

Summary
α-Neurexins are essential synaptic adhesion molecules implicated in autism spectrum disorder and schizophrenia. The α-neurexin extracellular domain consists of 6 LNS domains interspersed by 3 EGF-like repeats and interacts with many different proteins in the synaptic cleft. To understand how α-neurexins might function as synaptic organizers, we solved the structure of the neurexin 1α extracellular domain (n1α) to 2.65 Å. The L-shaped molecule can be divided into a flexible repeat I (LNS1-EGF-A-LNS2), a rigid horseshoe-shaped repeat II (LNS3-EGF-B-LNS4) with structural similarity to so-called reelin repeats, and an extended repeat III (LNS5-EGF-B-LNS6) with controlled flexibility. A 2.95 Å structure of n1α carrying splice insert SS#3 in LNS4 reveals that SS#3 protrudes as a loop and does not alter the rigid arrangement of repeat II. The global architecture imposed by conserved structural features enables α-neurexins to recruit and organize proteins in distinct and variable ways, influenced by splicing, thereby promoting synaptic function.
Introduction
Neurexins are synaptic adhesion molecules recently implicated in neuro-psychiatric diseases including autism spectrum disorder (ASD) and schizophrenia (SZ) (Südhof 2008, Betancur et al. 2009). Neurexins are located predominantly on the pre-synaptic membrane. Their extracellular domains interact with different proteins in the synaptic cleft, while their cytoplasmic tails interact with the pre-synaptic exocytotic machinery. Neurexins play a role in synapse maturation by fine-tuning synaptic properties and regulate synaptic transmission through a ‘trans-synaptic dialog’ (Südhof 2008, Missler et al. 2003, Kattenstroth et al. 2004, Zhang et al. 2005, Chubykin et al. 2007). When levels of neurexin 1α (or their ligands neuroligins and LRRTMs) are manipulated in animal models, excitatory and inhibitory transmission is altered, a balance that is thought to be disrupted in vivo in many neuro-psychiatric diseases (Missler et al. 2003, Kattenstroth et al. 2004, Levinson et al. 2005, Varoqueaux et al. 2006, Chubykin et al. 2007, Tabuchi et al. 2007, Hines et al. 2008, Gibson et al. 2009, Etherton et al. 2009, de Wit et al. 2009, Dahlhaus et al. 2010, Blundell et al. 2010). Manipulating levels of neurexins and their ligands in animal models also replicates many of the behavioral alterations seen in humans with ASD and SZ (Beglopoulos et al. 2005, Tabuchi et al. 2007, Hines et al. 2008, Jamain et al. 2008, Blundell et al. 2009, Etherton et al. 2009, Blundell et al. 2010, Dahlhaus et al. 2010). Neurexins and their protein partners are now believed to be central components of a synaptic protein network, which underlies a common biological pathway disrupted in many neuro-psychiatric disorders (Guilmatre 2009).
The extracellular domain of neurexin 1α (n1α) is composed of three neurexin repeats (I, II and III), which each contain the modules LNS-EGF-LNS (Fig. 1a). N1α binds endogenous ligands including neuroligins, LRRTM family members, neurexophilin, α-dystroglycan and GABAA-receptors (Südhof 2008, Ko et al. 2009a, de Wit et al. 2009, Zhang et al. 2010, Siddiqui et al. 2010). α-Neurexins also regulate the functionality of certain Ca2+-channels, though direct association has not been demonstrated (Missler et al. 2003, Zhang et al. 2005). Recent studies suggest that other, as yet unidentified, proteins interact with α-neurexins as well (Ko et al. 2009b). Neurexins are structurally and functionally diversified through alternative splicing of their mRNAs (Fig. 1a). Potentially, thousands of different neurexin splice forms are produced with different protein partner binding profiles, generating a diverse portfolio of synaptic organizers. N1α LNS domains contain two β-sheets stacked as a β-sandwich in a jelly-roll fold (Sheckler et al. 2006, Shen et al. 2008) (Fig. 1a). A functional region called the ‘hyper-variable surface’ is formed by loops that carry splice inserts SS#2, SS#3, and SS#4 and encompasses a Ca2+-binding site (experimentally shown for L2, L4, and L6) (Rudenko et al. 1999, Sheckler et al. 2006, Shen et al. 2008) (Fig. 1a). Some, but not all, protein partners interact with the ‘hyper-variable surfaces’. Crystal structures show that neuroligins bind to the hyper-variable surface of the LNS domain in n1β (identical to n1α L6) by sandwiching the central Ca2+-binding site between the two proteins (Araç et al. 2007, Fabrichny et al. 2007, Chen et al. 2008, Leone et al. 2010). LRRTM proteins compete with neuroligins to bind n1β, suggesting that they share overlapping binding sites (Ko et al. 2009a, Siddiqui et al. 2010). α-Dystroglycan binds the hyper-variable surface of L2 and its association is disrupted by destroying the Ca2+-binding site; α-dystroglycan also binds to the hyper-variable surface of L6 and is competed by exogenous α-latrotoxin, another ligand for L6 (Sugita et al. 2001). Single LNS domains can support ligand binding; binding of some, but not all, protein partners is regulated by alternative splice inserts and requires Ca2+.
Fig. 1.

N1α structure. a) Left: n1α is composed of 6 LNS or LG domains (laminin, neurexin, sex hormone binding globulin or laminin G domains) interspersed by three EGF-like repeats. LNS domains are indicated as grey squares labeled L1 through L6 and EGF-like repeats as black ovals labeled A, B and C. In full-length n1α, L1 is preceded by a signal peptide and L6 is tethered to the cell membrane via a transmembrane segment (tms). Splice inserts SS#2, SS#3 and SS#4 add small stretches of amino acids to L2, L4 and L6 respectively, while SS#1 inserts a stretch of residues between EGF-A and L2. Neurexin repeats I, II, and III are indicated. Right: nomenclature of secondary structure elements in neurexin LNS domains using L2 as a template. The concave β-sheet comprises β13, β2, β11, β4, β5, β6, and β7. The convex β-sheet comprises β1, β12, β3, β8, β9 and β10. The ‘hyper-variable surface’ surrounds a Ca2+-binding site (blue sphere) and includes loops β2-β3, β4-β5, β6-β7 and β10-β11. Some LNS domains deviate from L2 by replacing α1 with a turn accommodated in a loop β12-β13 or have an additional short β-strand β14 following β13.; b) Ribbon diagram of bovine n1α, domains L2–L6. β-Strands in neurexin LNS domains are depicted as yellow arrows, α-helices as yellow coils, loop β11-β12 is shown in cyan and the EGF-like repeats in magenta. The module boundaries are as follows: L2 (Glu281-Thr485), L3 (Leu486-Cys679), EGF-B (Ser680-Glu722), L4 (Ala723-Ile909), L5 (Ile910-Cys1087), EGF-C (Glu1088-Gly1130) and L6 (Thr1131-Val1337). An N-linked oligosaccharide chain attached to Asn1230 is shown in ball-and-stick representation (carbon atoms, white; nitrogen atoms, blue; oxygen atoms, red).
