree to which this accounts for leptin’s anti-obesity effects, however, has been unclear, especially since deletion of LEPRs from AgRP neurons produces only a small disturbance in energy balance (van de Wall et al., 2008).
In the present study, we show that the above-mentioned effect (i.e. leptin-mediated reduction of IPSC frequency in POMC neurons) is mediated by LEPRs on presynaptic GABAergic neurons, and that LEPRs on postsynaptic POMC neurons play no role. Furthermore, our data indicates that LEPRs on “non-AgRP” GABAergic neurons are predominantly responsible for this effect. The following three findings support this view: a) leptin-mediated reduction of IPSC frequency is minimally affected when LEPRs are deleted from AgRP neurons (Agrp-ires-Cre, Leprlox/lox mice), but is totally abrogated when LEPRs are deleted from all GABAergic neurons (Vgat-ires-Cre, Leprlox/lox mice, Figure 5B), b) this response is unimpaired in mice which cannot release GABA from AgRP neurons (Agrp-ires-Cre, Vgatlox/lox mice, Figure 5B), and c) deletion of LEPRs from GABAergic neurons (Vgat-ires-Cre, Leprlox/lox mice) markedly increases IPSC frequency and amplitude in POMC neurons, while in contrast, no effect is seen when LEPRs are deleted from AgRP neurons (Agrp-ires-Cre, Leprlox/lox mice, Figure 6A). These results clearly attest to the important role played by “non-AgRP” neurons in leptin-mediated disinhibition of POMC neurons, and, of interest, are congruent with the presence of massive obesity versus minimal obesity, respectively, in Vgat-ires-Cre, Leprlox/lox mice (Figure 2) versus Agrp-ires-Cre, Leprlox/lox mice (van de Wall et al., 2008). One notable caveat of the above analysis is the possibility of compensation, as was observed following diphtheria toxin-mediated ablation of AGRP neurons in neonates (Luquet et al., 2005). If such compensation were to occur following genetic deletion of LEPRs in AgRP neurons, then our approach could underestimate the contribution of AgRP GABAergic neurons. However, given that toxin ablation kills neurons, while LEPR deletion, on the other hand, leaves neurons largely intact, it is unclear if similar degrees or forms of compensation should be expected. To summarize, our results and those of others (Cowley et al., 2001) demonstrate that leptin reduces inhibitory tone to POMC neurons. This effect is mediated by LEPRs on presynaptic GABAergic neurons, some of which may express AgRP, many of which likely do not.
Leptin Action via GABAergic Neurons: Issues and Implications
It has previously been established that leptin’s anti-obesity effects require Tyr1138 of the LEPR, which allows for phosphorylation-dependent docking and activation (via subsequent phosphorylation) of the latent transcription factor, STAT3 (Bates et al., 2003). Of note, marked obesity, similar in magnitude to that observed in mice totally lacking LEPRs, occurs in mice homozygous for the LeprS1138 allele. This requirement for Tyr1138 strongly implicates STAT3-mediated gene expression in leptin’s anti-obesity effects. The relevant downstream transcriptional targets, however, are not yet known but are of great interest. Prior studies have focused on the Pomc gene (Munzberg et al., 2003). However, given the important role of leptin-responsive GABAergic neurons in regulating body weight, most of which do not express AgRP, and none of which appear to express POMC (Figure 3 and (Ovesjo et al., 2001; Yee et al., 2009), and for a dissenting view - (Hentges et al., 2009)), it will be important to reexamine this issue. Perhaps the key leptin-responsive target genes do not encode neuropeptides, but instead, proteins involved in modulating GABA release, or possibly proteins that regulate plasticity of GABAergic synapses (Pinto et al., 2004).
Related to the above, it has been reported that leptin’s ability to acutely decrease IPSC frequency in POMC neurons is unaltered in LeprS1138/S1138 mice (Munzberg et al., 2007). Specifically, intact inhibition was observed in 2 out of 5 POMC neurons (see Figure 3B of that prior study). Given that LeprS1138/S1138 mice are massively obese, this suggests that leptin-mediated acute suppression of IPSC frequency in POMC neurons, by itself, cannot prevent obesity, and, in addition, that it is not dependent on STAT3-mediated signaling. Alternatively, acute leptin suppression of IPSC frequency may still play an important role, as we hypothesize, but given the above, this role in regulating body weight may require intact STAT3 signaling as a necessary precondition. Resolution of these issues will require further investigation.
