

Abstract
A tandem gold-catalyzed cycloisomerization/Suzuki cross coupling sequence involving arylethynyl-N-methyliminodiacetic acid boronates is described. Combining the mildness of homogeneous gold catalysis with the versatility of N-methyliminodiacetic acid (MIDA) boronates, this tandem two-step method enables the rapid assembly of various aryl-substituted heterocycles without having to isolate or purify any heterocyclic MIDA boronate intermediates. Another major advantage of this method is that a wide range of heterocycles bearing different aryl groups may be made from a single MIDA boronate alkyne precursor.
Keywords: Gold catalysis, Electrophilic cyclization, Suzuki coupling, MIDA boronates, Heterocycles
1. Introduction
Given the biological activity that many benzofuran and indole-containing molecules possess, it comes as no surprise that these classes of molecules often represent important targets in the field of drug discovery and development.1 Accordingly, there exists ongoing interest in the development of novel synthetic routes towards these functionalized heterocycles, and in particular, their 2-substituted derivatives.2 In a bid to construct a range of such heterocycles in an expeditious fashion, we envisioned a tandem sequence depicted in Scheme 1 involving two catalytic processes, with the idea of generating heterocycles with diverse substituents from a single precursor. For this to work well, the Z moiety should be compatible with the cyclization event and also serve as a synthetic handle for a cross-coupling reaction.3 In addition, the first reaction (gold-catalyzed cycloisomerization) must give reasonably high yields of the intermediate, and proceed cleanly enough for next step to continue without prior intermediate purification and isolation.
Scheme 1.

Tandem process for the rapid construction of 2-substituted heterocycles.
In 2009, the laboratories of Prof. Martin Burke reported the use of air-stable MIDA boronates to address typical problems associated with the inherently unstable 2-heterocyclic boronic acids in Suzuki cross-coupling.4 More recently, the Burke group has also demonstrated that the highly versatile ethynyl MIDA boronate5 is amenable to Sonogashira coupling with aryl iodides and that the MIDA boronate functional group is tolerant of a wide variety of reaction conditions. We envisioned that the ethynyl MIDA boronate methodology could be applied to the synthesis of the aromatic substrates proposed in Scheme 1, which in turn could be cycloisomerized and coupled with commercial aryl halides to give substituted heterocycles. No compatibility issues were anticipated in view of the mildness of gold catalysis6 and the robustness of MIDA boronates. Additionally, with only the need for catalytic quantities of gold reagent, we expected that a Suzuki coupling could be carried out in a tandem fashion, without having to first isolate and purify the cycloisomerized intermediates. This tandem two-step7 process was likely to succeed if the 2-heterocyclic MIDA boronate could be generated cleanly without leaving behind any starting material in the post-reaction mixture. In addition to benzofurans8 and indoles,2e,9 we predicted that the same method could also be used to regioselectively construct additional heterocycles such as 1,3-dihydroisobenzofurans (phthalans)10 and isoindolines11 from benzylic alcohols and N-protected benzylic amines respectively. This secondary objective would take advantage of the directing ability of the electronegative MIDA boronate group to favor 5-exo-dig over 6-endo-dig products such as isochromenes12 and isoquinolines.13 Ultimately, the goal was to combine the ease of homogeneous gold catalysis with the versatility of Burke’s MIDA boronate chemistry to provide facile, quick one-pot access to a range of aryl-substituted heterocycles while bypassing cumbersome intermediate purification. Moreover, as stated earlier, this method would allow each class of heterocycles to be synthesized from a single arylethynyl MIDA boronate precursor, without the need to prepare individual alkynes for Sonogashira coupling. Instead, inexpensive and readily available aryl halides can simply be fed into the second step of the tandem sequence to generate a library of substituted heterocycles. Lastly, the ability of the MIDA boronate moiety to direct 5-exo-dig reactivity will prove itself a useful complement to the 6-endo-dig regioselectivity of several existing cyclization methods.12,14 The results of our investigations are described herein.