While isolated neurexin LNS domains have been extensively characterized, high resolution information on their arrangement in the full-length n1α extracellular domain is not known, though a recent study using electron microscopy has revealed a low resolution model of n1α (Comoletti et al. 2010). The global organization and conformational freedom between modules in the ectodomain of α-neurexins likely determines the ability of this family of proteins to organize protein partners in the synaptic cleft. To gain insight into n1α structure-function relationships, we solved the structure of bovine n1α by x-ray crystallography to a resolution of 2.65 Å and n1α+SS#3 to 2.95 Å. The crystal structure of n1α reveals an L-shaped molecule. The modules are tethered together in flexible and rigid arrangements encoded by characteristic structural features. Neurexin repeat I is flexible, repeat II adopts a horseshoe-shaped structure similar to reelin repeats, and repeat III adopts an extended arrangement with a putative hinge between L5 and EGF-C. The architecture of n1α is highly suited for a synaptic organizer that arranges different proteins into large macromolecule complexes in order to promote synaptic maturation and transmission efficiency.
Results
Overall shape of n1α
N1α (modules L1-L6, Leu31-…-Ser1339) is found as a monomer in the crystal structure with an elongated L-shape (Fig. 1b). Continuous electron density is visible for L2, L3, EGF-B, L4, L5, EGF-C and L6 (residues Glu281-Val1337), including interconnecting residues. L2 through L5 form a long rod-shaped assembly (the ‘back’ of the ‘L’). EGF-C and L6 form a shorter assembly (the ‘foot’ of the ‘L’). The dimensions of the L2–L6 array are ca. 137 Å × 100 Å × 59 Å. The two crystallographically independent copies of n1α in the asymmetric unit (space group P1) show an identical arrangement of modules (rmsd 0.295 Å for 1003 Cα pairs), suggesting their molecular organization is an intrinsic property. N1α appears increasingly flexible towards its N-terminus. L2 (Baverage 118.2 Å2) displays more thermal motion than its C-terminal neighbors L3 (Baverage 83.2 Å2), L4 (Baverage 31.8 Å2), L5 (Baverage 26.9 Å2), and L6 (Baverage 68.9 Å2) (as calculated by TLSANL (CCP4, 1994)). In addition, unlike for the rest of the molecule, the electron density for L2 and the concave β-sheet of L3 largely does not resolve the individual βstrands, though it clearly reveals the β-sheets. No electron density is observed for L1 and EGF-A, though present in the crystal, suggesting that these N-terminal modules are disordered and can adopt multiple conformations. Thus, the crystal structure of n1α reveals 7 out of 9 modules (1003 residues per molecule) organized in an L-shaped assembly. Prior to examining the architecture of n1α, and its impact on n1α function, the crystal contacts were analyzed (Supplemental Material, Fig. S1).
Modules of n1α
The individual modules of n1α display striking structural features that appear to have evolved to maintain the global architecture of the extracellular domain. Firstly, though neurexin LNS domains share a common jellyroll fold, characteristic loops extend from their β-sheets. Loop β11-β12 ranges from a short 10 residue loop in L6 to a long 16 and 22 residue loop in L3 and L5 with helical content, and fills the concave β-sheet in different ways (Fig. 1b, Fig. 2a). Loop β4-β5 of the hyper-variable surface is quite short, just 4 to 6 residues, except in L3 where it spans 18 residues (Fig. 2a). Secondly, in L2, L3, L4, and L5, but not the terminal L6, a cysteine is located just before the polypeptide chain continues on into the next module. These cysteine residues engage in a disulfide bond with a preceding cysteine located on or near strand β12, one of the last strands in the LNS domain fold (L2: Cys444-Cys480; L3: Cys650-Cys679; L4: Cys890-Cys898; L5: Cys1059-Cys1087). The disulfide bonds rigidly define each module from the interconnecting linker residues and module that follows. Thirdly, the EGF-like repeats B and C are structurally very similar to each other (1.0 Å rmsd for 35 matched Cα-atoms), even though they share only 34 % sequence identity (13 out of 38 residues). EGF-B and C both consist of 2 β-strands and loops held together by 3 disulfide bonds (connectivity C1–C3, C2–C4 and C5–C6). Their close structural similarity suggests that these modules are compact, very well-ordered domains that preserve their shape.
Fig. 2.

Neurexin repeat II (L3-EGF-B-L4). a) Variability of loop β11-β12 and loop β4-β5 in neurexin LNS domains. In a multiple structure alignment, the LNS modules superimpose in a range 1.09 to 1.37 Å for 177 to 194 Cα-atoms (see Supplemental Material). Loop β11-β12 is shown in cyan for all modules, except L6 shown in blue. Loop β4-β5 is shown in yellow for all modules, except L3 shown in red. The ‘concave’ and ‘convex’ β-sheets are indicated.; b) Ribbon diagram of the L3-EGF-B-L4 neurexin repeat II. Residues within 4 Å of each other forming the interfaces between modules are shown in ball-and-stick (carbon atoms, yellow, orange, cyan or magenta; oxygen atoms, red; nitrogen atoms, blue; sulfur atoms, green). Loop β11-β12 is shown in cyan, loop β4-β5 from L3 in orange. Molecular details are shown in the inset below. The insertion site for SS#3 (G790-K800) is indicated in green. Direct contacts are formed between L3 (primarily loop β4-β5 and loop β11-β12) and L4 (β2, loop β2-β3, β3, loop β4-β5, loop β6-β7, β8, loop β8-β9, loop β10-β11, β11 and loop β12-α1). Hydrophobic and charged interactions mediate the L3 and L4 interface. These include a hydrophobic cluster formed by Phe634, Trp639, Leu642, Leu643 from L3 that packs against hydrophobic areas of Met742, Thr744, Glu745, Ala746, Arg824 from L4, and many charged residues, in particular salt bridges between Asp536, Arg848, Glu637, Glu745 that ‘zig-zag’ between L3 and L4, and a striking number of histidine residues His539, His743, His852, and His846. Direct contacts are formed between EGF-B and L3 (loop β1-β2, loop β3-β4, loop β7-β8, loop β11-β12, and the β-strands of the convex sheet β1, β3, and β12, and linker residues connecting the two modules. Direct contacts are formed between EGF-B and L4 (primarily β3, β8, β12, loop β12-α1, α1, loop α1-β13) and linker residues between EGF-B and L4. Nomenclature as described in Fig. 1a. Asp546 of loop β4-β5 in L3, a conserved counterpart of D772 in L4, is shown in ball-and-stick representation.; c) Ribbon diagram of the reelin repeat R6 containing modules L1, EGF, and L2 (pdb id: 3A7Q, Yasui et al. 2010). L1 and L2 are shown in pink, EGF in magenta and the loop analogous to neurexin LNS domain loop β11-β12 is shown in cyan for both L1 and L2. The LA1 cysteine-rich repeat of the apoE receptor 2 binds to the bottom of L1, shown in green. Calcium ions are shown as blue spheres. Reelin L2 corresponds structurally more to n1α L3, and reelin L1 more to n1α L4 when using the concave sheet filling loop β11-β12 to orient the modules.