Finally, the subcellular site of action and molecular mechanism by which leptin modulates GABA release is unclear. Of interest, acute and chronic leptin modulation of GABA release, at least as detected in vitro in brain slices, is entirely independent of action potentials (i.e. occurs in the presence of TTX) (Figure S4A, Figure 6A and (Cowley et al., 2001; Pinto et al., 2004)). This, combined with our observation that leptin works on presynaptic GABAergic neurons to produce its effects, raises the distinct possibility that GABAergic axon terminals are the ultimate subcellular site of action for leptin’s effects. Whether this involves transcription/translation of proteins that subsequently affect the function of axon terminals or, alternatively, leptin receptor/signaling pathways that are self-contained within axon terminals, is presently unknown. Given the key role of leptin action on GABAergic neurons, further studies will be needed to address these interesting possibilities.
EXPERIMENTAL PROCEDURES
Animal Care
Care of all animals and procedures was approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Generation of Vgat-ires-Cre and Vglut2-ires-Cre mice
Vgat-ires-Cre and Vglut2-ires-Cre knockin mice were generated by gene targeting using the same approach as described previously (Tong et al., 2008). For details see Supplemental Information section.
Electrophysiological Studies
Slice preparation and whole-cell recordings
Brain slices were prepared from young adult mice (5–7 weeks old) as described previously (Dhillon et al., 2006) with the exception that 300 μM thick coronal sections were cut with a Leica VT1000S Vibratome.
Measuring inhibitory and excitatory post-synaptic currents
IPSCs and EPSCs were measured in whole-cell voltage clamp mode with a holding potential of -60 mV. The internal recording solution contained: 140 mM CsCl, 1 mM BAPTA, 10 mM HEPES, 5 mM MgCl2, 2 mM Mg-ATP, 0.3 mM Na-GTP (pH 7.35 with NaOH). Currents were amplified, filtered at 1kHz, and digitized at 20 kHz. EPSCs were measured in the presence of picrotoxin (100μM). IPSCs were measured in the presence of NBQX (10 μM) + D, L-APV (50μM) or Kynurenate (1mM). Miniature EPSCs and IPSCs were recorded with 1 μm tetrodotoxin in aCSF recording solution. Frequency and peak amplitude were measured using the Mini Analysis program (Synaptosoft, Inc.). Cumulative probability distribution for mIPSC amplitudes (Figure 6A) was measured for 3 minute periods (Figure 6A).
Measuring membrane potential and firing rate
Membrane potential and firing rate were measured by whole-cell current clamp recordings from POMC neurons in brain slices from Leprlox/lox mice and Vgat-ires-Cre, Leprlox/lox mice. Recording electrodes had resistances of 2.5–4 MΩ when filled with the K-gluconate internal solution (128 mM K-gluconate, 10 mM HEPES, 1 mM EGTA, 10 mM KCL, 1 mM MgCl2, 0.3 mM CaCl2, 2 mM Mg-ATP, 0.3 mM Na-GTP (pH 7.35 with KOH).
Supplementary Material
Acknowledgments
We would like to thank members of the Lowell lab for helpful discussions, C. B. Saper, C. Bjorbaek and B. P. Bean for advice, J. K. Elmquist and D. P. Olson for giving comments on the manuscript, and M. Herman for help with statistics. This work was supported by grants from the National Institute of Health/National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK089044, R01 DK075632, P30 DK046200, and P30 DK057521 to BBL; F32 DK078478 to LV).