2. Results and Discussion
2.1. Initial catalyst screening and optimization studies
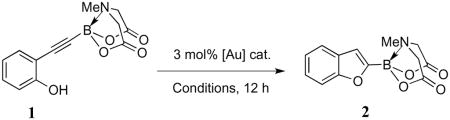
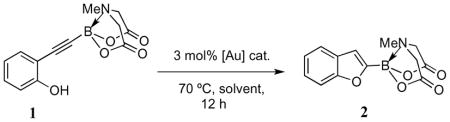
In our initial search for the optimal conditions to bring about the key 5-endo-dig electrophilic cyclization of phenol 1, a wide variety of gold catalysts and reaction conditions were screened (Table 1). The results of these preliminary screens indicated that the more electrophilic gold catalysts fared better in bringing about the necessary cycloisomerization. This was unsurprising in view of the fact that the C≡C in the substrate is electron-poor due to the inductive effect of the MIDA boronate moiety. The effect of solvent on the reaction was also investigated (Table 2) and it was found that the best yield was obtained when THF was employed. Thus, the use of 3 mol% of [(Ph3PAu)3O]BF4 in THF at 70 °C was established as a satisfactory set of conditions for achieving yields over 70%.15
Table 1.
Results of the initial catalyst screening.
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | Yield (%) |
| 1 | Ph3PAuCl, AgSbF6 | DMF | 70 | 0 |
| 2 | dppm(AuCl)2, AgSbF6 | DMF | 70 | 0 |
| 3 | IMes(AuCl), AgSbF6 | DMF | 70 | 0 |
| 4 | DTBM-Segphos(AuCl)2, AgSbF6 | DMF | 70 | 0 |
| 5 | (p-CF3C6H4)3PAuCl, AgSbF6 | DMF | 70 | 50 |
| 6 | [(Ph3PAu)3O]BF4 | DMF | 70 | 60 |
| 7 | (p-CF3C6H4)3PAuCl, AgSbF6 | MeNO2 | 55 | 0 |
| 8 | [(Ph3PAu)3O]BF4 | MeNO2 | 55 | 0 |
Table 2.
Solvent effects on the isolated yield.
 | |||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yield (%) |
| 1 | (p-CF3C6H4)3PAuCl, AgSbF6 | MeNO2 | 40 |
| 2 | (p-CF3C6H4)3PAuCl, AgSbF6 | 1,4-dioxane | 56 |
| 3 | (p-CF3C6H4)3PAuCl, AgSbF6 | Xylenes | 15 |
| 4 | (p-CF3C6H4)3PAuCl, AgSbF6 | PhF | 20 |
| 5 | (p-CF3C6H4)3PAuCl, AgSbF6 | 1,2-dichloroethane | 18 |
| 6 | (p-CF3C6H4)3PAuCl, AgSbF6 | i-PrNH2 | 0 |
| 7 | [(Ph3PAu)3O]BF4 | MeNO2 | 55 |
| 8 | [(Ph3PAu)3O]BF4 | THF | 75 |
Further optimization studies were subsequently carried out, and it was ultimately possible to synthesize the 2-benzofuryl MIDA boronate in 86% isolated yield after just 5 hours of reaction in THF at 65 °C. The same conditions were also found to be effective in cyclizing tosylated aniline 3 and benzyl alcohol 5, giving the N-protected indole 4 and phthalan 6 in 89% and 71% isolated yields respectively (Scheme 2). The issue of regioselectivity in the phthalan system will be discussed in section 2.3.
Scheme 2.

Gold-catalyzed cycloisomerizations of MIDA-alkynylboronates.