Organization of n1α
L2, L3, EGF-B, L4 and L5 form a rod-shaped array by packing sequentially against each other via extensive interdomain interactions. The β-sandwiches of L2, L3, L4, and L5 arrange so that the concave sheet of a preceding LNS domain interacts with the convex sheet of the next LNS domain (Fig. 1b). Loop β11-β12 that fills the concave β-sheet and in many structurally related legume lectins forms part of a carbohydrate binding site (Loris et al. 2002), plays an important role in mediating interactions between the concatenated LNS domains (Fig. 1b, Fig. 2a). Loop β11-β12 of L2 docks on L3 contacting central β-strands of its convex sheet (β3, β8) and the linker between L3 and EGF-B, (see Fig. 1a for nomenclature). Loop β11-β12 of L3, in turn, docks on central β-strands of the convex β-sheet of L4. Finally, the base of loop β11-β12 of L4 docks on L5 contacting central β-strands of the convex sheet of L5, though this interface is more complex and involves additional strands and loops of L4. Remarkably, L6 breaks away from the rod-like assembly, and veers off at ca. 75° angle mediated by EGF-C (Fig. 1b).
Although the protein sequence of n1α can be divided up in three repeats (I, II and III) (Fig. 1a), the spatial organizations of these repeats are dramatically different. The L1-EGF-A-L2 repeat appears flexible, as L1 and EGF-A are not visible in the crystal structure. L3-EGF-B-L4 adopts a globular horseshoe-shaped structure and L5-EGF-C-L6 forms an extended array. The compact EGF-like repeats play a crucial role in determining the different structural arrangements of the neurexin repeats and the overall shape of the n1α extracellular domain.
In L3-EGF-B-L4, EGF-B spans the top of the LNS domain pair providing extensive interactions to both (Fig. 2b, details in legend and summarized below). EGF-B interacts with loops connecting the convex and concave sheets of L3, and β-strands from the convex β-sheet of L3. EGF-B contacts β-strands from the convex β-sheet of L4, loop β12-β13 that contains helix α1, and the linker between EGF-B and L4. At the bottom of the L3–L4 pair, the long loop β4-β5 that is present only in L3 (refer back to Fig. 2a) arcs over and binds to the ‘hyper-variable surface’ of L4. Lys538 at the tip of this loop interacts with the side chain of Asp772 (loop β4-β5) and the backbone carbonyl of Leu789 (loop β6-β7), two key residues of the Ca2+-binding site in L4 (Fig. 2b). This site not only binds Ca2+-ions, but also competing positively charged amino-groups (Shen et al. 2008). Remarkably, L3-EGF-B-L4 resembles the jellyroll-EGF-jellyroll arrangement found in so-called ‘reelin repeats’ of reelin (Fig. 2c), a protein that directs neuronal migration during cortical development and is implicated in neuro-psychiatric disorders (Knuesel 2010, Yasui et al. 2010). Furthermore, the reelin-repeat R6 binds the LA1 module of one of its receptors, the apoE receptor 2, by exploiting a binding site that coincides structurally with the hyper-variable surface of L4 (Fig. 2c).
In L5-EGF-C-L6, EGF-C separates L5 from L6 (Fig. 3). EGF-C interacts with L5 in a fundamentally different way compared to L6, suggesting a structural mechanism has evolved to support neurexin function. EGF-C docks onto the terminal residues of the L5 polypeptide chain (Glu1084 from strand β14, Gly1086, and its final residue Cys1087). A very short ‘hinge’ connects L5 and EGF-C (Glu1088-Gly1089). The bulky side chains Arg1085 and Trp1109 pack on one side of the hinge, Glu1084 and Lys917 (that form a salt bridge together) as well as Pro1090 pack on the other side (Fig. 3). These residues likely limit the conformational freedom of the hinge. The hinge is further delineated by residues on either side in the polypeptide sequence: Cys1087-Glu1088-Gly1089-Pro1090-Ser1091. At one end, the disulfide bond between Cys1087 and Cys1059 covalently restricts motion. At the other end, Pro1090 restricts the polypeptide backbone conformational freedom through its covalent interaction with main chain atoms, while Ser1091 and subsequent residues are buried in EGF-C. L6 makes no direct interactions with L5 or the rest of the molecule, other than with the preceding EGF-C. Unlike the interaction between L5 and EGF-C, the interaction between EGF-C and L6 is extensive suggesting that the orientation between EGF-C and L6 is fixed (Fig. 3). The hinge in neurexin repeat III suggests that the ’foot’ of the L-shape (EGF-C-L6) can pivot with respect to the ‘back’ of the L-shape (L2–L5), though the range of motion is likely limited by the residues that pack on either side of the hinge.
Fig. 3.
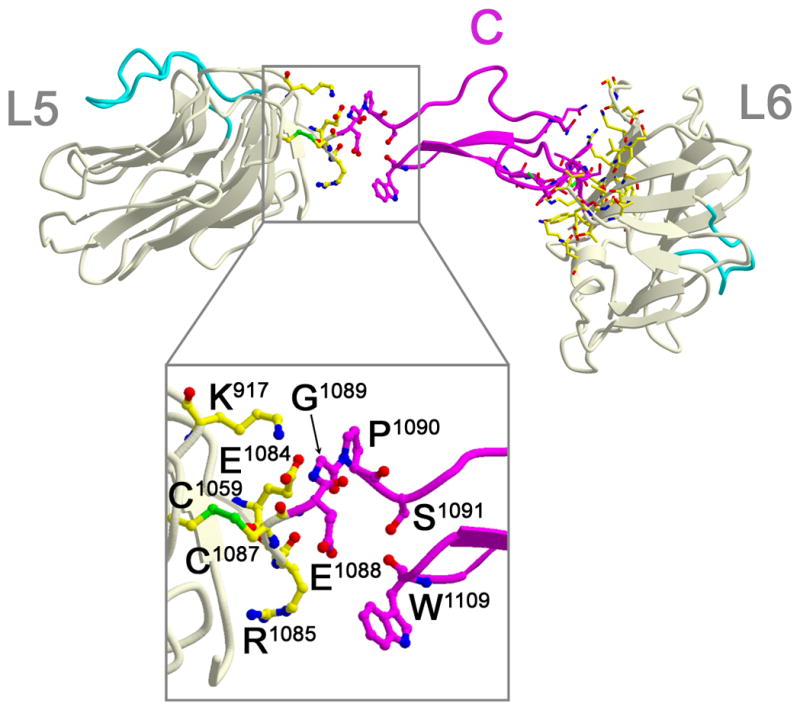
Neurexin repeat III (L5-EGF-C-L6). Residues within 4 Å of each other forming the interfaces between modules are shown in ball-and-stick. L5 and L6 are shown in light yellow, EGF-C in magenta. Loop β11-β12 is shown in cyan. Molecular details are shown in the inset below. EGF-C interacts with L6 by docking its C-terminal portion against the center of the convex sheet of L6 (β3, β8, β9, β12), as well as interacting with loop β7-β8, loop β9-β10, loop β12-α1, and the linker residues between EGF-C and L6.
