

Summary
Transfusion-related acute lung injury (TRALI) is the most common cause of serious morbidity and mortality due to hemotherapy. Although the pathogenesis has been related to the infusion of donor antibodies into the recipient, antibody negative TRALI has been reported. Changes in transfusion practices, especially the use of male-only plasma, have decreased the number of antibody-mediated cases and deaths; however, TRALI still occurs. The neutrophil appears to be the effector cell in TRALI and the pathophysiology is centered on neutrophil-mediated endothelial cell cytotoxicity resulting in capillary leak and ALI. This review will detail the pathophysiology of TRALI including recent pre-clinical data, provide insight into newer areas of research, and critically assess current practices to decrease it prevalence and to make transfusion safer.
Introduction
Transfusion-related acute lung injury (TRALI) is the most common cause of transfusion-related death world-wide. First described in the 1950s’ the clinical term was not coined until 1985 (1). The initial reports of TRALI occurred in relatively healthy patients with the first large series reported on patients who required transfusion after recent surgery. Newer animal models have been developed and in vitro assays using primary human cells to mimic the condition have been used. TRALI can certainly be caused by a number of mediators and each requires some specific constraints and must be thought of in context to the blood product from which it originates.
Diagnosis and Treatment
TRALI is a clinical diagnosis, and while laboratory data may support the diagnosis it is not required (2;3). TRALI occurs within 6 hours of transfusion with the majority of cases presenting during the transfusion or within the first 2 hours (1;4;5). TRALI is the insidious onset of acute pulmonary insufficiency presenting as tachypnea, cyanosis, and dyspnea with acute hypoxemia, PaO2/FiO2<300 mmHg, and decreased pulmonary compliance, despite normal cardiac function (1;4-8). Radiographic examination reveals diffuse, fluffy infiltrates consistent with pulmonary edema (1;7). TRALI is the new onset or worsening of pulmonary function with hypoxemia that satisfies the international criteria for ALI (PaO2/FiO2<300 mmHg) with a chest x-ray consistent with pulmonary edema occurring during or 6 hours within transfusion (2;3). What differs between the National Heart Lung and Blood Institute (NHLBI) and the Canadian Consensus Conference definitions is that the in the NHLBI definition other risk factors for ALI may be present, while the Canadian Consensus Conmference defitinition designates these conditions as “possible TRALI” (2;3). In any case, transfusion must be envisioned as the inciting event (2;3). All blood components have been implicated in TRALI; however, plasma containing blood components are most commonly implicated with fresh frozen plasma (FFP) and whole blood-derived platelet concentrates (WB-PLTs) having caused the largest number of reported cases (5;7;9). In addition, plasma is considered one of the most hazardous transfused components, mainly because of it association with TRALI, and at most centers the plasma is platelet-rich plasma because most blood collection facilities do not make platelet concentrates (PCs) from whole blood collections (10). This use of platelet-rich plasma is significant for it allows for the infusion of platelet fragments and all endogenous growth factors and other mediators which are platelet-derived. Many of these compounds are effective activators of PMNs and innate immunity including soluble CD40 ligand, from platelet membranes, ADP, ATP, and regulated on activation, normal T-cell expressed and secreted (RANTES) (11-16).
The treatment for TRALI is supportive and consists of aggressive respiratory support with supplemental oxygen and mechanical ventilation if required at low enough pressure and tidal volume to not induce barotrauma (4;5;17;18). Two separate consensus conferences have occurred to define TRALI, and in short, diuretics may cause decreases in intravascular volume and are not indicated (2;3).
Prevalence and Mortality
TRALI has been reported as commonly as 1/1,333-1/5,000 per unit transfused in North America with lesser rates in Europe (1;2;5;19;20). The reported mortality from TRALI is 5-35%, with lower mortality rates (5-10%) being more common (4;7;9;21;22). However, recent prospective data from critically ill patients in the intensive care units have documented TRALI rates as high as 8%; therefore these patitns appear to have the highest risk for dvelopping TRALI (23). Most patients recover within 72 hours; however, the data regarding TRALI are limited, and the attendant morbidity and mortality may be under appreciated due to both lack of recognition and under reporting (4;7;9;21). Autopsy specimens have demonstrated widespread PMN infiltration with interstitial and intra-alveolar pulmonary edema, hyaline membrane formation, and destruction of the normal lung parenchyma consistent with the acute respiratory distress syndrome (ARDS) (4;24-29). In addition, in epidemiological studies of ARDS, transfusion was implicated as the most common predisposing factor for ARDS, and a number of these cases may be TRALI (24;30-32). A recent analyses of all reported cases of TRALI concluded that antibody-mediated TRALI may represent a more clinically severe form as compared to those reported reactions secondary to lipids and other biologic response modifiers (BRMs) (33). However, because most centers require the presence of antibodies against HLA or granulocyte antigens to make the diagnosis, such an analysis of BRM-mediated TRALI may be invalid due to selection bias (33). Importantly, this bias probably reflects the availability of antibody (anti-HLA or anti-HNA) testing services and in contrast the scarcity of laboratories that investigate BRMs, such that investigation of the role of BRMs in TRALI should be promoted.
Neutrophils and TRALI
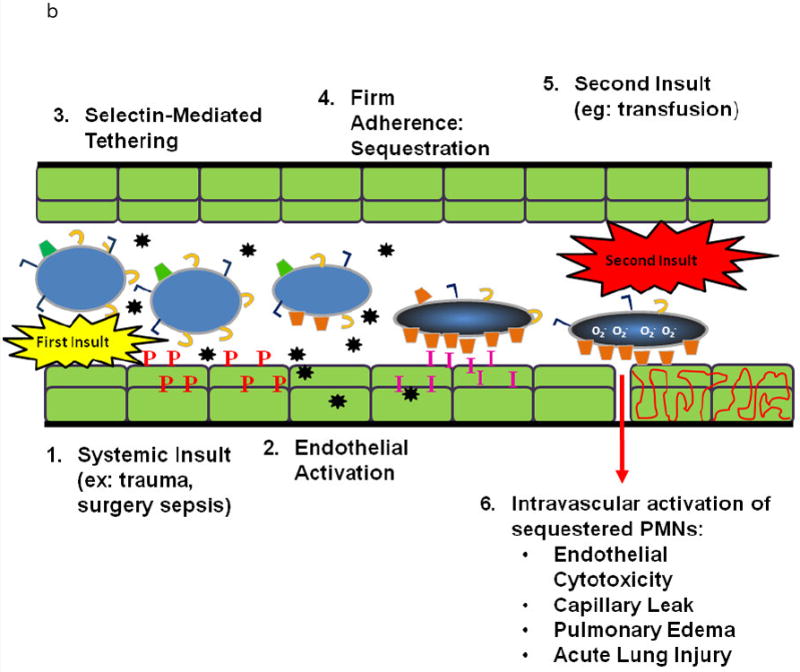
Popovsky and Moore first postulated that the PMN is the effector cell in TRALI and the murine and rat models of in vivo TRALI have reaffirmed that TRALI is PMN-mediated; thus, a review of PMN physiology is required (1;9;34;35). In response to an infection in the tissues signals are sent out that cause pro-inflammatory activation of the vascular endothelium in a concentration dependent fashion (Fig. 1A) (36-39). These signals induce the increased surface expression of the selectins on the endothelial cell (EC) surface and shedding of L-selectin on the PMNs, resulting in loose attachment of PMNs to the EC surface, known as capture in the pulmonary circulation (37;39-41). Closer to the nidus of the infection the ECs, in response to pro-inflammatory stimuli, synthesize and release chemokines, e.g. IL-8, which increase the surface expression of cellular adhesion molecules, especially intracellular adhesion moleducle-1 (ICAM-1) (37;39-41). The chemokines cause a rapid conformational change in the β2-integrins on the PMNs resulting in the firm adherence of PMNs to the vascular EC via PMN β2-integrins and ICAM-1 on the ECs (37;39-43). The change from a non-adherent to an adherent PMN is known as priming and not only involves PMN adhesion but also augments the release of microbicidal products from PMNs when activated to kill pathogens (37;39-44). In response to chemotaxins, the PMNs then diapedese through the EC layer and chemotax along a gradient of mediators to the nidus of infection where they phagocytize and destroy the invading microbes (36;39). In PMN-mediated ALI the first few steps are identical except that the stimulus is form the intravascular space and not the tissues (Fig.1B) (36;38;39). PMNs become firmly adherent (primed) but do not marginate due to the lack of a chemtoactic gradient (36;38;39) Therefore, these adherent, functionally hyperactive PMNs are sequestered in the lung and may predispose the host to ALI if a second stimulus, such as BRMs or antibodies in a blood transfusion, activates these primed granulocytes causing release of the microbicidal arsenal, EC damage, capillary leak, and ALI (36;38;39). Such “activating” agents may not have the capacity to cause activation of the microbicidal arsenal in “quiescent” PMNs but only have the ability to activate adherent, primed PMNs; otherwise, ALI would be a much more common event (36;38;39;44). Importantly, many clinical insults linked to TRALI may cause pro-inflammatory activation of vascular EC including: viral infections (adenovirus or cytomegalovirus), traumatic injury or major surgery, a “scheduled injury”, which release significant amounts of tumor necrosis factor–α(TNFα), and massive transfusion, which exposes the host to large amounts of neutral lipids which activate endothelial cells (42;45-48).
Figure 1.
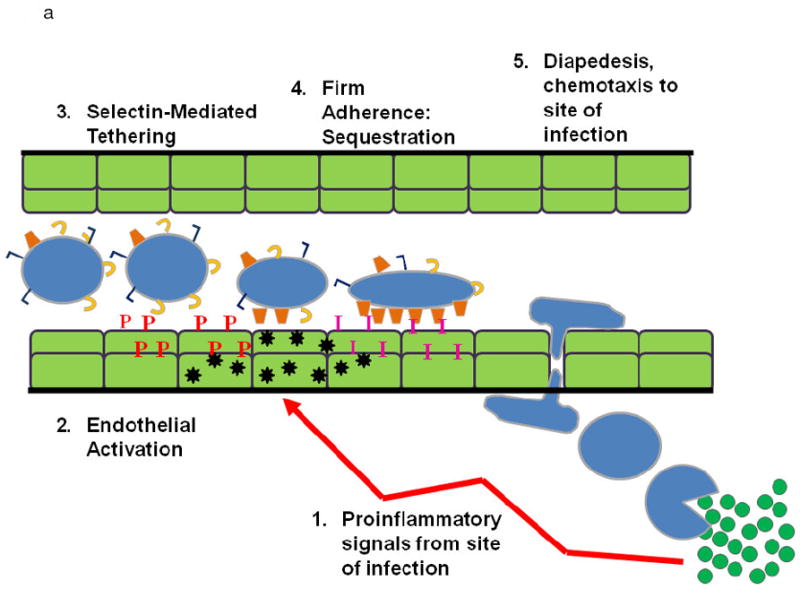
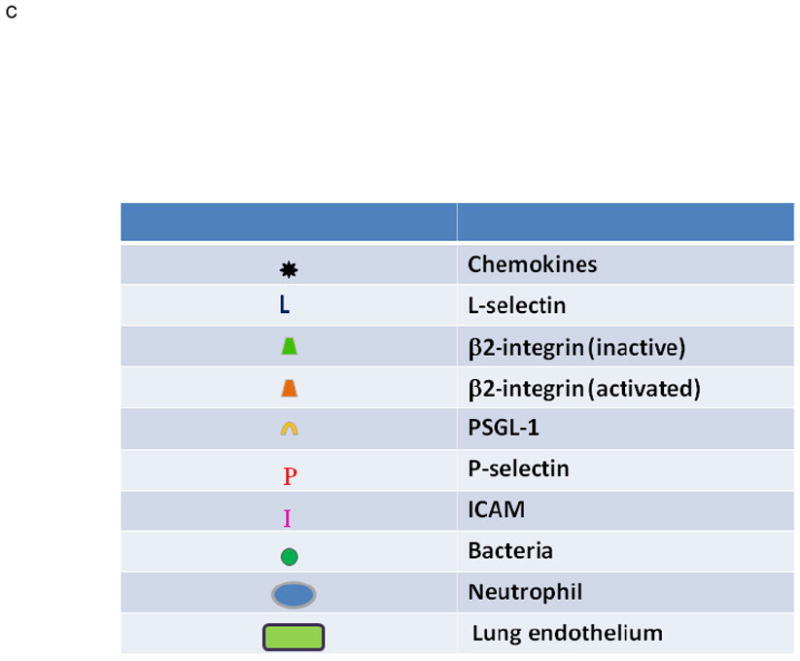
A. PMN Physiology: The normal response to infection. From an infection (green circles) in the tissues: (1) pro-inflammatory signals (LPS) are sent out (arrows) that activate ECs (2) causing chemokine synthesis and release (black stars), increases in P-selectin (red P’s) and increased surface expression of intercellular adhesion molecule-1 (ICAM-1, pink I’s). This activation elicits (3) PMN attraction and selectin-mediated tethering (capture) of PMNs between the PMN P=selectin glycoprotein ligand-1 (PSGL-1, tan C’s) and endothelial P-selectin. Capture is then followed by (4) firm PMN CD11b/CD18 (orange trapezoids) : ICAM-1 endothelial cell adherence resulting in PMN pulmonary sequestration. (5) PMNs diapedese through the endothelial cell layer and chemotax to the site of infection and kill the pathogens in the tissues.