2.2. Investigation of additional gold oxonium salt catalysts
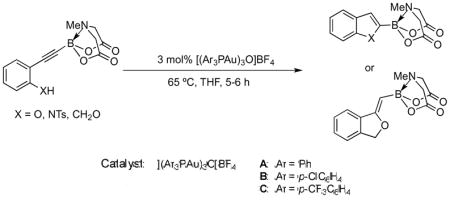
Having identified [(Ar3PAu)3O]BF4 as the optimal cyclization catalyst, further efforts to improve the yields were undertaken, by screening more reactive gold reagents of the same class. To that end, two catalysts B, [(Ar3PAu)3O]BF4 (Ar = p-ClC6H4), and C, [(Ar’3PAu)3O]BF4 (Ar’ = p-CF3C6H4) were investigated. In general, the electron-withdrawing substituents on the phosphine ligands render the catalysts more electrophilic, which we hoped would be more effective in activating the relatively electron-poor C≡C of the MIDA boronates towards intramolecular nucleophilic attack.16 This indeed proved to be the case with regards to benzofuran and indole formation, where the heterocycles were obtained in quantitative or near-quantitative yields. However, these enhanced catalysts were less successful in making phthalans from benzyl alcohols, with only 65% yield being obtained with catalyst B and complete decomposition being observed with catalyst C. The results are summarized in Table 3 below, with comparison to the less active but commercially available catalyst A, [(Ph3PAu)3O]BF4.
Table 3.
Comparison of three gold oxonium salt-based catalysts for cycloisomerization.
 | |||
|---|---|---|---|
| Entry | X | Catalyst | Isolated yield (%) |
| 1 | OH | A | 86 |
| 2 | OH | B | 100 |
| 3 | OH | C | 100 |
| 4 | NHTs | A | 89 |
| 5 | NHTs | B | 94 |
| 6 | NHTs | C | 98 |
| 7 | CH2OH | A | 71 |
| 8 | CH2OH | B | 65 |
| 9 | CH2OH | C | 0 |
2.3. Tandem cycloisomerization/Suzuki cross-coupling
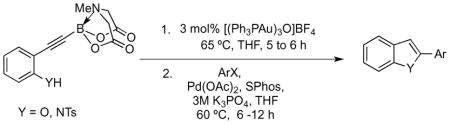
With the cycloisomerization of various substrates having been accomplished, we set out to develop the tandem two-step cycloisomerization/Suzuki cross-coupling protocol mentioned earlier. Owing to the mildness of the cyclization step, the complete absence of any starting material in the post-reaction mixture, there appeared to be few variables capable of interfering with the subsequent Suzuki coupling step. The only significant issue was the need for THF as the solvent, which was necessary for achieving high yields in the gold-catalyzed cyclization, whereas the Suzuki coupling of MIDA boronates as performed by Burke et al. had been optimized with dioxane. It was unfeasible to replace THF with dioxane in the cyclization step for two reasons: 1) yields were higher when the reaction was run in THF; 2) significant quantities of uncyclized alkyne MIDA boronate remain when dioxane is used, which would lead to undesired Suzuki coupling products in the final mixture. Consequently, the tandem two-step sequence involved performing the first reaction exactly as before, but instead of the usual workup and purification, the reaction vessel was opened up under positive pressure of nitrogen, into which the reagents for the Suzuki cross-coupling step were introduced. The second stage of the tandem process was then initiated immediately thereafter. Results of the cycloisomerization/cross-coupling sequence leading to benzofurans and N-protected indoles were encouraging and are summarized in Table 4. Isolated yields (over 2 steps) of 2-arylheterocycles were typically 50% or greater.
Table 4.
Tandem cycloisomerization/Suzuki coupling leading to 2-arylheterocycles.
 | ||||
|---|---|---|---|---|
| Entry | Y | ArX | Product | % isolated yield (over 2 steps) |
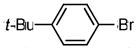
| 1 | OH |
|
7a |
64 |
| 2 | OH |
|
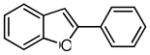 7b |
69 |
| 3 | OH |
|
7c |
48 |
| 4* | OH |
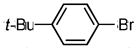
|
7d |
38 |
| 5 | NHTs |
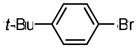
|
 8a |
58 |
| 6 | NHTs |
|
 8b |
70 |
| 7 | NHTs |
|
 8c |
56 |
Catalyst C (instead of A) was employed in this example.