N1α+SS#3
The remarkable horseshoe-shaped arrangement of L3-EGF-B-L4 and its similarity to reelin repeats prompted further investigation. The extra long loop β4-β5 from L3 that extends its tip into the Ca2+-binding site of L4 also docks close to the insertion site for splice insert SS#3 (for which no function is known yet). To answer the question if SS#3, which replaces Gly790 with a ten amino acid splice insert Asp790-Ser799 (refer back to Fig. 1a, Fig. 2b) changes the domain organization of L3-EGF-B-L4, we solved the structure of n1α+SS#3 to a resolution of 2.95 Å. L3-EGF-B-L4 with and without SS#3 superimpose within 0.27 Å for 414 Cα-atoms, indicating that SS#3 does not alter the arrangement of the modules (Fig. 4a). Both SS#3 as well as loop β6-β7 that hosts the splice insert are clearly visible in the electron density with the exception of a gap corresponding to Cys796-Asn797-Ser798-Ser799 of SS#3 (Fig. 4b, Fig. S2). SS#3 extends loop β6-β7 out into solution. A salt bridge forms between Arg793 of SS#3 and Asp845 of loop β10-β11 (Fig. 4c). Cys791 and Cys796 of SS#3 do not engage in a disulfide bridge. The presence of SS#3 does not rearrange the Ca2+-binding site of L4 significantly (i.e. the side chain of Asp772 and the backbone carbonyls of Arg848 and Leu789, the latter which is directly adjacent to the splice insert site). Strikingly, the presence of SS#3 does not prevent loop β4-β5 of L3 from binding to the hyper-variable surface of L4, and Lys538 is again found blocking the Ca2+-binding site of L4 (Fig. 4c). This suggests that the organization of repeat II is an intrinsic property of n1α that is tied to α-neurexin function.
Fig. 4.

Structure of n1α+SS#3. a) Superposition of neurexin repeat II (L3-EGF-B-L4) without SS#3 (in grey) and carrying SS#3 (in yellow and magenta). For the latter, SS#3 is shown in green with missing residues represented by dotted lines, and loop β4-β5 is shown in red. For clarity, the neurexin repeat is rotated 180° with respect to Fig. 2b. b) SigmaA-weighted 2Fo−Fc electron density map in the region surrounding the splice insert SS#3, contoured at 0.9 σ for molecule 1 (left) and molecule 2 (right). In n1α lacking SS#3, Asp790 is replaced by Gly790, and covalently connected to Lys800. Two residues have poor side chain density and have been built as alanine residues (Ile794 and Lys800). c) SS#3 (Asp790-Ile794, green carbon atoms) inserts between Leu789 and Lys800, and does not displace β4-β5 of L3 (orange carbon atoms). Residues are shown in ball-and-stick-representation with color scheme: oxygen, red; nitrogen, blue; and sulfur, yellow.
Discussion
Structure-function relationships of n1α
The remarkable shape of n1α is produced by interplay between structural elements within individual modules and interactions between modules leading to neurexin repeats with very different conformational arrangements and freedoms. The striking structural features that promote this architecture also suggest that the global organization of α-neurexins is fundamentally important for their function as synaptic organizers.
Before evaluating potential structure-function relationships, we assessed whether the crystal structure likely reflects the conformation of n1α in the synaptic cleft.
Does neurexin repeat II form a rigid core? L3-EGF-B-L4 forms the largest intramolecular interface in n1α, burying 2893.3 Å2 (1386 Å2 between L3 and L4, 870 Å2 between L3 and EGF-B, and 637 Å2 between EGF-B and L4). SS#3 does not disturb the organization of this repeat, nor prevent loop β4-β5 of L3 from inserting into the hyper-variable surface of L4. All mammalian α-neurexin genes share this long loop β4-β5 in L3, and a lysine or arginine residue is found at position Lys538 in fourteen mammalian n1α and n3α sequences, though ambiguous sequence alignments prevent an analogous residue from being identified in n2α. Reelin repeats show a similar spatial arrangement to neurexin repeat II, though they are further assembled into rod-shaped, linear assemblies, unlike n1α (Fig. 2c, Nogi et al. 2006, Yasui et al. 2007). Both n1α neurexin repeat II and reelin repeat R6 use a lysine residue at the hyper-variable surface (or its structural counterpart) to interact with a protein surface. In n1α, Lys538 interacts with Asp772 of L4 likely stabilizing neurexin repeat II. In reelin repeat R6, a lysine residue from jelly-roll L1 interacts with a protein partner (LA1 of the apoE receptor 2) via aspartate residues surrounding the LA1 Ca2+-binding site (Yasui et al. 2010, Fig. 2c). The situation is not completely analogous, because in n1α, Lys538 blocks the Ca2+-binding site of L4, whereas in reelin, Lys2467 likely requires the Ca2+-ion in LA1 to arrange the acidic residue cluster for interaction (Yasui et al. 2010). Nonetheless, the extensive buried surface, the conserved key structural features, undisturbed even by SS#3, and the structural similarity to reelin repeats all suggest that the horse-shoe shaped conformation of L3-EGF-B-L4 is an intrinsic property of the n1α architecture.
Does EGF-C keep L5 and L6 separated at a discrete distance? Though the interface between EGF-C and L6 (990 Å2) is smaller than that buried in the L3-EGF-B-L4 horseshoe, EGF-C likely packs solidly against L6. Its buried surface is on the same scale as that between L2 and L3 (994 Å2), L4 and L5 (1107 Å2), the direct interaction between L3 and L4 (1386 Å2), and the interaction site between reelin R6 and apoE receptor 2 LA1 (~ 700 Å2) with a reported affinity of ~100 nM (Yasui et al. 2010, Fig. 2c). In contrast, the buried surface between L5 and EGF-C is much smaller (289 Å2). Key structural features are identical in almost all 22 sequences from mammalian α-neurexin genes 1, 2 and 3, namely: the hinge residues (Glu1088 and Gly1089), stretches of 7 residues N- and C-terminal to the hinge, and the bulky side chains that pack spatially around the hinge. This suggests that neurexin repeat III enables the rigid EGF-C-L6 assembly to pivot with respect to the rest of the molecule to some (limited) extent.
Are the buried surfaces between modules in the n1α L2–L6 assembly true biological interfaces? Except for one, all the interfaces classify as relatively small biological interfaces (or very large crystal contacts) (see Supplemental Material). However, current methodologies do not accurately discriminate biological interfaces from fortuitous crystal contacts in crystal structures of proteins containing covalently connected modules, like n1α, because the steric and entropic contributions imparted by linking modules is not accounted for. Analysis of 22 mammalian α-neurexin sequences shows that almost 80% of all interface residues are either conserved or semi-conserved, and of these, ca. 75% of the Tyr, His, Phe, Trp, Leu, Ile, Val, Met, and Arg residues (residues that are often found at biological contacts (Lo Conte et al. 1999)). Therefore, while the sizes of the buried surfaces between modules in n1α do not unambiguously prove that these interfaces exist in solution, they are large enough and share high enough sequence conservation to indicate they form and that the organization of n1α in the crystal structure likely represents a physiologically relevant conformation.