B. PMN Physiology: PMN-mediated ALI. In response to intravascular stimuli due to a systemic (first) insult, ECs become (1) activated and synthesize and release chemokines (black stars) and increase the surface expression of P-selectin (red P’s) and ICAM-1 (pink I’s) (2). This endothelial activation causes tethering (3). of PMNs via the PMN PSGL-1 (tan C’s) and P-selectin followed by firm adherence (4) via the PMN CD11/CD18 : ICAM-1 from the endothelial surface resulting in pulmonary PMN sequestration. A second intravascular stimulus (insult), transfusion of specific antibodies against host PMN antigens or BRMs activate these primed, adherent PMNs (6) causing release of the microbicidal arsenal from the PMNs ,including O2-, resulting in EC damage, capillary leak, and TRALI.
There are also a number of important issues that are inherent to lung physiology: 1) the lung is the only organ in which PMN margination occurs in the capillaries (49-55). The capillaries are narrow and leukocytes, which have a larger diameter than the capillary, and need to “squeeze” through, such that mechanical sequestration may occur via the exposure of agents that stiffen PMNs (49-55). 2) There is controversy as to the mechanism of PMN sequestration whether firm adhesion, selectin-mediated tethering, or mechanical sequestration is required for PMN-induced ALI (49-55).
Pathogenesis
Three basic mechanisms have been proposed for the pathogenesis, and all require PMNs. Antibody-mediated TRALI: the infusion of donor antibodies specific for HLA class I and human neutrophil antigens (HNA) expressed by the recipient. The first clinical series of TRALI found donor antibodies against HLA class I antigens and granulocyte antigens in 89% and 72% of cases, respectively (1). Most of the granulocyte antibodies did not exhibit specificity but 59% of the HLA class I antibodies did, and the infusion of specific HLA antibodies has been confirmed in 50-65% of TRALI cases (1;7;56-58). However, in most investigation of antibody-linked TRALI, the investigators do not determine if the recipient expresses the cognate antigen, although it is a prerequisite for the proposed pathogenesis (1). Antibody-mediated TRALI requires that donor antibodies bind the cognate antigen in the host leading to the activation of complement, which causes pulmonary sequestration and activation of PMNs resulting in the release of cytotoxic agents, endothelial damage, capillary leak, and ALI (1;7;58). These antibodies are likely to be cytotoxic or pro-inflammatory, because HNA-3a is not cytotoxic, to confer such biologic activity upon binding the leukocyte antigen (1;5;59-62). Antibodies have caused TRALI in healthy humans and these clinical details supported this pathogenesis (63;64).
The in vivo relevance of antibodies to HLA class I antigens was confirmed in a murine model of TRALI in which a monoclonal antibody to mouse MHC class I antigens caused reproducible ALI at a dosage of 4.5 mg/kg (35). PMNs were required to recognize antigen:antibody complexes deposited onto the surface of the pulmonary EC and PMNs cytotoxicity was accomplished via activation of the murine Fc receptors because Fc knockout animals did not manifest TRALI when administered this antibody (35). This study mimicked the clinical time course for TRALI for the mice developed ALI within 2 hours of infusion; however the mortality was 50% (vs. 10-20% in clinical TRALI), and despite such a high mortality no blood pressure (BP) recordings or other physiologic parameters were included from the time of infusion the time of death/euthanasia. Furthermore, there was no dose-response to the infused antibody, i.e. only the one concentration caused ALI. It would be improbable for a human to be transfused with a specific antibody at a concentration of 4.5 mg/kg, for this specific antibody would have to comprise >10% of all IgG in 200 ml of FFP, the average dosage to a 70 kg adult, assuming the normal IgG concentrations (1060-1274 mg/dl) in the plasma and in the transfused host (35).
Interestingly, complement activation was not required for TRALI in this murine model, and the authors did state that the antibodies did not cause activation of the NADPH oxidase using a flow-based assay system. This last result is to be expected if the IgGs prime the PMN oxidase because priming agents, especially those implicated in TRALI, do not activate the respiratory burst (35;36). Lastly, a recent in vivo model employing the identical antibody from Looney et al demonstrated a requirement for the capture of platelets and a requirement of E-selectin to produce TRALI (65). Although this model represents an elegant mechanistic representation of antibody-induced inflammation, the pathogenesis represented differs widely from TRALI because of the aforementioned reasons and does not take into account the requirement for Fc receptor-mediated PMN activation through recognition of immune complexes on the endothelial surface (65).
Antibodies to granulocyte specific antigens have also been implicated in TRALI, and the in vivo relevance to these antibodies was confirmed in isolated, perfused rabbit lungs (62). ALI was characterized by pulmonary edema, and TRALI was precipitated by the infusion of a mixture of human PMNs (HNA-3a (5b) positive), HNA-3a antibodies, and rabbit plasma as a complement source (62). Pulmonary edema occurred 3-6 hours following the infusion of the admixture; however, if any one of the three components were deleted or antibodies without defined specificity were used, TRALI did not occur (62). The need for complement has been obviated in a rat model with the infusion of monoclonal antibodies (Mab) to HNA-2a (CD177) eliciting TRALI if they are infused with PMNs that express a high density of the HNA-2a antigen (66). The observed mild pulmonary edema with the Mab to HNA-2a and PMNs which express a high density of HNA-2a was significantly augmented with the addition of N-formyl-Met-Leu-Phe (66). In addition, using a flow based assay the Mabs to HNA-2a demonstrated the ability to activate the PMN oxidase in PMNs that express a high density of HNA-2a without affecting those PMNs with low density or lack of HNA-2a expression (66). Although eloquently done, one must consider the effects of the tubing on the infused PMNs in this ex vivo model because the tubing used to introduce PMNs is known to prime them, and such stiff leukocytes may become non-specifically sequestered due to their inability to “navigate” the pulmonary circulation (55;67;68). In addition one must be careful with antibodies to granulocyte specific antigens because animal models of human disease and inflammation routinely employ granulocyte specific antibodies to PMN deplete these rodents, and infusion of this antibody did not cause ALI (35;69). Transfusions PMN-reactive-antibodies to specific host antigens may result in alloimmune neutropenia depending upon the characteristic of the antibody:antigen interaction (70).
Antibody-mediated TRALI: the infusion of donor antibodies specific for HLA class II antigens expressed by the recipient
Antibodies specific for HLA class II antigens have been implicated in a large number of patients with TRALI who express the cognate antigens (17;23;71-73). Binding of specific HLA class II antibodies to their cognate antigens on monocytes in vitro resulted in intracellular synthesis of TNFα, IL-1β and tissue factor over a 4-hour time period as compared to identical monocytes incubated with control sera (74). Moreover, incubation (20 hours) of monocytes that express the cognate antigens with antibodies to these HLA class II antigens resulted in the production of both cytokines and chemokines (IL-8, GROα); the latter peptides could certainly activate PMNs sequestered in the lung (75). Recent in vitro studies demonstrated that co-cultures of human pulmonary microvascular endothelial cells (HMVECs) and monocytes stimulated with antibodies to HLA class II antigens expressed on the monocytes lead to the release of leukotriene B4 (LTB4) and TNFα into the supernatant with concomitant apoptosis of HMVECs (76;77). These experiments also identified the importance of monocytes and the HMVECs together because if alone minimal LTB4 was produced, and if an interfering anti-Dr antibody was introduced, HMVEC apoptosis and mediator release was inhibited (76;77). Lastly, because HLA class II antigens may be expressed on ECs the infusion of class II antibodies into a recipient with the cognate antigen present on the pulmonary EC may manifest TRALI due to antibody-mediated EC activation, fenestration, and mild leak (74).
Although attractive, this model of TRALI raises a number of issues: 1) while the synthesis of cytokines by circulating monocytes has the potential to cause TRALI, there is a significant time delay (4-20 hours) for the synthesis of these cytokines and chemokines, and at 4 hours these cytokines were never released extracellularly (74;75). By definition, TRALI occurs within 6 hours of transfusion with most of the reactions occurring during or 1-2 hours following transfusion; thus, the synthesis of chemokines and cytokines over a 20 hour time period may have little to do with TRALI (2;3;74;75). However, the production of LTB4 and TNFα in the model of Nishimura et al is temporally consistent with TRALI. This model requires further elucidation because the number of circulating monocytes in contact with the pulmonary EC must be relatively high as monocytes contain the LTA4 hydrolase to synthesize LTB4 from LTA4; whereas, ECs alone can only synthesize the cysteinyl leukotrienes via activation of the LTA4 synthetase and glutathione (LTC4, LTD4 & LTE4) (78-82). 2) Pathologic examination of lungs from a fatal TRALI reaction attributed to HLA class II antibodies documented that there were no HLA class II antigens on the pulmonary EC (25). In addition, although prolonged (72 hrs) in vitro cytokine exposure has lead to the surface expression of HLA class II molecules on PMNs, HLA class II antigen expression on the PMN surface has only been demonstrated in patients treated with G-CSF or GM-CSF, and although such cytokine exposure may predispose these individuals to TRALI, this group of patients is largely neutropenic from chemotherapy and may not manifest PMN-mediated ALI (4;83-88). These data are important because some interaction must occur between the pulmonary EC and the monocytes to produce clinically relevant amounts of LTB4 and the expression of HLA class II molecules is one potential factor.
The two-event model of TRALI
TRALI has been documented in cases in which antibodies have not been detected in either the donor or the recipient (1;4;5;7;9); moreover, if the infusion of specific anti-leukocyte antibodies were sufficient to cause TRALI, then blood from donors with specific antibodies against common leukocyte antigens should elicit TRALI reactions in the vast majority of recipients who express the cognate antigen. In “look back” studies of donors with known high-titer “TRALI-implicated” antibodies to a) HLA class I alone, b) HNA-3a, c) HLA class I and class II, or d) HLA class II alone, few of the transfused patients who expressed the cognate antigens developed TRALI (18;57;89-92). Therefore, the clinical condition of the patient appears important and may serve as the first event in the two-event model (4;5;57). The second event is inherent to the blood product and may be either donor-derived anti-leukocyte antibodies which recognize a cognate antigen in the recipient or BRMs that accumulate during blood storage (4;36;93). During storage of cellular components, effective PMN priming agents accumulate that are lipids as determined by solubility in chloroform (94;95). Identification of these lipids determined that a mixture of lysophosphatidylcholines (lyso-PCs) accumulates in the plasma fraction of all cellular components and reaches a relative maximal concentration on the day of component outdate (day 42) in the 100 units of packed red blood cells (PRBCs) tested (94;95). These compounds effectively prime the PMN oxidase, activate primed, adherent PMNs in vitro, and may serve as the second event in a two-event models of PMN cytotoxicity and PMN-mediated ALI (44;94-97).