2.4. Regioselectivity
The regioselectivity observed in the cyclization of benzyl alcohol 5 is particularly interesting in that 5-exo-dig reactivity is favored over 6-endo-dig, exclusively resulting in phthalan 6 and none of the 6-membered isochromene MIDA boronate (Scheme 2). In contrast, when a 4-methoxyphenyl substituent was used in place of the electron-withdrawing and sterically demanding MIDA boronate group, a gold-catalyzed 6-endo-dig cyclization occurred exclusively to afford a known isochromene (Scheme 3), the NMR data of which agreed with published literature results. The tandem cyclization/Suzuki coupling sequence of benzyl alcohol 5, involving 4-bromoanisole in the Suzuki step, afford phthalan 9 in 53% yield (over 2 steps). Thus, with the 5-exo-dig regioselectively conferred by the MIDA boronate provides complementary reactivity to various electrophilic cyclizations of boron-free substrates.
Scheme 3.

Regioselective formation of phthalan and isochromene using complementary methods.
3. Summary and conclusions
The ubiquity of heterocycles in fields as diverse as drug discovery, total synthesis, agricultural chemistry and materials science highlights the need for more efficient synthetic methods for their construction. The results revealed herein show that by combining the advantages of modern gold catalysis, MIDA boronate chemistry, and palladium-catalyzed cross couplings, a wide range of aryl-substituted heterocycles can be rapidly and easily assembled in a tandem fashion, bypassing the need to isolate and purify any intermediates. Furthermore, using this method, each class of heterocycles may be made starting from a single arylethynyl MIDA boronate precursor, without having to individually synthesize every unique Sonogashira coupling partner. Lastly, the MIDA boronate is also capable of directing gold-catalyzed 5-exo-dig reactivity, complementing the 6-endo-dig regioselectivity of many existing methods.
4. Experimental
4.1. Materials
Unless otherwise noted, all commercial reagents were purchased from Sigma-Aldrich, Alfa Aesar, Fisher Scientific, and TCI America and used without further purification. Palladium(II) acetate, trans-dichlorobis(triphenylphosphine)palladium(II), copper(I) iodide, and tris[triphenylphosphinegold(I)] oxonium tetrafluoroborate were purchased from Strem Chemicals Inc. and used without further purification. Tris[triphenylphosphinegold(I)] oxonium tetrafluoroborate synthesized using published procedures17 was also used. Anhydrous organic solvents were purified and obtained via passage through packed columns as described by Pangborn and coworkers.18 Deionized water employed as a reagent in the Suzuki cross-coupling reactions was obtained from the College of Chemistry, University of California, Berkeley house deionized water supply. Solvents used for chromatographic purification procedures (HPLC grade hexane and ethyl acetate, ACS grade dichloromethane, chloroform and ethanol) were obtained from Fisher Scientific.
4.2. General experimental procedures
Unless otherwise stated, all reactions were performed in oven-dried glassware under an inert atmosphere of nitrogen gas using standard Schlenk techniques. Thin-layer chromatography (TLC) analysis of reaction mixtures was performed on E. Merck silica gel 60 F254 TLC plates, with UV light (λ = 254 nm), potassium permanganate stain, and/or elemental iodine to visualize the post-reaction components. Flash chromatography was performed on ICN SiliTech 32–63 D 60 Å silica gel according to standard procedures. The MIDA boronates are compatible with standard silica gel column chromatography, including the standard loading techniques.
4.3. Structural analysis
All 1H and 13C NMR spectra were recorded at ambient temperature on the Bruker AVB-400 and AVQ-400 NMR spectrometers. Chemical shifts (δ) are reported in parts per million (ppm) and referenced to residual proton and carbon resonances in the NMR solvent (1H: CDCl3, δ = 7.26; CD2Cl2, δ = 5.32, center line; acetone-d6, δ = 2.04, center line; 13C: CDCl3, δ = 77.2, center line; CD2Cl2, δ = 54.0, center line; acetone-d6, δ = 29.8, center line). Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = septet, m = multiplet, b = broad, app = apparent), and coupling constant (J) in Hertz (Hz). Carbons attached to boron substituents were typically not observed due to quadrupolar relaxation. High resolution mass spectra (HRMS) and analytical data were obtained via the QB3/Chemistry Mass Spectrometry Facility operated by the QB3 Institute and the College of Chemistry, University of California, Berkeley.