The shape of n1α shows similarities with molecular envelopes of n1α produced recently by electron microscopy (EM). N1α was visualized as a Y-shape. L1–L4 were assigned to the long base of the Y, while L5–L6 were assigned to the two arms of the Y (Comoletti et al. 2010). Two regions of flexibility were pinpointed, one between EGF-A and L2 and a second between L5 and L6. The resolution of the EM data did not permit the orientation of the LNS domains to be determined or the position of the EGF-like repeats, thereby limiting further comparison. Nevertheless, the global architecture of n1α, i.e. an elongated molecule with L6 veering spatially away from the L2–L5 array, is consistent between the EM and crystallographic studies suggesting it is a physiological feature.
Functional consequences of the n1α architecture
The domain arrangement of n1α has fundamental consequences for α-neurexin function. The hyper-variable surfaces of L2, L3, L4, L5, and L6 line up on one side of the molecule (Fig. 5a). Because these surfaces are putative binding sites for protein partners (discussed in the Introduction), interacting ligands would assemble on just one side of the extracellular domain, a feature that would facilitate pre-synaptically tethered n1α to recruit multiple partners exposed on the opposing post-synaptic membrane. The n1α arrangement also suggests that multiple modes of protein partner recognition are possible. Ligands interacting with different single LNS domains could be recruited simultaneously by α-neurexins and spatially organized into large macromolecular complexes in the synaptic cleft (Fig. 5b). Protein partners might also interact with two LNS domains (Fig. 5c). Though it is not known how multiple domains of n1α might interact with α-dystroglycan, the structurally related laminin α2 LG4–LG5 tandem binds α-dystroglycan using surfaces that surround a Ca2+-binding site on each module, analogous to the Ca2+-binding sites found at neurexin hyper-variable surfaces. Although the two LG domains adopt a fixed V-shape not seen in n1α, their arrangement constrains the putative α-dystroglycan binding surfaces to the same side of the molecule at the tips of the V (Tisi et al. 2000). Likewise, a protein partner interacting with neurexin L2 and L3, or L3 and L4, would subject itself to the spatial constraints imposed by n1α. In contrast, proteins interacting with L1 and L2, or L5 and L6 would either need to be flexible themselves or rigid molecules that imposed their own molecular architecture on n1α. Lastly, large protein partners might contact more than two adjacent or non-adjacent n1α modules, enabling multiple splice inserts in n1α to potentially modulate protein partner binding and recognition (Fig. 5d). Molecular recognition would be influenced by the rigid and flexible connections within the n1α architecture. The relatively large distances between the splice insert sites (> 26 Å) and the relatively short stretches of polypeptide added (< 30 a.a.) suggest that it is unlikely that splice inserts or insert sites end up directly side-by-side each other. However, a portfolio of different composite binding sites could be formed (like mosaics made up of varying pieces) as a function of the different splice inserts present or absent (Fig. 5d).
Fig. 5.

Protein partner recognition by n1α. a) The hyper-variable surfaces of L2, L3, L4, L5 and L6 align on one side of n1α (loops shown in light orange). Conserved residues Asp329 (L2), Asp546 (L3), Asp772 (L4), Asp958 (L5) and Asp1183 (L6) central to each hyper-variable surface (and in L2, L4 and L6 can ligand Ca2+) are shown in ball-and-stick with distances indicated. Insertion sites for splice insert SS#2, SS#3, and SS#4 are indicated with yellow, green and magenta spheres respectively. The loop β4-β5 of L3 is shown in dark orange. Possible mechanisms by which n1α (L1–L6) recognizes different protein partners (‘p1’ – ‘p6’) are shown in b–d. Variability introduced by alternative splice inserts is shown for L2, L4, and L6 with carets.; b) single LNS domains could interact with different protein partners using splice insert dependent and/or independent mechanisms.; c) combinations of two LNS domains could bind protein partners potentially influenced by a splice insert.; d) multiple modules of n1α could bind a protein partner potentially influenced by one or more splice inserts.
With the different binding modes in mind, we modeled the interaction between n1α and neuroligin 1 (NL1), using the structure of the n1β:NL1 complex as a guide (Araç et al. 2007, Chen et al. 2008). Like neurexins, the family of post-synaptic adhesion molecules, neuroligins, is diversified through alternative splicing at a Site A (which can carry ca. 20 a.a. inserts) and in NL1 also at a Site B (that can carry a 9 a.a. insert) (Südhof 2008). Though n1α appears to bind NL2 preferentially over NL1, NL3 or NL4 (Kang et al. 2008, suggested by Chih et al. 2006), subsequent reports have shown that n1α also binds NL1 if the Site B insert is absent (Boucard et al. 2005, Ko et al. 2009a, Siddiqui et al. 2010, Comoletti et al. 2010, Reissner et al. 2008). Superimposition of n1α L6 on the identical domain of n1β of the n1β:NL1 complex reveals the spatial location of the n1α specific modules with respect to NL1. The docking studies show that when L6 binds NL1, EGF-C functions as a spacer, placing the L2–L5 assembly alongside each NL1 subunit of the dimer (Fig. 6). In the n1α:NL1 model, Site B of NL1 is close to the interface between L6 and NL1, and Site A of NL1 in close to L3 and L4 (Fig. 6). The hinge between L5 and EGF-C would permit optimal alignment of L3 and/or L4 with NL1. Our docking studies therefore suggest that it is possible for Site B in NL1 to cooperate with SS#4 of L6 in n1α (as it does in n1β, Araç et al. 2007, Chen et al. 2008) following the concept of a splice code centered around Site B and SS#4 that governs the interactions between neurexins and neuroligins (Südhof 2008). In addition, it is possible that Site A in neuroligins cooperates with SS#3 of L4. If true, interaction between Site B and SS#4 would constitute one part of the splice code, and Site A and SS#3 another for regulating binding between α-neurexins and neuroligins.
Fig. 6.

Molecular modeling of the interaction between n1α and NL1. The NL1 dimer (blue) is shown with an n1α molecule (coloring scheme as in Fig. 5a) bound to each subunit. On the left, the n1α:NL1 interaction is displayed as a ribbon diagram with key structural features labeled. On the right, the n1α:NL1 interaction is shown as a ribbon diagram covered by a solvent accessible surface. Site A in NL1 is indicated by two red spheres connected by a dotted line, Site B by a single, red sphere. Splice insert sites SS#2, SS#3 and SS#4 in n1α are indicated by yellow, green and magenta spheres respectively. The putative hinge between L5 and EGF-C is indicated. A dotted double-headed arrow depicts potential movement of the L2–L5 assembly with respect to EGF-C-L6 controlled by the hinge.
Our model of the n1α:NL1 complex is consistent with biochemical data describing the interaction of n1α with different partners.