The two-event model of TRALI was initially verified in an isolated perfused lung model and then re-confirmed in an in vivo rat model (28;69;98). A two-event rat model was employed with saline (NS) or endotoxin (LPS) as the first event and the infusion of plasma from PRBCs or antibodies (OX18 and OX27) against MHC class I antigens as the second event (69). The plasma from stored PRBCs, both pre-storage leukoreduced and unmodified [10% D28 and 5-10% D42]FINAL, and antibodies directed against MHC class I antigens caused ALI as the second event in this two-event in vivo model. ALI was demonstrated in a concentration-dependent manner in multiple ways including Evans Blue Dye (EBD) leak, lung histology, increases in total protein and CINC-1 in the bronchoalveolar lavage (BAL), and for antibody-mediated TRALI increases in EBD in the pulmonary interstitium. Importantly, in NS-injected rats neither the plasma from stored PRBCs nor the antibodies caused ALI, even when the concentration of OX27 was increased to 4.5 mg/kg, a level at which a monoclonal antibody induced ALI alone in vivo corroborating a two-event mechanism (35). In addition, lipopolysaccharide (LPS) was not lethal and did not cause 1) EBD leak into the BAL or the pulmonary interstitium, 2) increases in total protein or CINC-1 in the BAL or histological evidence of ALI, but did induce pulmonary sequestration of PMNs (28). LPS was employed as the first event because acute, active infection (bacterial or viral) was implicated as a predisposing clinical event in TRALI (4). Bacterial and viral infections induce pro-inflammatory activation of the vascular endothelium which leads to adherence/sequestration of PMNs (42;44;99-101). Other first events have been implicated including: cytokine administration, massive transfusion, recent surgery (especially cardiovascular surgery), and induction chemotherapy, but only infections have been studied to date in vivo (4-6;38;102-105). PMNs were also required because granulocyte depletion inhibited ALI. Furthermore, the OX18 and OX27 antibodies demonstrated both antigen recognition and the ability to prime the fMLP-activated respiratory burst of rat PMNs, which were not affected by Fc receptor blockade, implying that these interactions were specific to antibody:antigen binding (69). Lastly, a third antibody, which was granulocyte-(PMN) specific, caused immunodepletion of PMNs from rats, did not elicit ALI, when used in the two-event model, nor it prime rat PMNs (69). Both clinical TRALI and this model are similar with the onset of ALI within 6 hours, mortality of 5-10%, identical histological evidence of ALI, and a dose-response relationship of antibodies or plasma from stored PRBCs to elicit TRALI as the second event such that lower concentrations did not cause TRALI or elicited milder ALI (7;34;39). PMN reactive antibodies have varying effects on PMN physiology in vivo for they may prime or immunodeplete PMNs. A number of donor “look-back” studies which demonstrated that transfusion of a donor antibody to a recipient that expressed the cognate antigen caused TRALI in the minority of cases; thus, these antibodies appear to induce TRALI as a “second event”; hence, the clinical condition of the patient may be determinant for its genesis (18;57;89). In addition, these antibodies were found on the surface of PMNs that were sequestered in the lung, but not on the surface of pulmonary vascular endothelium as demonstrated in the murine model (35;69). Both OX18 and OX27 primed the rat PMN oxidase in vitro, identical to previous data which demonstrated that antibodies to HNA-3a could prime PMNs that expressed HNA-3a on their surface; moreover, Fc receptor blockade did not affect priming activity or cognate antigen immunoreactivity, implying specificity for these antigen:antibody interactions (69). Therefore, a number of specific prerequisites appear to be required for antibody-induced TRALI which may explain its relatively low prevalence: 1) the recipient has an underlying clinical condition that predisposes to TRALI, 2) the donor antibody must recognize the cognate antigen on the host’s leukocytes which are sequestered in the lung, and 3) the antigen:antibody interaction must cause a pro-inflammatory change in the PMN (69). The presented model is superior to other in vivo modeling because it better approximates clinical TRALI: 1) the first event, LPS, approximates acute active infection, a proposed risk factor for TRALI and causes pro-inflammatory activation of the pulmonary capillary bed without causing death or other organ injury (28;37;98;106-109). LPS has been used in many animal models and even injected into human volunteers and mimics infection (28;37;98;106-111). 2) ALI was induced by two separate second events, like clinical TRALI, and the second events demonstrated a concentration:response relationship in ALI using multiple parameters, unlike other in vivo models (35). 3) The MHC class I antibodies localized to the antigen on the PMN membrane, identical to Popovsky’s proposed antibody pathophysiology, and did not implicate Fc-mediated indirect activation, reminiscent of immune complex disease, not currently implicated in clinical TRALI (1;7;35). 4) The novel pathophysiology of antibody- and BRM-mediated TRALI in this model are due to direct pro-inflammatory changes (priming) of PMNs induced by the second events, synonymous with in vitro studies using human pulmonary endothelium, PMNs, granulocyte antibodies, sCD40L or lipids (28;44;94;95;112). This pro-inflammatory requirement may explain why not all antibodies may cause TRALI and induce TRAIN. 5) Disparate from the murine model, this model used antibody concentrations 15- to 30-fold less and did not require supra-physiologic concentrations of a specific antibody. If one calculates the amount of this specific antibody needed to cause clinical TRALI, then this antibody must comprise 10% of all IgG in 200 ml of transfused FFP (35;69).
Two clinical studies support the two-event model. The first identified 4 separate first events that may predispose patients to TRALI that were not present in a control group of patients with febrile or urticarial transfusion reactions including: 1) active infection, 2) recent surgery, 3) cytokine therapy, and 4) massive transfusion (4). These first events have been documented by other investigators in both antibody-mediated TRALI and TRALI caused by BRMs, including the seminal article of Popovsky and Moore in which all TRALI patients had a surgical operation 24 hours prior to the transfusion (1;104;105;113-117). In addition, a nested case control study documented that two patient groups were at particular risk for TRALI: patients who had cardiac surgery and patients with hematological malignancies in the induction phase of therapy (all with absolute PMN counts > 500/μl) (5). The second event was the infusion of bioactive lipids from the stored products as supported by the following data: 1) the implicated units were stored longer than control units that did not cause transfusion reactions, 2) the implicated blood products demonstrated significant plasma PMN priming activity as compared to the identically stored control units, and 3) there was PMN priming activity in all TRALI patients at the time of recognition, which consisted of neutral lipids and lyso-PCs, compared to pre-transfusion plasma (5). These cases of TRALI were not antibody-mediated because of the donors tested, only 1/28 exhibited a leukocyte antibody with specificity (HLA-A26) (5). Furthermore, patients with hematological malignancies and patients requiring cardiac surgery comprised the majority of patients with fatal TRALI in a recent FDA report on transfusion-related mortality (102). “Healthy” patients who experience TRALI would seem to disprove the two-event model; however, by definition, patients who require transfusion are not healthy. Lastly, a case of autologous TRALI has been reported in a patient who required a radical prostatectomy and transfusion. No leukocyte antibodies were detected but significant amounts of lipid priming activity were present in his PRBC units and in the post-transfusion plasma at the time TRALI was recognized (6).
Merging the Pathogenesis
Lipids, sCD40L, antibodies to HNA-3a, and antibodies to MHC class I antigens that cause TRALI in vivo have been linked to clinical TRALI and prime isolated PMNs that exhibit the cognate ligand whether it be a G-protein coupled receptor, lipids and sCD40L, or the cognate antigen, HNA3a or OX18 or OX27 (28;69;96;112;118). Importantly, immunoglobulins used to immunodeplete rats recognize the rat PMNs but do not induce TRALI in the two-event model in vivo, and do not prime PMNs. Moreover these BRMs and antibodies that primed PMNs caused PMN cytotoxicity of pulmonary endothelial cells as the second event in an in vitro model of two-event PMN-mediated cytotoxicity or TRALI in vivo (28;44;69;96;112;118). Thus, antibodies that cause pro-inflammatory changes in PMNs may induce TRALI and those that do not may cause immunodepletion and lead to immune neutropenia.
Laboratory Work-up of TRALI
The combination of the granulocyte immunofluorescence test (GIFT) and the granulocyte agglutination test (GAT) is an effective approach for detecting PMN reactive antibodies in serum or plasma (Fig.2) (119;120). These methods detect antibodies to both HNA and HLA class I, and possibly HLA class II, although further work is needed (119;121-123); moreover if antibodies to HNA-3a are suspected GAT is required (120). GAT provides the best indication as their physiologic relevance because it demonstrates direct antibody agglutination which is the results of PMN chemotaxis and homotypic PMN:PMN interactions, distinct from passive IgM cross-liking (124;125). The combination of GAT and GIFT applied to panels of phenotyped PMNs enable identification of HNA antibody specificity. These techniques are limited to reference laboratories in the Granulocyte Working Party (GWP) of the ISBT (www.isbt-web.org) is a necessary reference for finding the appropriate laboratories in different geographic locales.
Figure 2.
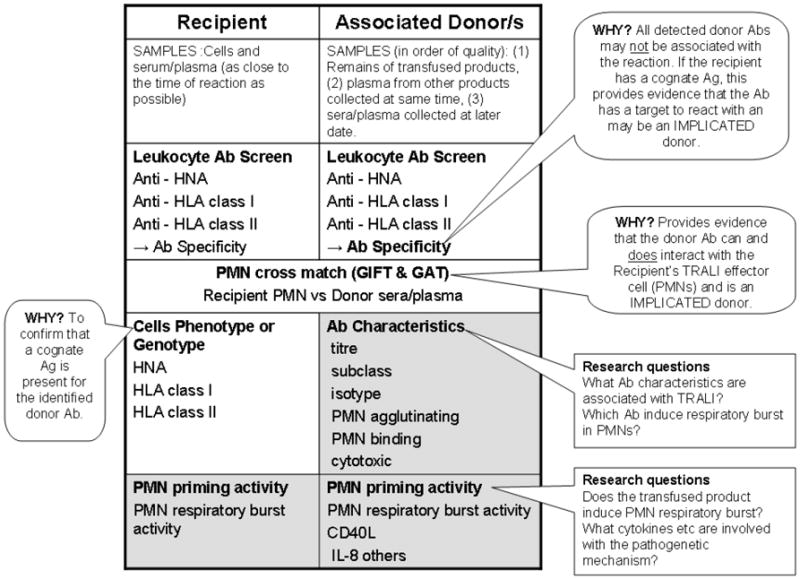
Laboratory investigations into the antibody (Ab) mediated and priming mechanism of TRALI. Patient samples (cellular, serum or plasma) should be as close to the time of the TRALI event as possible to reflect the physiological circumstances of the event. The remains of the transfused blood product provide the most ideal sample for the investigation (primary donor sample), followed by samples from other products manufactured at same collection date from that donor (secondary donor sample) or finally a new sample from the donor (tertiary donor sample). This is because antibody titers and priming/biological response modifiers (BRM) effect can be significantly different between various blood products from the same donor e.g. FFP versus PRBCs, and antibody titers of a donor can vary between blood collected on different dates. Laboratory investigation for Ab mediated TRALI begins with screening for leukocyte antibodies in associated donations and the recipient. Specificities of donor antibodies need to be determined as typing of recipient cells will confirm if there is a cognate Ag on the recipient the donor Ab to interact with. A PMN cross-match (GIFT and GAT) provides in vitro evidence that the donor Ab does react with the recipient’s cells and reveals they type of interaction – agglutination or binding. Primary donor samples should be investigated for their PMN priming activity, especially if leukocyte Abs are not detected.