4.4. General procedure A: Sonogashira coupling
A 50-mL round-bottom flask equipped with a magnetic stir bar was charged with an aryl iodide (3.57 mmol, 1.1 equiv), ethynyl MIDA boronate (3.24 mmol, 1.0 equiv), CuI (9 mol %), PdCl2(PPh3)2 (5 mol %), and placed under nitrogen atmosphere. Then, anhydrous DMF (15 mL) and Et3N (1.4 mL) were introduced via syringe, and the reaction mixture was allowed to stir for 6–7 h at room temperature, after which the mixture was poured into a separatory funnel containing deionized water. Extraction with ethyl acetate (3 × 25 mL) was carried out, and the combined organic extracts were concentrated in vacuo to give a crude residue. The crude product was subsequently loaded onto a silica gel column and purified by flash column chromatography (on this scale, 2–3 rounds of column chromatography was typically required for complete purification) to afford the desired compound.
4.5. General procedure B: Gold-catalyzed cycloisomerization
A 25-mL two-neck round-bottom flask equipped with a magnetic stir bar was charged with the arylethynyl-2-MIDA boronate (0.090 mmol, 1.0 equiv), [(Ph3PAu)3O]BF4 (0.0027 mmol, 3 mol %) and THF (2.5 mL). The solution was stirred at 65 °C for 5–7 h under nitrogen, after which the mixture was cooled down to room temperature, diluted with ethyl acetate and poured into a separatory funnel containing aqueous NaCl solution. Extraction with ethyl acetate (3 × 20 mL) was carried out, and the combined organic extracts were concentrated in vacuo to give a crude product that was subsequently loaded onto a silica gel column and purified by flash chromatography to afford the target heterocycle as a white solid. (Note: Post-reaction workup and purification were only done in cases where the heterocyclic MIDA boronate was to be isolated and characterized.)
4.6. General procedure C: Tandem two-step preparation of aryl substituted heterocycles
The first step was carried out exactly as detailed in General procedure B, except that the no workup/purification/isolation of the intermediate heterocyclic MIDA boronate was carried out. Instead, following the completion of the first reaction, the reaction vessel was simply opened up briefly under positive nitrogen pressure, whereupon Pd(OAc)2 (0.9 mg, 0.0045 mmol, 5 mol %), SPhos (3.1 mg, 0.0090 mmol, 10 mol %), 3.0 M aqueous K3PO4 (0.23 mL, 7.5 equiv of base), and the aryl halide (0.090 mmol, 1 equiv) were introduced. The mixture was then stirred under nitrogen at 60 °C for 6–15 h depending on the specific substrate, before being poured into a separatory funnel containing aqueous NaCl solution. Extraction with ethyl acetate (3 × 20 mL) was carried out, and the combined organic extracts were concentrated in vacuo to give a crude product that was subsequently loaded onto a silica gel column and purified by flash chromatography to afford the 2-arylheterocycle.