Footnotes
Supplemental information includes five supplemental figures, a table, supplemental experimental procedures, supplemental text, and supplemental references.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acuna-Goycolea C, Tamamaki N, Yanagawa Y, Obata K, van den Pol AN. Mechanisms of neuropeptide Y, peptide YY, and pancreatic polypeptide inhibition of identified green fluorescent protein-expressing GABA neurons in the hypothalamic neuroendocrine arcuate nucleus. J Neurosci. 2005;25:7406–7419. doi: 10.1523/JNEUROSCI.1008-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- Bagnol D, Lu XY, Kaelin CB, Day HE, Ollmann M, Gantz I, Akil H, Barsh GS, Watson SJ. Anatomy of an endogenous antagonist: relationship between Agouti-related protein and proopiomelanocortin in brain. J Neurosci. 1999;19:RC26. doi: 10.1523/JNEUROSCI.19-18-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Jr, Elmquist JK, Lowell BB. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Baskin DG, Breininger JF, Schwartz MW. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes. 1999a;48:828–833. doi: 10.2337/diabetes.48.4.828. [DOI] [PubMed] [Google Scholar]
- Baskin DG, Hahn TM, Schwartz MW. Leptin sensitive neurons in the hypothalamus. Horm Metab Res. 1999b;31:345–350. doi: 10.1055/s-2007-978751. [DOI] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- Berthoud HR. Multiple neural systems controlling food intake and body weight. Neurosci Biobehav Rev. 2002;26:393–428. doi: 10.1016/s0149-7634(02)00014-3. [DOI] [PubMed] [Google Scholar]
- Bewick GA, Gardiner JV, Dhillo WS, Kent AS, White NE, Webster Z, Ghatei MA, Bloom SR. Post-embryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype. FASEB J. 2005;19:1680–1682. doi: 10.1096/fj.04-3434fje. [DOI] [PubMed] [Google Scholar]
- Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology. 2008;149:2138–2148. doi: 10.1210/en.2007-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–281. [PubMed] [Google Scholar]
- Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, et al. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol. 1998;402:442–459. [PubMed] [Google Scholar]
- Faouzi M, Leshan R, Bjornholm M, Hennessey T, Jones J, Munzberg H. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology. 2007;148:5414–5423. doi: 10.1210/en.2007-0655. [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 2003;964:107–115. doi: 10.1016/s0006-8993(02)04087-8. [DOI] [PubMed] [Google Scholar]
- Friedman JM. Leptin at 14 y of age: an ongoing story. Am J Clin Nutr. 2009;89:973S–979S. doi: 10.3945/ajcn.2008.26788B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, Maratos-Flier E, Flier JS. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51:811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Kaplan JM. The neuroanatomical axis for control of energy balance. Front Neuroendocrinol. 2002;23:2–40. doi: 10.1006/frne.2001.0224. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, Baskin DG. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology. 2002;143:239–246. doi: 10.1210/endo.143.1.8589. [DOI] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C, Chen P, Li C, Chang K, Smith MS, Cameron JL, Cone RD. Characterization of the neuroanatomical distribution of agouti-related protein immunoreactivity in the rhesus monkey and the rat. Endocrinology. 1999;140:1408–1415. doi: 10.1210/endo.140.3.6544. [DOI] [PubMed] [Google Scholar]
- Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ. Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci. 2009;29:13684–13690. doi: 10.1523/JNEUROSCI.3770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, Cho YR, Chuang JC, Xu Y, Choi M, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C. Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res. 1997;756:283–286. doi: 10.1016/s0006-8993(97)00184-4. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Naftolin F, Kalra SP, Leranth C. Neuropeptide-Y innervation of beta-endorphin-containing cells in the rat mediobasal hypothalamus: a light and electron microscopic double immunostaining analysis. Endocrinology. 1992;131:2461–2467. doi: 10.1210/endo.131.5.1425443. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, et al. Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. 2009;10:89–98. doi: 10.1016/j.cmet.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinninger GM, Myers MG., Jr LRb signals act within a distributed network of leptin-responsive neurones to mediate leptin action. Acta Physiol (Oxf) 2008;192:49–59. doi: 10.1111/j.1748-1716.2007.01784.x. [DOI] [PubMed] [Google Scholar]
- Leshan RL, Louis GW, Jo YH, Rhodes CJ, Munzberg H, Myers MG., Jr Direct innervation of GnRH neurons by metabolic- and sexual odorant-sensing leptin receptor neurons in the hypothalamic ventral premammillary nucleus. J Neurosci. 2009;29:3138–3147. doi: 10.1523/JNEUROSCI.0155-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, McGarry JD, Parker KL. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143:607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Arai Y. Morphologic evidence for intranuclear circuits in the hypothalamic arcuate nucleus. Exp Neurol. 1978;59:404–412. doi: 10.1016/0014-4886(78)90232-7. [DOI] [PubMed] [Google Scholar]
- McMinn JE, Liu SM, Dragatsis I, Dietrich P, Ludwig T, Eiden S, Chua SC., Jr An allelic series for the leptin receptor gene generated by CRE and FLP recombinase. Mamm Genome. 2004;15:677–685. doi: 10.1007/s00335-004-2340-1. [DOI] [PubMed] [Google Scholar]
- Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996;387:113–116. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- Mercer JG, Moar KM, Hoggard N. Localization of leptin receptor (Ob-R) messenger ribonucleic acid in the rodent hindbrain. Endocrinology. 1998;139:29–34. doi: 10.1210/endo.139.1.5685. [DOI] [PubMed] [Google Scholar]
- Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and [corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47:294–297. doi: 10.2337/diab.47.2.294. [DOI] [PubMed] [Google Scholar]
- Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- Munzberg H. Differential leptin access into the brain--a hierarchical organization of hypothalamic leptin target sites? Physiol Behav. 2008;94:664–669. doi: 10.1016/j.physbeh.2008.04.020. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. 2003;144:2121–2131. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Jobst EE, Bates SH, Jones J, Villanueva E, Leshan R, Bjornholm M, Elmquist J, Sleeman M, Cowley MA, Myers MG., Jr Appropriate inhibition of orexigenic hypothalamic arcuate nucleus neurons independently of leptin receptor/STAT3 signaling. J Neurosci. 2007;27:69–74. doi: 10.1523/JNEUROSCI.3168-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr, Munzberg H, Leinninger GM, Leshan RL. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 2009;9:117–123. doi: 10.1016/j.cmet.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- Ovesjo ML, Gamstedt M, Collin M, Meister B. GABAergic nature of hypothalamic leptin target neurones in the ventromedial arcuate nucleus. J Neuroendocrinol. 2001;13:505–516. doi: 10.1046/j.1365-2826.2001.00662.x. [DOI] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutter JR, Graham M, Kinsey AC, Scully S, Luthy R, Stark KL. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes Dev. 1997;11:593–602. doi: 10.1101/gad.11.5.593. [DOI] [PubMed] [Google Scholar]
- Smart JL, Tolle V, Low MJ. Glucocorticoids exacerbate obesity and insulin resistance in neuron-specific proopiomelanocortin-deficient mice. J Clin Invest. 2006;116:495–505. doi: 10.1172/JCI25243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi KA, Cone RD. Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology. 2005;146:1043–1047. doi: 10.1210/en.2004-1397. [DOI] [PubMed] [Google Scholar]
- Takamori S. VGLUTs: ‘exciting’ times for glutamatergic research? Neurosci Res. 2006;55:343–351. doi: 10.1016/j.neures.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5:383–393. doi: 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wall E, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, Jo YH, MacKenzie RG, Allison DB, Dun NJ, et al. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology. 2008;149:1773–1785. doi: 10.1210/en.2007-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Cassidy JR. The hypothalamic arcuate nucleus of rat--a quantitative Golgi analysis. J Comp Neurol. 1982;204:65–98. doi: 10.1002/cne.902040108. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Yao Y, Fu LY, Foo K, Huang H, Coppari R, Lowell BB, Broberger C. Neuromedin B and gastrin-releasing peptide excite arcuate nucleus neuropeptide Y neurons in a novel transgenic mouse expressing strong Renilla green fluorescent protein in NPY neurons. J Neurosci. 2009;29:4622–4639. doi: 10.1523/JNEUROSCI.3249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Top M, Lee K, Whyment AD, Blanks AM, Spanswick D. Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat Neurosci. 2004;7:493–494. doi: 10.1038/nn1226. [DOI] [PubMed] [Google Scholar]
- Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF, Elmquist JK. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. 2010;30:2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BD, Bagnol D, Kaelin CB, Ollmann MM, Gantz I, Watson SJ, Barsh GS. Physiological and anatomical circuitry between Agouti-related protein and leptin signaling. Endocrinology. 1999;140:2387–2397. doi: 10.1210/endo.140.5.6728. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Wu Q, Boyle MP, Palmiter RD. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell. 2009;137:1225–1234. doi: 10.1016/j.cell.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Morton GJ, Ogimoto K, Stanhope K, Graham J, Baskin DG, Havel P, Schwartz MW, Barsh GS. Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol. 2005;3:e415. doi: 10.1371/journal.pbio.0030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav VK, Oury F, Suda N, Liu ZW, Gao XB, Confavreux C, Klemenhagen KC, Tanaka KF, Gingrich JA, Guo XE, et al. A Serotonin-Dependent Mechanism Explains the Leptin Regulation of Bone Mass, Appetite, and Energy Expenditure. Cell. 2009;138:976–989. doi: 10.1016/j.cell.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- Yee CL, Wang Y, Anderson S, Ekker M, Rubenstein JL. Arcuate nucleus expression of NKX2.1 and DLX and lineages expressing these transcription factors in neuropeptide Y(+), proopiomelanocortin(+), and tyrosine hydroxylase(+) neurons in neonatal and adult mice. J Comp Neurol. 2009;517:37–50. doi: 10.1002/cne.22132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Bakke M, Hanley NA, Majdic G, Stallings NR, Jeyasuria P, Parker KL. Tissue-specific knockouts of steroidogenic factor 1. Mol Cell Endocrinol. 2004;215:89–94. doi: 10.1016/j.mce.2003.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.