The laboratory investigation of HLA class I and II antibodies will not be discussed in detail as the techniques are widely and there are excellent, recent reviews available (126;127). It is important to note that many of the assays employed are highly sensitive techniques to detect the presence of any antibodies in donor or recipient sera that could be clinically relevant leading to graft rejection and or graft versus host disease. Because HLA antibody screening is widely available it is often the first step in many TRALI investigations when screening implicated donors; however, two reports revealed that a disproportionately small number of HLA class I antibodies actually induce TRALI (89;128). With this in mind and the high sensitivity of current HLA techniques, one questions the clinical relevance of all of the HLA antibodies detected in TRALI because most of the current flow-based assays are so sensitive that a relatively high percentage of non-transfused males demonstrated antibodies (129;130). Similarly, flow based assays used to detect antibodies to HLA class II antigens in donors are designed to detect very small amounts of antibodies using a PRA luminex bead assay (One Lambda, Canoga Park, CA). These tests may overestimate the role of antibodies to HLA class II antigens in TRALI because: a) no titers of antibody concentration are employed, b) without sufficient modeling of HLA class II antibody-mediated TRALI it is not known what concentration is applicable and c) the role of the antibody has been thought to be PMN-dependent although PMNs do not “naturally” express HLA class II antigens (1;35;69). Recently, the current Leukocyte Antibody Prevalence Study (LAPS) determined the prevalence of HLA class I and II antibodies in large cohorts of non-transfused males, transfused males, nulliparous females, and parous females (130). Separate analyses were generated for both antibodies to HLA class I and class II antigens and stratified by donor group, and the data was log transformed such that positives were defined as three standard deviations above the mean. Samples were considered antibody positive with normalized background ratios of >10.8 for HLA class I antibodies and >6.9 for HLA class II antibodies. Antibody positivity for transfused males (1.7%) was similar to that of non-transfused males (1.0%) and nulliparous, non-transfused females (1.7%) (130). Such criteria of a less sensitive cut-off designed to detect passively transfused antibodies rather than an amamnestic response has never been implemented with respect to the prior data which implicated antibodies to HLA class II antigens in TRALI that employed a 8-10% shifts by flow as positive or shifts as low as 8% (17;25;74). These very small flow shifts may be of import for successful organ transplantation but their relevance is not defined in TRALI. Effective HLA antibody screening methods for blood donors from various testing sites reveal challenges in differentiating background noise and defining suitable cut offs such that if these new normal range of cutoffs are employed, some antibodies implicated in TRALI never reach a positive titer (130-133). Furthermore, the clinical effects of transfused HLA antibodies may be attenuated due to absorption by lymphocytes or platelets, and neutralization by soluble HLA class I molecules which comprise the majority of HLA class I antigens in the blood (134). One notable exception is HLA-A2 which has been implicated in many TRALI cases and thus should be included in any first line TRALI investigation screen (1;135-137).
It is important to determine if the cognate antigen is present in the transfused host, and currently there is great variability in how TRALI cross-matches are performed. Techniques employed include the lymphocytotoxicity test (LCT) (1), GIFT alone or GIFT and GAT combination (123). The LCT is based on lymphocytes which are stable and storable making them investigator friendly, however lymphocytes are not the primary target cell in TRALI and do not express PMN-specific antigens (HNA-1 and 2) and thus. will not detect any incompatibilities with these antigens. Therefore, reports using the LCT cross-match alone need to be interpreted with these potential limitations in mind. Importantly, while confirmation of the presence of a cognate antigen implies that the donor antibody has a target or substrate to react with, it does not confirm that the antibody contributed etiologically to the TRALI reaction, for in three look-back reports the minority of confirmed antigen:antibody pairs developed TRALI (74;138;139).
In the majority of cases, antibodies implicated in TRALI are donor-derived. Consequently, an appropriate cross-match strategy involves only the testing of the associated donor serum/plasma with the recipient’s effector cells (PMNs). In the rare case where the antibody is present in the TRALI-affected transfusion recipient, the cross-match should, in addition, test recipient serum/plasma with the associated donor’s PMNs, especially for TRALI linked to granulocyte transfusions (137). In a PMN cross-match, serum or plasma from associated donors should be incubated with the recipient’s PMNs using both the GIFT and GAT techniques (123;137;140). The PMN cross-match is very valuable because positive cross-matches or incompatibilities provide in-vitro evidence of a reaction between recipient PMNs and associated donor serum/plasma antibodies. This is especially useful in cases where antibody specificities cannot be defined, a common phenomenon in PMN serology, because of the very small number of well defined and characterized HNAs. In determining if a donor antibody did cause TRALI a number of important points need to be considered and should be the subject of ongoing research including: 1) the method of detection, is the antibody an agglutinating (GAT) or a binding antibody (GIFT), 2) the antibody titer, 3) epitope specificity, and 4) antibody isotype, 5) its ability to prime PMNs that express the cognate antigen. Such detailed antibody information may shed more light on the role of allo-antibodies in the TRALI mechanism.
Investigation of BRMs implicated in TRALI
These PMN priming assays are only performed in a few laboratories world wide including the Research Department at Bonfils Blood Center, Denver, CO and by the Australian Red Cross Blood Service, Brisbane, Australia. Such assays have their inherent quirks and require trained personnel, but the assays themselves are not difficult. These and comparable laboratories usually will test samples as part of their research endeavors. When priming activity is found then further identification is required including lipid identification or measurement of chemokines or sCD40L by commercial ELISA.
TRALI Avoidance
Manipulation of Blood products
Washing of cellular blood products removes all of the implicated mediators in TRALI, which are present in the plasma fraction; however, washing is expensive, time consuming, and the time constraints in critically ill patients may not allow for the time to remove the plasma from stored cellular components. Decreases in the plasma in cellular blood products has been employed with some success; however, in a number of TRALI cases only small amounts of plasma are required to elicit TRALI (141;142). Although pre-storage leukoreduction does decrease a number of leukocyte and platelet-derived mediators, it was not effective in inhibiting BRM-mediated TRALI in vivo (69). In Norway no TRALI cases have been reported since the use of solvent-detergent plasma (SDP), a product manufactured from enormous pools of plasma in which leukocyte antibodies are thought to be diluted and neutralized during processing, and cellular fragments are removed during filtration (143).
Donor selection
The use of male predominant plasma in Great Britain has significantly decreased the number of the total cases of TRALI particularly the cases of fatal TRALI (19). Although there may be selection bias in these studies for antibody-mediated TRALI as nicely demonstrated by Dr. Hume, this intervention appears to be effective (19;144). Not all antibody-mediated TRALI is caused by female donors as emphasized by the look-back study of reported TRALI cases by Middleburg et al that demonstrated that 48% of the implicated donors who gave antibody positive units were male (145). Rapid methods of screening for donor HLA and HNA antibodies appears pertinent, especially for apheresis donors; however, the cost and time required is not insignificant and many of the assays employed have a level of sensitivity related to organ transplantation and have not been validated for Transfusion Medicine. In addition, HNA antibody testing presents a major hurdle as there is currently no mass screening technology available.
Management of implicated donors
Look-back studies have confirmed that donors of implicated products do cause TRALI in other recipients (18;131). As discussed previously, screening implicated donors for HLA class I, class II and HNA antibodies, followed by confirmation of the cognate antigen in the recipient or incompatible PMN crossmatches between donor and recipient identifies TRALI-associated donors. Blood centers need to have effective systems to identify implicated donors so that future donations do not cause TRALI. If the antibodies recognize common leukocyte antigens the donors may be excluded or their products used for plasma poor products.
Conclusions
TRALI is the leading cause of transfusion-related morbidity and mortality (102). Although much of this review has focused on immunocompetent hosts, TRALI has been reported in neutropenic patients and the etiology is thought to involve the transfusion of permeability agents, such as vascular endothelial growth factor, VEGF, or antibody-mediated activation and fenestration of the pulmonary endothelium both resulting in mild lung leak (146;147). Proper evaluation of antibodies and BRMs implicated in TRALI using in vivo and in vitro models may lead to novel clinical strategies to preclude patient exposures or methods to identify TRALI risk factors and enable their removal to make transfusions safer and to not needlessly disqualify blood donors. Other than the presence of antibodies, little convincing evidence supporting the danger of female plasma has been presented. Recently, Palfi et al demonstrated a statistical decrease in blood oxygenation in patients following transfusion of female plasma versus the transfusion of male plasma, but this “decrease” was not out of the normal range for human subjects (148). Gajic et al prospectively investigated the transfusion of blood components from male donors versus blood transfusion with one or more female donors to patients in the intensive care unit and found a statistical decrease in oxygenation in the female containing transfusion group. Interestingly, there was more ALI in the male-only transfusion group with none being diagnosed with TRALI (149). Other investigators have refuted the inherent danger of female plasma; however more work is required to determine if male-predominant plasma transfusion is clinically warranted (150;151). In addition, if the new transfusion guidelines for military trauma, which included a 1:1 ratio of FFP units to PRBC units for optimal survival, are implemented in civilian trauma, there will not be nearly enough male-only plasma to transfuse injured patients (152). Although the use of male-only plasma transfusion may decrease a handful of fatalities, this practice may be disastrous to locales in which multiparous women comprise a large proportion of the donor pool, such as in the western United States (153;154), and newer methods to remove antibodies and other BRMs from the plasma/ plasma fraction of blood components are required.
Practice Points.
TRALI is a complex clinical syndrome that appears to require at least two clinical events for its development.
TRALI is a clinical diagnosis and should be made on clinical grounds although laboratory test may be supportive.
Antibody testing via flow cytometry may be too sensitive and requires modification for its use in Transfusion Medicine.
Male predominant plasma transfusion appears effective to reduce TRALI and further work is needed to include all cases and not just those with donor antibodies.
Research Agenda.
TRALI appears to affect the critically ill and further work is needed to determine appropriate transfusion strategies in this patient population.
Further work is need with regard to the role of antibodies to HLA class II antigens in TRALI for the pathophysiology is unknown.
The at-risk populations for TRALI remain undefined.