Neuroligins
Because L6 protrudes as an isolated domain from n1α extending its hyper-variable surface out to the solvent (Fig. 1b, Fig. 6), it is likely that the binding mode between n1β and neuroligins is largely preserved in the interactions between L6 of n1α and neuroligins. N1β and NL1 splice forms bind with nanomolar affinity (ca. 10–95 nM, Comoletti et al. 2006, Araç et al. 2007, Chen et al. 2008, Siddiqui et al. 2010, though much weaker affinities have been reported, Koehnke et al. 2010). For β-neurexins, binding is weakest when the site B insert and SS#4 are present simultaneously and binding between β-neurexins and neuroligins is not affected by Site A (Boucard et al. 2005, Ko et al. 2009a, Siddiqui et al. 2010). For α-neurexins, however, SS#4 in n1α reduces binding to NL1, as it does for n1β, but the Site B insert in NL1 completely disrupts binding to n1α, unlike to n1β (Boucard et al. 2005, Ko et al. 2009a, Reissner et al. 2008, Siddiqui et al. 2010). Furthermore, Site A seems to promote binding between n1α and NL1 (Boucard et al. 2005), a curious result given the large distance between Site A and the L6:neuroligin interface. The n1α crystal structure suggests the following insights. Firstly, Site B is located at the L6:neuroligin interface, near the hinge between L5 and EGF-C (Fig. 6). The B site insert, which also carries an N-linked glycosylation site, likely produces steric hindrance when brought in proximity with the n1α specific L2–L5 assembly. Indeed, enzymatic deglycosylation of NL1 (A+, B+) largely restores its binding to n1α and this activation through deglycosylation requires the Site B insert to be present (Boucard et al. 2005). Site B and its N-linked glycosylation site are believed to be functionally important, because deletion of this glycosylation site shifts NL1 (A+, B+ N303A) to GABA-ergic synapses (Chih et al. 2006), potentially because binding to n1α splice forms at inhibitory synapses is now more viable. Secondly, our n1α:NL1 model suggests that n1α-specific modules can interact with Site A explaining why it modulates α-neurexin binding but not β-neurexin binding. While Site B has received much attention, Site A is biologically important as well. The presence of Site A inserts in NL1 (-B) and NL2 (not spliced at B) shifts these neuroligins from excitatory synapses to inhibitory synapses (Chih et al. 2006), though it must now be demonstrated that this is due to n1α-specific interactions and not other interacting proteins. Lastly, α-neurexin specific modules were shown to reduce binding to neuroligins, at least for the combinations of splice forms tested so far. Deletion mutagenesis pinpointed L3-EGF-B-L4 as the main suppressors of n1α:NL2 interaction compared to n1β-mediated binding (Kang et al. 2008). If n1α were a linear concatenation of modules, as often depicted in the literature, these results would be difficult to explain. However, our n1α:NL1 model predicts that modules N-terminal to L6 are able to influence neuroligin binding because the extracellular domain is not linear, and L3 and/or L4 may interact directly with neuroligins (regulated by the combination of Site A and SS#3).
α-Latrotoxin
α- and β-neurexins bind α-latrotoxin in a Ca2+-dependent manner via L6. α-Latrotoxin binds n1α with low nanomolar affinity, competes pre-bound α-dystroglycan off, and the presence of SS#4 in n1α and n1β disrupts its binding (Davletov et al. 1995, Sugita et al. 1999). At low salt concentration (ca. 100 mM) n1β-SS#4 binds α-latrotoxin as well as n1α (+SS#1/−SS#2/+SS#3/−SS#4) (Sugita et al. 1999). However, the interaction between α-latrotoxin and n1β does not withstand a high salt concentration, unlike n1α, which still binds α-latrotoxin at unusually high salt concentrations (1.5 M NaCl) (Sugita et al. 1999). This strongly suggests that the interaction between n1β (or n1α L6) and α-latrotoxin is predominantly governed by electrostatic interactions, while additional interactions must be present in n1α that contribute hydrophobic surfaces to the interaction site with α-latrotoxin. Our n1α model suggests that not only L6, but also modules N-terminal to L6 could interact directly with α-latrotoxin because the mobility of the L5-EGF-C hinge and the spacer role of EGF-C would provide them access.
Therefore, while binding of n1α to neuroligins and α-latrotoxin could be described by model Fig. 5d, binding to other protein partners, for example neurexophilin binding to L2 (Missler et al. 1998) and LRRTM proteins that seem to bind n1α and n1β similarly well (Siddiqui et al. 2010, de Wit et al. 2009, Ko et al. 2009a) might best be described by model Fig. 5b.
Future directions
The n1α architecture likely impacts how many of the different α-neurexin protein partners are recruited and organized. Likewise, protein partner binding may impact the conformation of n1α, underscoring the importance of determining structures of n1α in complex with different ligands. The crystal structure of n1α suggests that the modules preceding L6 may regulate the interaction of ligands bound to L6 (positively or negatively). This must now be experimentally addressed. Given that α-neurexins are increasingly implicated in neuro-psychiatric diseases, it is fascinating to speculate that their lesion destroys the spatial organization of an array of synaptic proteins who are functional on their own but must be properly assembled into large macromolecular complexes to release their full potential. The strategically positioned structural elements of n1α and a protein partner profile that can be regulated by different splice inserts distributed along its length, suggest that α-neurexins are extremely well suited to function as scaffolding molecules in the synaptic cleft and that α-neurexins may host a diverse set of protein interactions, some of which might even be targeted therapeutically in future with small molecule compounds.
Experimental Procedures
Protein expression and purification
The extracellular domain of bovine neurexin1α (called n1α, nm_174404) was expressed in insect cells. N1α contains residues Met1-Ser1339 including the endogenous signal peptide, but not SS#1, SS#2, SS#3 or SS#4 though they are accommodated in the numbering scheme. Crystals of mutant n1α_N190Q (no splice inserts) diffracted past 2.65Å. Crystals of n1α+SS#3, identical to n1α but containing SS#3 (DCIRINCNSS), diffract similarly to n1α without the optimizing N190Q mutation. N1α was also expressed as a seleno-methionyl form replacing 19 out of 22 methionines per molecule on average as determined by mass spectrometry.
Protein crystallization
Crystals of n1α (native and seleno-methionyl forms), n1α_N190Q and n1α+SS#3 grow in a broad range of conditions containing ca. 5–10% Peg 8000 or Peg 10000, 100 mM Bicine pH 8.0–9.0, 2.5 mM CaCl2 at 20°C, and were harvested under similar conditions containing 5 mM CaCl2 and flash cooled with glycerol. All four proteins produce crystals with space group P1 and similar cell dimensions (for n1α_N190Q, a=60.953 Å, b=114.543 Å, c=159.58, α=90.61 Å, β=90.87°, γ=92.18°) that are radiation sensitive and diffract weakly.