Acknowledgments
This work was supported by Bonfils Blood Center, and grants GM49222 from NIGMS and HL59355 from NHLBI, NIH (CCS) and the Australian Red Cross Blood Service,
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Popovsky MA, Moore SB. Diagnostic and pathogenetic considerations in transfusion-related acute lung injury. Transfusion. 1985 November;25(6):573–7. doi: 10.1046/j.1537-2995.1985.25686071434.x. [DOI] [PubMed] [Google Scholar]
- 2.Kleinman S, Caulfield T, Chan P, Davenport R, McFarland J, McPhedran S, Meade M, Morrison D, Pinsent T, Robillard P, Slinger P. Toward an understanding of transfusion-related acute lung injury: statement of a consensus panel. Transfusion. 2004 December;44(12):1774–89. doi: 10.1111/j.0041-1132.2004.04347.x. [DOI] [PubMed] [Google Scholar]
- 3.Toy P, Popovsky MA, Abraham E, Ambruso DR, Holness LG, Kopko PM, McFarland JG, Nathens AB, Silliman CC, Stroncek D. Transfusion-related acute lung injury: definition and review. Crit Care Med. 2005 April;33(4):721–6. doi: 10.1097/01.ccm.0000159849.94750.51. [DOI] [PubMed] [Google Scholar]
- 4.Silliman CC, Paterson AJ, Dickey WO, Stroneck DF, Popovsky MA, Caldwell SA, Ambruso DR. The association of biologically active lipids with the development of transfusion-related acute lung injury: a retrospective study. Transfusion. 1997 July;37(7):719–26. doi: 10.1046/j.1537-2995.1997.37797369448.x. [DOI] [PubMed] [Google Scholar]
- 5.Silliman CC, Boshkov LK, Mehdizadehkashi Z, Elzi DJ, Dickey WO, Podlosky L, Clarke G, Ambruso DR. Transfusion-related acute lung injury: epidemiology and a prospective analysis of etiologic factors. Blood. 2003 January 15;101(2):454–62. doi: 10.1182/blood-2002-03-0958. [DOI] [PubMed] [Google Scholar]
- 6.Covin RB, Ambruso DR, England KM, Kelher MR, Mehdizadehkashi Z, Boshkov LK, Masuno T, Moore EE, Kim FJ, Silliman CC. Hypotension and acute pulmonary insufficiency following transfusion of autologous red blood cells during surgery: a case report and review of the literature. Transfus Med. 2004 October;14(5):375–83. doi: 10.1111/j.0958-7578.2004.00529.x. [DOI] [PubMed] [Google Scholar]
- 7.Popovsky MA. Transfusion related acute lung injury. In: Popovsky MA, editor. Transfusion Reactions. 2. Bethesda, MD: AABB Press; 2001. [Google Scholar]
- 8.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, LeGall JR, Morris A, Spragg R. Report of the American-European Consensus conference on acute respiratory distress syndrome: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Consensus Committee. J Crit Care. 1994 March;9(1):72–81. doi: 10.1016/0883-9441(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 9.Silliman CC. Transfusion-related acute lung injury. Transfus Med Rev. 1999 July;13(3):177–86. doi: 10.1016/s0887-7963(99)80031-5. [DOI] [PubMed] [Google Scholar]
- 10.MacLennan S, Williamson LM. Risks of fresh frozen plasma and platelets. J Trauma. 2006 June;60(6 Suppl):S46–S50. doi: 10.1097/01.ta.0000199546.22925.31. [DOI] [PubMed] [Google Scholar]
- 11.Boehlen F, Clemetson KJ. Platelet chemokines and their receptors: what is their relevance to platelet storage and transfusion practice? Transfus Med. 2001 December;11(6):403–17. doi: 10.1046/j.1365-3148.2001.00340.x. [DOI] [PubMed] [Google Scholar]
- 12.Khan H, Belsher J, Yilmaz M, Afessa B, Winters JL, Moore SB, Hubmayr RD, Gajic O. Fresh-frozen plasma and platelet transfusions are associated with development of acute lung injury in critically ill medical patients. Chest. 2007 May;131(5):1308–14. doi: 10.1378/chest.06-3048. [DOI] [PubMed] [Google Scholar]
- 13.Phipps RP, Kaufman J, Blumberg N. Platelet derived CD154 (CD40 ligand) and febrile responses to transfusion. Lancet. 2001 June 23;357(9273):2023–4. doi: 10.1016/s0140-6736(00)05108-4. [DOI] [PubMed] [Google Scholar]
- 14.Power CA, Clemetson JM, Clemetson KJ, Wells TN. Chemokine and chemokine receptor mRNA expression in human platelets. Cytokine. 1995 August;7(6):479–82. doi: 10.1006/cyto.1995.0065. [DOI] [PubMed] [Google Scholar]
- 15.Schroder JM, Kameyoshi Y, Christophers E. RANTES, a novel eosinophil-chemotactic cytokine. Ann N Y Acad Sci. 1994 May 28;725:91–103. doi: 10.1111/j.1749-6632.1994.tb39793.x. [DOI] [PubMed] [Google Scholar]
- 16.Wakamoto S, Fujihara M, Kuzuma K, Sato S, Kato T, Naohara T, Kasai M, Sawada K, Kobayashi R, Kudoh T, Ikebuchi K, Azuma H, Ikeda H. Biologic activity of RANTES in apheresis PLT concentrates and its involvement in nonhemolytic transfusion reactions. Transfusion. 2003 August;43(8):1038–46. doi: 10.1046/j.1537-2995.2003.00458.x. [DOI] [PubMed] [Google Scholar]
- 17.Kopko PM, Popovsky MA, MacKenzie MR, Paglieroni TG, Muto KN, Holland PV. HLA class II antibodies in transfusion-related acute lung injury. Transfusion. 2001 October;41(10):1244–8. doi: 10.1046/j.1537-2995.2001.41101244.x. [DOI] [PubMed] [Google Scholar]
- 18.Kopko PM, Marshall CS, MacKenzie MR, Holland PV, Popovsky MA. Transfusion-related acute lung injury: report of a clinical look-back investigation. JAMA. 2002 April 17;287(15):1968–71. doi: 10.1001/jama.287.15.1968. [DOI] [PubMed] [Google Scholar]
- 19.Chapman CE, Stainsby D, Jones H, Love E, Massey E, Win N, Navarrete C, Lucas G, Soni N, Morgan C, Choo L, Cohen H, Williamson LM. Ten years of hemovigilance reports of transfusion-related acute lung injury in the United Kingdom and the impact of preferential use of male donor plasma. Transfusion. 2008 October 28; doi: 10.1111/j.1537-2995.2008.01948.x. [DOI] [PubMed] [Google Scholar]
- 20.Wallis JP, Lubenko A, Wells AW, Chapman CE. Single hospital experience of TRALI. Transfusion. 2003 August;43(8):1053–9. doi: 10.1046/j.1537-2995.2003.00466.x. [DOI] [PubMed] [Google Scholar]
- 21.Wallis JP. Transfusion-related acute lung injury (TRALI)--under-diagnosed and under-reported. Br J Anaesth. 2003 May;90(5):573–6. doi: 10.1093/bja/aeg101. [DOI] [PubMed] [Google Scholar]
- 22.Renaudier PC, Vo Mai MP, Azanowsky JM, Breton P, Cheze S, Girard A, Huaser L, Legras JF, Rebibo D, Waller C, Ounnoughene N. Epidemiology of transfusion related acute lung injury in Gifit, the Frech Hemovigilance Database. a study of the French Hemovigilance Network. Transfusion. 2004;44(9S):23A. [Google Scholar]
- 23.Gajic O, Rana R, Winters JL, Yilmaz M, Mendez JL, Rickman OB, O’byrne MM, Evenson LK, Malinchoc M, Degoey SR, Afessa B, Hubmayr RD, Moore SB. Transfusion Related Acute Lung Injury in the Critically Ill: Prospective Nested Case-Control Study. Am J Respir Crit Care Med. 2007 July 12; doi: 10.1164/rccm.200702-271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fowler AA, Hamman RF, Good JT, Benson KN, Baird M, Eberle DJ, Petty TL, Hyers TM. Adult respiratory distress syndrome: risk with common predispositions. Ann Intern Med. 1983 May;98(5 Pt 1):593–7. doi: 10.7326/0003-4819-98-5-593. [DOI] [PubMed] [Google Scholar]
- 25.Kao GS, Wood IG, Dorfman DM, Milford EL, Benjamin RJ. Investigations into the role of anti-HLA class II antibodies in TRALI. Transfusion. 2003 February;43(2):185–91. doi: 10.1046/j.1537-2995.2003.00285.x. [DOI] [PubMed] [Google Scholar]
- 26.Kollef MH, Schuster DP. The acute respiratory distress syndrome. N Engl J Med. 1995 January 5;332(1):27–37. doi: 10.1056/NEJM199501053320106. [DOI] [PubMed] [Google Scholar]
- 27.Pepe PE, Potkin RT, Reus DH, Hudson LD, Carrico CJ. Clinical predictors of the adult respiratory distress syndrome. Am J Surg. 1982 July;144(1):124–30. doi: 10.1016/0002-9610(82)90612-2. [DOI] [PubMed] [Google Scholar]
- 28.Silliman CC, Voelkel NF, Allard JD, Elzi DJ, Tuder RM, Johnson JL, Ambruso DR. Plasma and lipids from stored packed red blood cells cause acute lung injury in an animal model. J Clin Invest. 1998 April 1;101(7):1458–67. doi: 10.1172/JCI1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dry SM, Bechard KM, Milford EL, Churchill WH, Benjamin RJ. The pathology of transfusion-related acute lung injury. Am J Clin Pathol. 1999 August;112(2):216–21. doi: 10.1093/ajcp/112.2.216. [DOI] [PubMed] [Google Scholar]
- 30.Chaiwat O, Lang JD, Vavilala MS, Wang J, MacKenzie EJ, Jurkovich GJ, Rivara FP. Early packed red blood cell transfusion and acute respiratory distress syndrome after trauma. Anesthesiology. 2009 February;110(2):351–60. doi: 10.1097/ALN.0b013e3181948a97. [DOI] [PubMed] [Google Scholar]
- 31.Silverboard H, Aisiku I, Martin GS, Adams M, Rozycki G, Moss M. The role of acute blood transfusion in the development of acute respiratory distress syndrome in patients with severe trauma. J Trauma. 2005 September;59(3):717–23. [PubMed] [Google Scholar]
- 32.Zilberberg MD, Carter C, Lefebvre P, Raut M, Vekeman F, Duh MS, Shorr AF. Red blood cell transfusions and the risk of acute respiratory distress syndrome among the critically ill: a cohort study. Crit Care. 2007;11(3):R63. doi: 10.1186/cc5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bux J. Transfusion-related acute lung injury (TRALI): a serious adverse event of blood transfusion. Vox Sang. 2005 July;89(1):1–10. doi: 10.1111/j.1423-0410.2005.00648.x. [DOI] [PubMed] [Google Scholar]
- 34.Looney MR, Gropper MA, Matthay MA. Transfusion-related acute lung injury: a review. Chest. 2004 July;126(1):249–58. doi: 10.1378/chest.126.1.249. [DOI] [PubMed] [Google Scholar]
- 35.Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their Fc gamma receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest. 2006 June;116(6):1615–23. doi: 10.1172/JCI27238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silliman CC, Ambruso DR, Boshkov LK. Transfusion-related acute lung injury. Blood. 2005 March 15;105(6):2266–73. doi: 10.1182/blood-2004-07-2929. [DOI] [PubMed] [Google Scholar]
- 37.Silliman CC, Kelher M. The role of endothelial activation in the pathogenesis of transfusion-related acute lung injury. Transfusion. 2005 August;45(2 Suppl):109S–16S. doi: 10.1111/j.1537-2995.2005.00531.x. [DOI] [PubMed] [Google Scholar]
- 38.Silliman CC, McLaughlin NJ. Transfusion-related acute lung injury. Blood Rev. 2006 May;20(3):139–59. doi: 10.1016/j.blre.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Silliman CC. The two-event model of transfusion-related acute lung injury. Crit Care Med. 2006 May;34(5 Suppl):S124–S131. doi: 10.1097/01.CCM.0000214292.62276.8E. [DOI] [PubMed] [Google Scholar]
- 40.Albelda SM, Smith CW, Ward PA. Adhesion molecules and inflammatory injury. FASEB J. 1994 May;8(8):504–12. [PubMed] [Google Scholar]
- 41.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994 October 1;84(7):2068–101. [PubMed] [Google Scholar]
- 42.Grundy JE, Lawson KM, MacCormac LP, Fletcher JM, Yong KL. Cytomegalovirus-infected endothelial cells recruit neutrophils by the secretion of C-X-C chemokines and transmit virus by direct neutrophil- endothelial cell contact and during neutrophil transendothelial migration. J Infect Dis. 1998 June;177(6):1465–74. doi: 10.1086/515300. [DOI] [PubMed] [Google Scholar]
- 43.Vedder NB, Harlan JM. Increased surface expression of CD11b/CD18 (Mac-1) is not required for stimulated neutrophil adherence to cultured endothelium. J Clin Invest. 1988 March;81(3):676–82. doi: 10.1172/JCI113372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wyman TH, Bjornsen AJ, Elzi DJ, Smith CW, England KM, Kelher M, Silliman CC. A two-insult in vitro model of PMN-mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol Cell Physiol. 2002 December;283(6):C1592–C1603. doi: 10.1152/ajpcell.00540.2001. [DOI] [PubMed] [Google Scholar]
- 45.Cinat ME, Waxman K, Granger GA, Pearce W, Annas C, Daughters K. Trauma causes sustained elevation of soluble tumor necrosis factor receptors. J Am Coll Surg. 1994 November;179(5):529–37. [PubMed] [Google Scholar]
- 46.Li S, Jiao X, Tao L, Liu H, Cao Y, Lopez BL, Christopher TA, Ma XL. Tumor necrosis factor-{alpha} in mechanic trauma plasma mediates cardiomyocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007 September;293(3):H1847–H1852. doi: 10.1152/ajpheart.00578.2007. [DOI] [PubMed] [Google Scholar]
- 47.Maier B, Lefering R, Lehnert M, Laurer HL, Steudel WI, Neugebauer EA, Marzi I. EARLY VERSUS LATE ONSET OF MULTIPLE ORGAN FAILURE IS ASSOCIATED WITH DIFFERING PATTERNS OF PLASMA CYTOKINE BIOMARKER EXPRESSION AND OUTCOME AFTER SEVERE TRAUMA. Shock. 2007 July 19; Publish Ahead of Print. [PubMed] [Google Scholar]
- 48.Tan PH, Xue SA, Manunta M, Beutelspacher SC, Fazekasova H, Alam AK, McClure MO, George AJ. Effect of vectors on human endothelial cell signal transduction: implications for cardiovascular gene therapy. Arterioscler Thromb Vasc Biol. 2006 March;26(3):462–7. doi: 10.1161/01.ATV.0000200083.95349.9e. [DOI] [PubMed] [Google Scholar]
- 49.Burns JA, Issekutz TB, Yagita H, Issekutz AC. The alpha 4 beta 1 (very late antigen (VLA)-4, CD49d/CD29) and alpha 5 beta 1 (VLA-5, CD49e/CD29) integrins mediate beta 2 (CD11/CD18) integrin-independent neutrophil recruitment to endotoxin-induced lung inflammation. J Immunol. 2001 April 1;166(7):4644–9. doi: 10.4049/jimmunol.166.7.4644. [DOI] [PubMed] [Google Scholar]
- 50.Doerschuk CM. Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation. 2001 April;8(2):71–88. [PubMed] [Google Scholar]
- 51.Downey GP, Worthen GS. Neutrophil retention in model capillaries: deformability, geometry, and hydrodynamic forces. J Appl Physiol. 1988 October;65(4):1861–71. doi: 10.1152/jappl.1988.65.4.1861. [DOI] [PubMed] [Google Scholar]
- 52.Downey GP, Doherty DE, Schwab B, III, Elson EL, Henson PM, Worthen GS. Retention of leukocytes in capillaries: role of cell size and deformability. J Appl Physiol. 1990 November;69(5):1767–78. doi: 10.1152/jappl.1990.69.5.1767. [DOI] [PubMed] [Google Scholar]
- 53.Downey GP, Worthen GS, Henson PM, Hyde DM. Neutrophil sequestration and migration in localized pulmonary inflammation. Capillary localization and migration across the interalveolar septum. Am Rev Respir Dis. 1993 January;147(1):168–76. doi: 10.1164/ajrccm/147.1.168. [DOI] [PubMed] [Google Scholar]
- 54.Lien DC, Henson PM, Capen RL, Henson JE, Hanson WL, Wagner WW, Jr, Worthen GS. Neutrophil kinetics in the pulmonary microcirculation during acute inflammation. Lab Invest. 1991 August;65(2):145–59. [PubMed] [Google Scholar]
- 55.Worthen GS, Schwab B, III, Elson EL, Downey GP. Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science. 1989 July 14;245(4914):183–6. doi: 10.1126/science.2749255. [DOI] [PubMed] [Google Scholar]
- 56.Gans RO, Duurkens VA, van Zundert AA, Hoorntje SJ. Transfusion-related acute lung injury. Intensive Care Med. 1988;14(6):654–7. doi: 10.1007/BF00256772. [DOI] [PubMed] [Google Scholar]
- 57.Van Buren NL, Stroncek DF, Clay ME, McCullough J, Dalmasso AP. Transfusion-related acute lung injury caused by an NB2 granulocyte- specific antibody in a patient with thrombotic thrombocytopenic purpura. Transfusion. 1990 January;30(1):42–5. doi: 10.1046/j.1537-2995.1990.30190117629.x. [DOI] [PubMed] [Google Scholar]
- 58.Yomtovian R, Kline W, Press C, Clay M, Engman H, Hammerschmidt D, McCullough J. Severe pulmonary hypersensitivity associated with passive transfusion of a neutrophil-specific antibody. Lancet. 1984 February 4;1(8371):244–6. doi: 10.1016/s0140-6736(84)90124-7. [DOI] [PubMed] [Google Scholar]
- 59.Boshkov LK. Transfusion-associated acute lung injury (TRALI): an evolving understanding of the role of anti-leukocyte antibodies. Vox Sang. 2002 August;83(Suppl 1):299–303. doi: 10.1111/j.1423-0410.2002.tb05322.x. [DOI] [PubMed] [Google Scholar]
- 60.Popovsky MA, Chaplin HC, Jr, Moore SB. Transfusion-related acute lung injury: a neglected, serious complication of hemotherapy. Transfusion. 1992 July;32(6):589–92. doi: 10.1046/j.1537-2995.1992.32692367207.x. [DOI] [PubMed] [Google Scholar]
- 61.Popovsky MA. Transfusion-related acute lung injury. In: Popovsky MA, editor. Transfusion Reactions. first ed. Bethesda, MD: American Association of Blood Banks Press; 1996. pp. 167–83. [Google Scholar]
- 62.Seeger W, Schneider U, Kreusler B, von Witzleben E, Walmrath D, Grimminger F, Neppert J. Reproduction of transfusion-related acute lung injury in an ex vivo lung model. Blood. 1990 October 1;76(7):1438–44. [PubMed] [Google Scholar]
- 63.BRITTINGHAM TE, CHAPLIN H., Jr Febrile transfusion reactions caused by sensitivity to donor leukocytes and platelets. J Am Med Assoc. 1957 October 19;165(7):819–25. doi: 10.1001/jama.1957.02980250053013. [DOI] [PubMed] [Google Scholar]
- 64.Dooren MC, Ouwehand WH, Verhoeven AJ, von dem Borne AE, Kuijpers RW. Adult respiratory distress syndrome after experimental intravenous gamma-globulin concentrate and monocyte-reactive IgG antibodies. Lancet. 1998 November 14;352(9140):1601–2. doi: 10.1016/s0140-6736(05)61049-5. [DOI] [PubMed] [Google Scholar]
- 65.Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009 April;15(4):384–91. doi: 10.1038/nm.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sachs UJ, Hattar K, Weissmann N, Bohle RM, Weiss T, Sibelius U, Bux J. Antibody-induced neutrophil activation as a trigger for transfusion-related acute lung injury in an ex vivo rat lung model. Blood. 2006 February 1;107(3):1217–9. doi: 10.1182/blood-2005-04-1744. [DOI] [PubMed] [Google Scholar]
- 67.Partrick DA, Moore EE, Fullerton DA, Barnett CC, Jr, Meldrum DR, Silliman CC. Cardiopulmonary bypass renders patients at risk for multiple organ failure via early neutrophil priming and late neutrophil disability. J Surg Res. 1999 September;86(1):42–9. doi: 10.1006/jsre.1999.5702. [DOI] [PubMed] [Google Scholar]
- 68.Ward RA, McLeish KR. Hemodialysis with cellulose membranes primes the neutrophil oxidative burst. Artif Organs. 1995 August;19(8):801–7. doi: 10.1111/j.1525-1594.1995.tb02431.x. [DOI] [PubMed] [Google Scholar]
- 69.Kelher MR, Masuno T, Moore EE, Damle S, Meng X, Song Y, Liang X, Niedzinski J, Geier SS, Khan SY, Gamboni-Robertson F, Silliman CC. Plasma from stored packed red blood cells and MHC class I antibodies causes acute lung injury in a 2-event in vivo rat model. Blood. 2009 February 26;113(9):2079–87. doi: 10.1182/blood-2008-09-177857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wallis JP, Haynes S, Stark G, Green FA, Lucas GF, Chapman CE. Transfusion-related alloimmune neutropenia: an undescribed complication of blood transfusion. Lancet. 2002 October 5;360(9339):1073–4. doi: 10.1016/S0140-6736(02)11132-9. [DOI] [PubMed] [Google Scholar]
- 71.Varela M, Mas A, Nogues N, Escorsell A, Mazzara R, Lozano M. TRALI associated with HLA class II antibodies. Transfusion. 2002 August;42(8):1102. doi: 10.1046/j.1537-2995.2002.00225.x. [DOI] [PubMed] [Google Scholar]
- 72.Win N, Brown C, Navarrete C. TRALI associated with HLA class II antibodies. Transfusion. 2003 April;43(4):545–6. doi: 10.1046/j.1537-2995.2003.00363.x. [DOI] [PubMed] [Google Scholar]
- 73.Eder AF, Herron R, Strupp A, Dy B, Notari EP, Chambers LA, Dodd RY, Benjamin RJ. Transfusion-related acute lung injury surveillance (2003-2005) and the potential impact of the selective use of plasma from male donors in the American Red Cross. Transfusion. 2007 April;47(4):599–607. doi: 10.1111/j.1537-2995.2007.01102.x. [DOI] [PubMed] [Google Scholar]
- 74.Kopko PM, Paglieroni TG, Popovsky MA, Muto KN, MacKenzie MR, Holland PV. TRALI: correlation of antigen-antibody and monocyte activation in donor-recipient pairs. Transfusion. 2003 February;43(2):177–84. doi: 10.1046/j.1537-2995.2003.00307.x. [DOI] [PubMed] [Google Scholar]
- 75.Sakagawa H, Miyazaki T, Fujihara M, Sato S, Yamaguchi M, Fukai K, Morioka M, Kato T, Azuma H, Ikeda H. Generation of inflammatory cytokines and chemokines from peripheral blood mononuclear cells by HLA Class II antibody-containing plasma unit that was associated with severe nonhemolytic transfusion reactions. Transfusion. 2007 January;47(1):154–61. doi: 10.1111/j.1537-2995.2007.01078.x. [DOI] [PubMed] [Google Scholar]
- 76.Nishimura M, Hashimoto S, Satake M, Okazaki H, Tadokoro K. Interference with TRALI-causing anti-HLA DR alloantibody induction of human pulmonary microvascular endothelial cell injury by purified soluble HLA DR. Vox Sang. 2007 July;93(1):78–82. doi: 10.1111/j.1423-0410.2007.00925.x. [DOI] [PubMed] [Google Scholar]