Structure determination
N1α was solved by a combination of molecular replacement and single anomalous dispersion (SAD) techniques using PHASER (McCoy et al. 2007). Briefly, the seleno-methionyl n1α data to 4 Å was used to iteratively place 8 out of 12 possible LNS domains. Model phases generated from the partial molecular packing were then used by PHASER to locate 37 out of 44 possible selenium (Se) atoms producing a phase set with figure of merit (fom) 0.507 to 4 Å. Subsequent rounds of model building and refinement with REFMAC (Murshudov et al. 1997) generated an improved model that enabled additional Se atoms to be located by PHASER. In total, 42 potential Se sites were refined with SHARP (Bricogne et al. 2003) using the best available model phases as an external source of phase information and they produced a phase set with fom 0.678 to 3.26 Å. The model was completed and refined to 2.65 Å with the n1α_N190Q data set. Of the 40 ordered methionine residues in the n1α molecules, 39 match a Se atom position. Difference Fourier analysis suggests that only L2 may contain a Ca2+-ion. The model of n1α has a Molprobity Score (Chen et al. 2010) of 2.78 (66th percentile) and a clashscore of 16.45 (84th percentile); 92.2 % of the residues are in the favored region of the Ramachandran plot and 1.6 % of the residues are in the unfavorable region. The atomic coordinates of n1α_N190Q have been deposited (pdbid:3QCW). N1α+SS#3 was solved by molecular replacement with PHASER using the n1α monomer as a search model (and, as a control, single LNS domains). The model of n1α+SS#3 refined to 2.95 Å has a Molprobity Score of 3.25 (54th percentile) and a clashscore of 25.82 (85th percentile); 89.9 % of the residues are in the favored region of the Ramachandran plot and 1.8 % of the residues are in the unfavorable region (pdbid:3R05).
Model analysis
The contact analysis was carried out using ePISA (www.ebi.ac.uk/msd-srv/prot_int/pistart.html, Krissinel & Henrick 2007).
Details of the structure determinations are summarized in Table 1 and additional details are provided in the Supplemental materials.
Table 1.
Data and Refinement Statistics Summary for n1α_N190Q, SMet-n1α and n1α+SS#3
| Crystal | n1α_N190Q | SMet-n1α | n1α+SS#3 |
|---|---|---|---|
| Data Statistics | |||
| Spacegroup | P1 | P1 | P1 |
| Resolution (Å) a | 39.9-2.65 (2.70 - 2.65) | 35.0-3.25 (3.31 - 3.25) | 48.51-2.95 (3.00-2.95) |
| Reflections total/unique | 418903/124701 | 267276/67601 | 306149/89668 |
| Multiplicity | 3.4 | 4.0 | 3.4 |
| Completeness (%) | 98.5 (98.2) | 98.6 (97.9) | 98.4 (95.6) |
| Rmerge (%) b | 7.9 (52.6) | 13.5 (80.2) | 9.0 (53.2) |
| I/σ | 17.8 (3.0) | 14.6 (1.9) | 14.1 (2.1) |
| Fom from PHASER (8 LNS domains + 37 Se atoms) to 4.0 Å: 0.507 | |||
| Fom from SHARP (model + 42 Se atoms) to 3.26 Å: 0.678 | |||
| Refinement Statistics | |||
| Resolution (all reflections |F|/σ ≥ 0.0) | 30.0 – 2.65 Å | 30.0 – 2.95 Å | |
| Protein atoms (including carbohydrate) | 15440 | 15522 | |
| Protein residues | 2006 (2× 1003) | 2016 (2× 1008) | |
| Solvent atoms | 0 | 0 | |
| Molecules in the asymmetric unit | 2 | 2 | |
| Unique reflections | 123177 | 89495 | |
| Working set/Test set c | 116726/6451 | 84841/4654 | |
| Rwork | 20.82 % | 20.83 % | |
| Rfree | 22.96 % | 23.54 % | |
| Rmsd bond lengths | 0.018 Å | 0.026 Å | |
| Rmsd bond angles | 1.730° | 2.335° | |
| Rmsd NCS-related molecules(Cα atoms) | 0.295 Å (1003) | 0.203 Å (1008) | |
| Total Bave/residual Bave (atomsmainchain) d | 65.3 Å2/47.2 Å2 (8024) | 91.7 Å2/79.4 Å2 (8064) | |
| Total Bave/residual Bave (atomssidechain) d | 65.7 Å2/47.9 Å2 (7360) | 92.4 Å2/79.8 Å2 (7402) | |
Outer shell statistics in parentheses;
Rmerge=Σ (|(I − <I>)|/Σ (I);
Test set for n1α+SS#3 constructed using reflections present in the n1α test set to avoid bias.
Total Bave reported by TLSANL/residual Bave reported after TLS and positional refinement by REFMAC
Supplementary Material
Acknowledgments
This work was funded by NIMH RO1 MH077303 and would not have been possible without funds from the American Recovery and Reinvestment Act 3R01MH077303-04S1. We thank Dr. T. Südhof for the n1α cDNA, Dr. D. Borek for advice on data processing, Dr. S. Anderson for excellent beam-line support, I. Wu for initial crystallization experiments and Dr. C. Brown for reagents. Diffraction data was collected at LS-CAT (beam-line 21-ID-D) at the Advanced Photon Source, Argonne National Laboratories.