- 77.Nishimura M, Hashimoto S, Takanashi M, Okazaki H, Satake M, Nakajima K. Role of anti-human leucocyte antigen class II alloantibody and monocytes in development of transfusion-related acute lung injury. Transfus Med. 2007 April;17(2):129–34. doi: 10.1111/j.1365-3148.2006.00721.x. [DOI] [PubMed] [Google Scholar]
- 78.Gronert K, Clish CB, Romano M, Serhan CN. Transcellular regulation of eicosanoid biosynthesis. Methods Mol Biol. 1999;120:119–44. doi: 10.1385/1-59259-263-5:119. [DOI] [PubMed] [Google Scholar]
- 79.Krump E, Picard S, Mancini J, Borgeat P. Suppression of leukotriene B4 biosynthesis by endogenous adenosine in ligand-activated human neutrophils. J Exp Med. 1997 October 20;186(8):1401–6. doi: 10.1084/jem.186.8.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Palmantier R, Rocheleau H, Laviolette M, Mancini J, Borgeat P. Characteristics of leukotriene biosynthesis by human granulocytes in presence of plasma. Biochim Biophys Acta. 1998 January 23;1389(3):187–96. doi: 10.1016/s0005-2760(97)00149-5. [DOI] [PubMed] [Google Scholar]
- 81.Sala A, Bolla M, Zarini S, Muller-Peddinghaus R, Folco G. Release of leukotriene A4 versus leukotriene B4 from human polymorphonuclear leukocytes. J Biol Chem. 1996 July 26;271(30):17944–8. doi: 10.1074/jbc.271.30.17944. [DOI] [PubMed] [Google Scholar]
- 82.Serhan CN. Cell-cell interactions in the generation of eicosanoids: charting the routes and products of transcellular biosynthesis. J Lab Clin Med. 1993 March;121(3):372–4. [PubMed] [Google Scholar]
- 83.Gosselin EJ, Wardwell K, Rigby WF, Guyre PM. Induction of MHC class II on human polymorphonuclear neutrophils by granulocyte/macrophage colony-stimulating factor, IFN-gamma, and IL-3. J Immunol. 1993 August 1;151(3):1482–90. [PubMed] [Google Scholar]
- 84.Lei L, Altstaedt J, von der OM, Proft T, Gross U, Rink L. Induction of interleukin-8 in human neutrophils after MHC class II cross-linking with superantigens. J Leukoc Biol. 2001 July;70(1):80–6. [PubMed] [Google Scholar]
- 85.Matsumoto S, Takei M, Moriyama M, Imanishi H. Enhancement of Ia-like antigen expression by interferon-gamma in polymorphonuclear leukocytes. Chem Pharm Bull (Tokyo) 1987 January;35(1):436–9. doi: 10.1248/cpb.35.436. [DOI] [PubMed] [Google Scholar]
- 86.Reinisch W, Lichtenberger C, Steger G, Tillinger W, Scheiner O, Gangl A, Maurer D, Willheim M. Donor dependent, interferon-gamma induced HLA-DR expression on human neutrophils in vivo. Clin Exp Immunol. 2003 September;133(3):476–84. doi: 10.1046/j.1365-2249.2003.02245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Spagnoli GC, Juretic A, Rosso R, Van BJ, Harder F, Heberer M. Expression of HLA-DR in granulocytes of polytraumatized patients treated with recombinant human granulocyte macrophage-colony-stimulating factor. Hum Immunol. 1995 May;43(1):45–50. doi: 10.1016/0198-8859(94)00131-9. [DOI] [PubMed] [Google Scholar]
- 88.Zarco MA, Ribera JM, Villamor N, Balmes A, Urbano IA, Feliu E. Phenotypic changes in neutrophil granulocytes after G-CSF administration in patients with acute lymphoblastic leukemia under chemotherapy. Haematologica. 1998 June;83(6):573–5. [PubMed] [Google Scholar]
- 89.Toy P, Hollis-Perry KM, Jun J, Nakagawa M. Recipients of blood from a donor with multiple HLA antibodies: a lookback study of transfusion-related acute lung injury. Transfusion. 2004 December;44(12):1683–8. doi: 10.1111/j.0041-1132.2004.04193.x. [DOI] [PubMed] [Google Scholar]
- 90.Aysola AE, Deisting B, Mair DC. Review of 16 blood components from a donor with HLA class II antibodies. Transfusion. 2005;45(Supplement):83A. [Google Scholar]
- 91.Maslanka K, Michur H, Zupanska B, Uhrynowska M, Nowak J. Leucocyte antibodies in blood donors and a look back on recipients of their blood components. Vox Sang. 2007 April;92(3):247–9. doi: 10.1111/j.1423-0410.2007.00890.x. [DOI] [PubMed] [Google Scholar]
- 92.Zupanska B, Uhrynowska M, Michur H, Maslanka K, Zajko M. Transfusion-related acute lung injury and leucocyte-reacting antibodies. Vox Sang. 2007 July;93(1):70–7. doi: 10.1111/j.1423-0410.2007.00920.x. [DOI] [PubMed] [Google Scholar]
- 93.Silliman CC, Kelher M, Hess JR, England KM, Gorman J, McLaughlin-Malaxecheberria N, Greenwalt TJ. Experimental additive solution-64 inhibits the accumulation of neutrophil priming activity and interleukin-18 during storage of packed red blood cells. Blood. 2002;100(supplement) [Google Scholar]
- 94.Silliman CC, Clay KL, Thurman GW, Johnson CA, Ambruso DR. Partial characterization of lipids that develop during the routine storage of blood and prime the neutrophil NADPH oxidase. J Lab Clin Med. 1994 November;124(5):684–94. [PMC free article] [PubMed] [Google Scholar]
- 95.Silliman CC, Dickey WO, Paterson AJ, Thurman GW, Clay KL, Johnson CA, Ambruso DR. Analysis of the priming activity of lipids generated during routine storage of platelet concentrates. Transfusion. 1996 February;36(2):133–9. doi: 10.1046/j.1537-2995.1996.36296181925.x. [DOI] [PubMed] [Google Scholar]
- 96.Silliman CC, Elzi DJ, Ambruso DR, Musters RJ, Hamiel C, Harbeck RJ, Paterson AJ, Bjornsen AJ, Wyman TH, Kelher M, England KM, McLaughlin-Malaxecheberria N, Barnett CC, Aiboshi J, Bannerjee A. Lysophosphatidylcholines prime the NADPH oxidase and stimulate multiple neutrophil functions through changes in cytosolic calcium. J Leukoc Biol. 2003 April;73(4):511–24. doi: 10.1189/jlb.0402179. [DOI] [PubMed] [Google Scholar]
- 97.Aiboshi J, Moore EE, Ciesla DJ, Silliman CC. Blood transfusion and the two-insult model of post-injury multiple organ failure. Shock. 2001 April;15(4):302–6. doi: 10.1097/00024382-200115040-00009. [DOI] [PubMed] [Google Scholar]
- 98.Silliman CC, Bjornsen AJ, Wyman TH, Kelher M, Allard J, Bieber S, Voelkel NF. Plasma and lipids from stored platelets cause acute lung injury in an animal model. Transfusion. 2003 May;43(5):633–40. doi: 10.1046/j.1537-2995.2003.00385.x. [DOI] [PubMed] [Google Scholar]
- 99.Arnold R, Konig W. Respiratory syncytial virus infection of human lung endothelial cells enhances selectively intercellular adhesion molecule-1 expression. J Immunol. 2005 June 1;174(11):7359–67. doi: 10.4049/jimmunol.174.11.7359. [DOI] [PubMed] [Google Scholar]
- 100.Talavera D, Castillo AM, Dominguez MC, Gutierrez AE, Meza I. IL8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. J Gen Virol. 2004 July;85(Pt 7):1801–13. doi: 10.1099/vir.0.19652-0. [DOI] [PubMed] [Google Scholar]
- 101.Vilela MC, Mansur DS, Lacerda-Queiroz N, Rodrigues DH, Arantes RM, Kroon EG, Campos MA, Teixeira MM, Teixeira AL. Traffic of leukocytes in the central nervous system is associated with chemokine up-regulation in a severe model of herpes simplex encephalitis: an intravital microscopy study. Neurosci Lett. 2008 November 7;445(1):18–22. doi: 10.1016/j.neulet.2008.08.072. [DOI] [PubMed] [Google Scholar]
- 102.Holness L, Knippen MA, Simmons L, Lachenbruch PA. Fatalities caused by TRALI. Transfus Med Rev. 2004 July;18(3):184–8. doi: 10.1016/j.tmrv.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 103.Keifer JC, Kingsley CP, Roth MT, Abt AB, Romano PJ. Transfusion-related acute lung injury (TRALI) complicating colectomy for ulcerative colitis. Anesthesiology. 1998 October;89(4):1020–3. doi: 10.1097/00000542-199810000-00027. [DOI] [PubMed] [Google Scholar]
- 104.Medeiros BC, Kogel KE, Kane MA. Transfusion-related acute lung injury (TRALI) following platelet transfusion in a patient receiving high-dose interleukin-2 for treatment of metastatic renal cell carcinoma. Transfus Apheresis Sci. 2003 August;29(1):25–7. doi: 10.1016/S1473-0502(03)00094-6. [DOI] [PubMed] [Google Scholar]
- 105.Roffey P, Thangathurai D, Mikhail M, Riad M, Mogos M. TRALI and massive transfusion. Resuscitation. 2003 July;58(1):121. doi: 10.1016/s0300-9572(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 106.Han I, Saito H, Fukatsu K, Inoue T, Yasuhara H, Furukawa S, Matsuda T, Lin MT, Ikeda S. Ex vivo fluorescence microscopy provides simple and accurate assessment of neutrophil-endothelial adhesion in the rat lung. Shock. 2001 August;16(2):143–7. doi: 10.1097/00024382-200116020-00010. [DOI] [PubMed] [Google Scholar]
- 107.Salzer WL, McCall CE. Primed stimulation of isolated perfused rabbit lung by endotoxin and platelet activating factor induces enhanced production of thromboxane and lung injury. J Clin Invest. 1990 April;85(4):1135–43. doi: 10.1172/JCI114545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wagner JG, Roth RA. Neutrophil migration during endotoxemia. J Leukoc Biol. 1999 July;66(1):10–24. doi: 10.1002/jlb.66.1.10. [DOI] [PubMed] [Google Scholar]
- 109.Woodman RC, Teoh D, Payne D, Kubes P. Thrombin and leukocyte recruitment in endotoxemia. Am J Physiol Heart Circ Physiol. 2000 September;279(3):H1338–H1345. doi: 10.1152/ajpheart.2000.279.3.H1338. [DOI] [PubMed] [Google Scholar]
- 110.Abraham E. Effects of recombinant human activated protein C in human models of endotoxin administration. Proc Am Thorac Soc. 2005;2(3):243–7. doi: 10.1513/pats.200501-004AC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O’Brien JM, Jr, Abraham E. Human models of endotoxemia and recombinant human activated protein C. Crit Care Med. 2004 May;32(5 Suppl):S202–S208. doi: 10.1097/01.ccm.0000126123.34119.98. [DOI] [PubMed] [Google Scholar]
- 112.Khan SY, Kelher MR, Heal JM, Blumberg N, Boshkov LK, Phipps R, Gettings KF, McLaughlin NJ, Silliman CC. Soluble CD40 ligand accumulates in stored blood components, primes neutrophils through CD40, and is a potential cofactor in the development of transfusion-related acute lung injury. Blood. 2006 October 1;108(7):2455–62. doi: 10.1182/blood-2006-04-017251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bueter M, Thalheimer A, Schuster F, Bock M, von EC, Meyer D, Fein M. Transfusion-related acute lung injury (TRALI)--an important, severe transfusion-related complication. Langenbecks Arch Surg. 2006 September;391(5):489–94. doi: 10.1007/s00423-006-0072-2. [DOI] [PubMed] [Google Scholar]
- 114.Fung YL, Williams BA. TRALI in 2 cases of leukemia. J Pediatr Hematol Oncol. 2006 June;28(6):391–4. doi: 10.1097/00043426-200606000-00014. [DOI] [PubMed] [Google Scholar]
- 115.Higgins S, Fowler R, Callum J, Cartotto R. Transfusion-Related Acute Lung Injury in Patients With Burns. J Burn Care Res. 2007 January;28(1):56–64. doi: 10.1097/BCR.0b013E31802C88EC. [DOI] [PubMed] [Google Scholar]
- 116.Li GS, Ye QF, Xia SS, Chen ZS, Zeng FJ, Lin ZB, Gong NQ, Zhang WJ, Wen ZX, Sha P, Jiang JP. Acute respiratory distress syndrome after liver transplantation: etiology, prevention and management. Hepatobiliary Pancreat Dis Int. 2002 August;1(3):330–4. [PubMed] [Google Scholar]
- 117.Sanchez R, Bacchetti P, Toy P. Transfusion-related acute lung injury: a case-control pilot study of risk factors. Am J Clin Pathol. 2007 July;128(1):128–34. doi: 10.1309/HC4PVY24NJXMQ884. [DOI] [PubMed] [Google Scholar]
- 118.Silliman CC, Curtis BR, Kopko PM, Khan SY, Kelher MR, Schuller RM, Sannoh B, Ambruso DR. Donor antibodies to HNA-3a implicated in TRALI reactions prime neutrophils and cause PMN-mediated damage to human pulmonary microvascular endothelial cells in a two-event in vitro model. Blood. 2007 February 15;109(4):1752–5. doi: 10.1182/blood-2006-05-025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bux J, Stroncek D. Human neutrophil antigens. Transfusion. 2002 November;42(11):1523. doi: 10.1046/j.1537-2995.2002.00265.x. [DOI] [PubMed] [Google Scholar]
- 120.Lucas G, Rogers S, de HM, Porcelijn L, Bux J. Report on the Fourth International Granulocyte Immunology Workshop: progress toward quality assessment. Transfusion. 2002 April;42(4):462–8. doi: 10.1046/j.1525-1438.2002.00053.x. [DOI] [PubMed] [Google Scholar]
- 121.Bierling P, Bux J, Curtis B, Flesch B, Fung L, Lucas G, Macek M, Muniz-Diaz E, Porcelijn L, Reil A, Sachs U, Schuller R, Tsuno N, Uhrynowska M, Urbaniak S, Valentin N, Wikman A, Zupanska B. Recommendations of the ISBT Working Party on Granulocyte Immunobiology for leucocyte antibody screening in the investigation and prevention of antibody-mediated transfusion-related acute lung injury. Vox Sang. 2008 December 25; doi: 10.1111/j.1423-0410.2008.01144.x. [DOI] [PubMed] [Google Scholar]
- 122.Bux J. Challenges in the determination of clinically significant granulocyte antibodies and antigens. Transfus Med Rev. 1996 July;10(3):222–32. doi: 10.1016/s0887-7963(96)80061-7. [DOI] [PubMed] [Google Scholar]
- 123.Fung YL, Goodison KA, Wong JK, Minchinton RM. Investigating transfusion-related acute lung injury (TRALI) Intern Med J. 2003 July;33(7):286–90. doi: 10.1046/j.1445-5994.2003.00352.x. [DOI] [PubMed] [Google Scholar]
- 124.Lalezari P, Radel E. Neutrophil-specific antigens: immunology and clinical significance. Semin Hematol. 1974 July;11(3):281–90. [PubMed] [Google Scholar]
- 125.Verheugt FW, von dem Borne AE, van Noord-Bokhorst JC, van Elven EH, Engelfriet CP. Serological, immunochemical and immuoncytological properties of granulocyte antibodies. Vox Sang. 1978;35(5):294–303. doi: 10.1111/j.1423-0410.1978.tb02938.x. [DOI] [PubMed] [Google Scholar]
- 126.Fuggle SV, Martin S. Tools for human leukocyte antigen antibody detection and their application to transplanting sensitized patients. Transplantation. 2008 August 15;86(3):384–90. doi: 10.1097/TP.0b013e31817c90f5. [DOI] [PubMed] [Google Scholar]
- 127.Zeevi A, Girnita A, Duquesnoy R. HLA antibody analysis: sensitivity, specificity, and clinical significance in solid organ transplantation. Immunol Res. 2006;36(1-3):255–64. doi: 10.1385/IR:36:1:255. [DOI] [PubMed] [Google Scholar]
- 128.Cooling L. Transfusion-related acute lung injury. JAMA. 2002 July 17;288(3):315–6. doi: 10.1001/jama.288.3.315. [DOI] [PubMed] [Google Scholar]
- 129.Fadeyi EA, De Los Angeles MM, Wayne AS, Klein HG, Leitman SF, Stroncek DF. The transfusion of neutrophil-specific antibodies causes leukopenia and a broad spectrum of pulmonary reactions. Transfusion. 2007 March;47(3):545–50. doi: 10.1111/j.1537-2995.2006.01148.x. [DOI] [PubMed] [Google Scholar]
- 130.Triulzi DJ, Kleinman S, Kakaiya RM, Busch MP, Norris PJ, Steele WR, Glynn SA, Hillyer CD, Carey P, Gottschall JL, Murphy EL, Rios JA, Ness PM, Wright DJ, Carrick D, Schreiber GB. The effect of previous pregnancy and transfusion on HLA alloimmunization in blood donors: implications for a transfusion-related acute lung injury risk reduction strategy. Transfusion. 2009 May 18; doi: 10.1111/j.1537-2995.2009.02206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fadeyi E, Adams S, Peterson B, Hackett J, Byrne P, Klein HG, Marincola FM, Leitman SF, Stroncek DF. Analysis of a high-throughput HLA antibody screening assay for use with platelet donors. Transfusion. 2008 June;48(6):1174–9. doi: 10.1111/j.1537-2995.2008.01684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Powers A, Stowell CP, Dzik WH, Saidman SL, Lee H, Makar RS. Testing only donors with a prior history of pregnancy or transfusion is a logical and cost-effective transfusion-related acute lung injury prevention strategy. Transfusion. 2008 August 20; doi: 10.1111/j.1537-2995.2008.01902.x. [DOI] [PubMed] [Google Scholar]
- 133.Sachs UJ, Link E, Hofmann C, Wasel W, Bein G. Screening of multiparous women to avoid transfusion-related acute lung injury: a single centre experience. Transfus Med. 2008 December;18(6):348–54. doi: 10.1111/j.1365-3148.2008.00889.x. [DOI] [PubMed] [Google Scholar]
- 134.Kao KJ, Scornik JC, Riley WJ, McQueen CF. Association between HLA phenotype and HLA concentration in plasma or platelets. Hum Immunol. 1988 February;21(2):115–24. doi: 10.1016/0198-8859(88)90086-9. [DOI] [PubMed] [Google Scholar]
- 135.Jensen S, Jersild C, Grunnet N. Severe transfusion reaction to donor plasma containing HLA antibody. Vox Sang. 1990;59(1):62. doi: 10.1111/j.1423-0410.1990.tb02119.x. [DOI] [PubMed] [Google Scholar]
- 136.Kopko PM, Holland PV. Transfusion-related acute lung injury. Br J Haematol. 1999 May;105(2):322–9. doi: 10.1111/j.1365-2141.1999.01357.x. [DOI] [PubMed] [Google Scholar]
- 137.Sachs UJ, Bux J. TRALI after the transfusion of cross-match-positive granulocytes. Transfusion. 2003 December;43(12):1683–6. doi: 10.1111/j.0041-1132.2003.00568.x. [DOI] [PubMed] [Google Scholar]
- 138.Muniz M, Sheldon S, Schuller RM, Young NS, Klein HG, Leitman SF, Stroncek DF. Patient-specific transfusion-related acute lung injury. Vox Sang. 2008 January;94(1):70–3. doi: 10.1111/j.1423-0410.2007.00982.x. [DOI] [PubMed] [Google Scholar]
- 139.Nicolle AL, Chapman CE, Carter V, Wallis JP. Transfusion-related acute lung injury caused by two donors with anti-human leucocyte antigen class II antibodies: a look-back investigation. Transfus Med. 2004 June;14(3):225–30. doi: 10.1111/j.0958-7578.2004.00504.x. [DOI] [PubMed] [Google Scholar]
- 140.Bux J, Becker F, Seeger W, Kilpatrick D, Chapman J, Waters A. Transfusion-related acute lung injury due to HLA-A2-specific antibodies in recipient and NB1-specific antibodies in donor blood. Br J Haematol. 1996 June;93(3):707–13. doi: 10.1046/j.1365-2141.1996.d01-1703.x. [DOI] [PubMed] [Google Scholar]
- 141.Reil A, Keller-Stanislawski B, Gunay S, Bux J. Specificities of leucocyte alloantibodies in transfusion-related acute lung injury and results of leucocyte antibody screening of blood donors. Vox Sang. 2008 November;95(4):313–7. doi: 10.1111/j.1423-0410.2008.01092.x. [DOI] [PubMed] [Google Scholar]
- 142.Insunza A, Romon I, Gonzalez-Ponte ML, Hoyos A, Pastor JM, Iriondo A, Hermosa V. Implementation of a strategy to prevent TRALI in a regional blood centre. Transfus Med. 2004 April;14(2):157–64. doi: 10.1111/j.0958-7578.2004.00492.x. [DOI] [PubMed] [Google Scholar]
- 143.Flesland O. A comparison of complication rates based on published haemovigilance data. Intensive Care Med. 2007 June;33(Suppl 1):S17–S21. doi: 10.1007/s00134-007-2875-1. [DOI] [PubMed] [Google Scholar]
- 144.Hume HA. TRALI: moving toward prevention. Transfusion. 2009 March;49(3):402–5. doi: 10.1111/j.1537-2995.2008.02090.x. [DOI] [PubMed] [Google Scholar]
- 145.Middelburg RA, van Stein D, Zupanska B, Uhrynowska M, Gajic O, Silliman CC, Reif A, Bux J, Wallis JP, Briet E, van der Bom JG. Inconsistent association between donor gender and clinically defined TRALI. J Thromb Haenostas. 2008;6(supplement 1) [Google Scholar]
- 146.Boshkov LK, Maloney J, Bieber S, Silliman CC. Two cases of TRALI from the same platelet unit: implications for pathophysiology and the role of PMNs and VEGF. Blood. 2000;96(11):655a. [Google Scholar]
- 147.Kopko PM. Review: transfusion-related acute lung injury: pathophysiology, laboratory investigation, and donor management. Immunohematol. 2004;20(2):103–11. [PubMed] [Google Scholar]
- 148.Palfi M, Berg S, Ernerudh J, Berlin G. A randomized controlled trial oftransfusion-related acute lung injury: is plasma from multiparous blood donors dangerous? Transfusion. 2001 March;41(3):317–22. doi: 10.1046/j.1537-2995.2001.41030317.x. [DOI] [PubMed] [Google Scholar]
- 149.Gajic O, Yilmaz M, Iscimen R, Kor DJ, Winters JL, Moore SB, Afessa B. Transfusion from male-only versus female donors in critically ill recipients of high plasma volume components. Crit Care Med. 2007 July;35(7):1645–8. doi: 10.1097/01.CCM.0000269036.16398.0D. [DOI] [PubMed] [Google Scholar]
- 150.Benjamin RJ, Roggeveen B, Reeves C, Kelly ME. TRALI: components from female donors are not disproportionately involved in common transfusion reactions. Transfusion. 2004;44(9S):34A. [Google Scholar]
- 151.Brumit MC, Welsby I, Phillips-Bute B, Ramsey R, Lin S, Boyd T, Stafford-Smith M. Relationship between FFP donor gender and outome in 2,157 transfused cardiac surgery patients. Transfusion. 2007;47(Suppl):6A. [Google Scholar]
- 152.Gonzalez EA, Moore FA, Holcomb JB, Miller CC, Kozar RA, Todd SR, Cocanour CS, Balldin BC, McKinley BA. Fresh frozen plasma should be given earlier to patients requiring massive transfusion. J Trauma. 2007 January;62(1):112–9. doi: 10.1097/01.ta.0000250497.08101.8b. [DOI] [PubMed] [Google Scholar]
- 153.Landro L. New roles may shrink banks of blood donors. The Wall Street Journal. 2007 Jan 10;:D1–D8. Sect. Personal Journal. [Google Scholar]
- 154.Neergaard L. Gender matters in plasma transfusion. Associated Press Wire Service. 2007 Jan 22; [Google Scholar]