Footnotes
Author contributions: F.C. performed molecular biology, protein purification and crystallization. V.V. performed crystallization, and structure determination of n1α+SS#3. B.M. performed molecular biology. G.R. conceived the research plan, performed protein purification, crystallization, structure determination of n1α and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Araç D, Boucard AA, Őzkan E, Strop P, Newell E, Südhof TC, Brunger AT. Structures of neuroligin-1 and the neuroligin-1/neurexin-1β complex reveal specific protein:protein and protein-Ca2+ interactions. Neuron. 2007;56:1–12. doi: 10.1016/j.neuron.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Beglopoulos V, Montag-Sallaz M, Rohlmann A, Piechotta K, Ahmad M, Montag D, Missler M. Neurexophilin 3 is highly localized in cortical and cerebellar regions and is functionally important for sensorimotor gating and motor coordination. Mol Cell Biol. 2005;25:7278–7288. doi: 10.1128/MCB.25.16.7278-7288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur C, Sakurai T, Buxbaum JD. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 2009;32:402–412. doi: 10.1016/j.tins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Tabuchi K, Bolliger MF, Blaiss CA, Brose N, Liu X, Südhof TC, Powell CM. Increased anxiety-like behavior in mice lacking the inhibitory synapse cell adhesion molecule neuroligin 2. Genes Brain Behav. 2009;8:114–126. doi: 10.1111/j.1601-183X.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, Bolliger MF, Südhof TC, Powell CM. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci. 2010;30:2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Südhof TC. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron. 2005;48:229–236. doi: 10.1016/j.neuron.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Cryst. 2003;D59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- Chen X, Liu H, Shim AH, Focia PJ, He X. Structural basis for synaptic adhesion mediated by neuroligin-neurexin interactions. Nat Struct Mol Biol. 2008;15:50–56. doi: 10.1038/nsmb1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Gollan L, Scheiffele P. Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron. 2006;51:171–178. doi: 10.1016/j.neuron.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Südhof TC. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Comoletti D, Flynn RE, Boucard AA, Demeler B, Schirf V, Shi J, Jennings LL, Newlin HR, Südhof TC, Taylor P. Gene selection, alternative splicing, and post-translational processing regulate neuroligin selectivity for beta-neurexins. Biochemistry. 2006;45:12816–12827. doi: 10.1021/bi0614131. [DOI] [PubMed] [Google Scholar]
- Comoletti D, Miller MT, Jeffries CM, Wilson J, Demeler B, Taylor P, Trewhella J, Nakagawa T. The macromolecular architecture of extracellular domain of alphaNRXN1: domain organization, flexibility, and insights into trans-synaptic disposition. Structure. 2010;18:1044–1053. doi: 10.1016/j.str.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhaus R, El-Husseini A. Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav Brain Res. 2010;208:96–110. doi: 10.1016/j.bbr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Davletov BA, Krasnoperov V, Hata Y, Petrenko AG, Südhof TC. High affinity binding of alpha-latrotoxin to recombinant neurexin I alpha. J Biol Chem. 1995;270:23903–23905. doi: 10.1074/jbc.270.41.23903. [DOI] [PubMed] [Google Scholar]
- de Wit J, Sylwestrak E, O’Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, 3rd, Comoletti D, Taylor P, Ghosh A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64:799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton MR, Blaiss CA, Powell CM, Südhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci USA. 2009;106:17998–18003. doi: 10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrichny IP, Leone P, Sulzenbacher G, Comoletti D, Miller MT, Taylor P, Bourne Y, Marchot P. Structural analysis of the synaptic protein neuroligin and its β-neurexin complex: determinants for folding and cell adhesion. Neuron. 2007;56:979–991. doi: 10.1016/j.neuron.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Huber KM, Südhof TC. Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J Neurosci. 2009;29:13883–13897. doi: 10.1523/JNEUROSCI.2457-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilmatre A. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines RM, et al. Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J Neurosci. 2008;28:6055–6067. doi: 10.1523/JNEUROSCI.0032-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci USA. 2008;105:1710–1715. doi: 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhang X, Dobie F, Wu H, Craig AM. Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J Biol Chem. 2008;283:2323–2334. doi: 10.1074/jbc.M703957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattenstroth G, Tantalaki E, Südhof TC, Gottmann K, Missler M. Postsynaptic N-methyl-D-aspartate receptor function requires alpha-neurexins. Proc Natl Acad Sci USA. 2004;101:2607–2612. doi: 10.1073/pnas.0308626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel I. Reelin-mediated signaling in neuropsychiatric and neurodegenerative diseases. Prog Neurobiol. 2010;91:257–274. doi: 10.1016/j.pneurobio.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Ko J, Fuccillo MV, Malenka RC, Südhof TC. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009a;64:791–798. doi: 10.1016/j.neuron.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Zhang C, Arac D, Boucard AA, Brunger AT, Südhof TC. Neuroligin-1 performs neurexin-dependent and neurexin-independent functions in synapse validation. EMBO J. 2009b;28:3244–3255. doi: 10.1038/emboj.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J, Katsamba PS, Ahlsen G, Bahna F, Vendome J, Honig B, Shapiro L, Jin X. Splice form dependence of beta-neurexin/neuroligin binding interactions. Neuron. 2010;67:61–74. doi: 10.1016/j.neuron.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Leone P, Comoletti D, Ferracci G, Conrod S, Garcia SU, Taylor P, Bourne Y, Marchot P. Structural insights into the exquisite selectivity of neurexin/neuroligin synaptic interactions. EMBO J. 2010;29:2461–2471. doi: 10.1038/emboj.2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson JN, El-Husseini A. Building excitatory and inhibitory synapses: balancing neuroligin partnerships. Neuron. 2005;48:171–174. doi: 10.1016/j.neuron.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- Loris R. Principles of structures of animal and plant lectins. Biochim Biophys Acta. 2002;1572:198–208. doi: 10.1016/s0304-4165(02)00309-4. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M, Hammer RE, Südhof TC. Neurexophilin binding to alpha-neurexins. A single LNS domain functions as an independently folding ligand-binding unit. J Biol Chem. 1998;273:34716–34723. doi: 10.1074/jbc.273.52.34716. [DOI] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC. Alpha-neurexins couple Ca2+-channels to synaptic vesicle exocytosis. Nature. 2003;423:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nogi T, Yasui N, Hattori M, Iwasaki K, Takagi J. Structure of a signaling-competent reelin fragment revealed by X-ray crystallography and electron tomography. EMBO J. 2006;25:3675–3683. doi: 10.1038/sj.emboj.7601240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner C, Klose M, Fairless R, Missler M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc Natl Acad Sci USA. 2008;105:15124–15129. doi: 10.1073/pnas.0801639105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko G, Nguyen T, Chelliah Y, Südhof TC, Deisenhofer J. The structure of the ligand-binding domain of neurexin I beta: regulation of LNS domain function by alternative splicing. Cell. 1999;99:93–101. doi: 10.1016/s0092-8674(00)80065-3. [DOI] [PubMed] [Google Scholar]
- Sheckler LR, Henry L, Sugita S, Südhof TC, Rudenko G. Crystal structure of the second LNS/LG domain from neurexin 1alpha: Ca2+ binding and the effects of alternative splicing. J Biol Chem. 2006;281:22896–22905. doi: 10.1074/jbc.M603464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KC, Kuczynska DA, Wu IJ, Murray BH, Sheckler LR, Rudenko G. Regulation of neurexin 1beta tertiary structure and ligand binding through alternative splicing. Structure. 2008;16:422–431. doi: 10.1016/j.str.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci. 2010;30:7495–7506. doi: 10.1523/JNEUROSCI.0470-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Khvochtev M, Südhof TC. Neurexins are functional alpha-latrotoxin receptors. Neuron. 1999;22:489–496. doi: 10.1016/s0896-6273(00)80704-7. [DOI] [PubMed] [Google Scholar]
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Südhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisi D, Talts JF, Timpl R, Hohenester E. Structure of the C-terminal laminin G-like domain pair of the laminin alpha2 chain harbouring binding sites for alpha-dystroglycan and heparin. EMBO J. 2000;19:1432–1440. doi: 10.1093/emboj/19.7.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Yasui N, Nogi T, Kitao T, Nakano Y, Hattori M, Takagi J. Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proc Natl Acad Sci USA. 2007;104:9988–9993. doi: 10.1073/pnas.0700438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui N, Nogi T, Takagi J. Structural basis for specific recognition of reelin by its receptors. Structure. 2010;18:320–331. doi: 10.1016/j.str.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang W, Rohlmann A, Sargsyan V, Aramuni G, Hammer RE, Südhof TC, Missler M. Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+-channels. J Neurosci. 2005;25:4330–4342. doi: 10.1523/JNEUROSCI.0497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Atasoy D, Araç D, Yang X, Fucillo MV, Robison AJ, Ko J, Brunger AT, Südhof TC. Neurexins physically and functionally interact with GABA(A) receptor. Neuron. 2010;66:403–416. doi: 10.1016/j.neuron.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.