2-hydroxyphenylethynyl MIDA boronate (1)
Prepared according to General Procedure A using 2-iodophenol (0.393 g, 1.79 mmol), ethynyl MIDA boronate (0.294 g, 1.62 mmol), CuI (0.027 g, 0.142 mmol), PdCl2(PPh3)2 (0.057 g, 0.081 mmol), DMF (6 mL) and Et3N (1.5 mL). The reaction was stirred for 6 h. The crude product, a reddish-brown oil, was purified by flash column chromatography (SiO2; ethyl acetate) to afford the 1 as an off-white crystalline solid (0.221 g, 50%). 1H NMR (400 MHz, acetone-d6, δ ppm): 7.36 (1H, d, J = 7.6 Hz), 7.22 (1H, t, J = 8.4 Hz), 6.91 (1H, d, J = 8.4 Hz), 6.83 (1H, t, J = 7.6 Hz), 4.32 (2H, d, J = 17.2 Hz), 4.16 (2H, d, J = 16.8 Hz), 3.32 (3H, s). 13C NMR (400 MHz, acetone-d6, δ ppm): 169.0, 159.6, 134.3, 131.4, 120.8, 116.7, 111.5, 62.7, 49.0. HRMS: calcd: 273.0809 (M)+, found: 296.0701 (M + Na)+.
2-benzofuryl MIDA boronate (2)
Prepared according to General Procedure B using compound 1 (24.5 mg, 0.09 mmol), [(Ph3PAu)3O]BF4 (4 mg, 0.00269 mmol) and anhydrous THF (2.5 mL). The reaction was stirred at 65 °C for 5 h. The crude product was purified by flash column chromatography (SiO2; ethyl acetate) to afford 2 as a white solid (21 mg, 86 %). 1H NMR (400 MHz, acetone-d6, δ ppm): 7.26 (1H, d, J = 7.6 Hz), 7.11 (1H, d, J = 8.4 Hz), 6.91 (1H, t, J = 7.2 Hz), 6.83 (1H, t, J = 7.2 Hz), 6.72 (1H, s), 4.06 (2H, d, J = 17.2 Hz), 3.85 (2H, d, J = 16.8 Hz), 2.56 (3H, s). 13C NMR (400 MHz, acetone-d6, δ ppm): 169.4, 158.7, 129.6, 125.8, 123.8, 122.6, 115.9, 112.5, 63.0, 48.4. HRMS: calcd: 273.0809 (M)+, found: 273.0811.
2-(N-tosylamino)phenylethynyl MIDA boronate (3)
Prepared according to General Procedure A using N-tosyl-2-iodoaniline (0.666 g, 1.79 mmol), ethynyl MIDA boronate (0.294 g, 1.62 mmol), CuI (0.027 g, 0.11 mmol), PdCl2(PPh3)2 (0.057 g, 0.081 mmol), DMF (8 mL) and Et3N (0.66 mL). The reaction mixture was allowed to stir for 6 h. The crude product was purified by flash column chromatography (SiO2; gradient: 1:9 → 1:1 → 3:2 ethyl acetate/dichloromethane) to afford 3 as an off-white solid (0.311 g, 45 %). 1H NMR (400 MHz, CDCl3, δ ppm): 7.75 (1H, s), 7.68 (2H, d, J = 8.4 Hz), 7.39 (1H, d, J = 8.0 Hz), 7.1–7.3 (4H, m), 6.97 (1H, t, J = 7.6 Hz), 4.23 (2H, d, J = 17.2 Hz), 4.00 (2H, d, J = 16.8 Hz), 3.14 (3H, s), 2.28 (3H, s). 13C NMR (400 MHz, CDCl3, δ ppm): 168.6, 144.3, 138.3, 136.3, 133.0, 130.1, 129.9, 127.5, 124.8, 121.0, 115.0, 62.2, 60.6, 48.5, 21.7. HRMS: calcd: 426.1057 (M)+, found: 449.0948 (M + Na)+.
1-tosyl-2-indole MIDA boronate (4)
Prepared according to General Procedure B using compound 3 (38.4 mg, 0.09 mmol), ([Ph3PAu)3O]BF4 (4 mg, 0.00269 mmol) and anhydrous THF (2.5 mL). The reaction was stirred at 65 °C for 6 h. The crude product was purified by flash column chromatography (SiO2; gradient: 3:7 → 2:3 ethyl acetate/dichloromethane) to afford 4 as a white solid (42.7 mg, 89 %). 1H NMR (400 MHz, acetone-d6, δ ppm): 8.26 (1H, d, J = 8.4 Hz), 8.03 (2H, d, J = 8.4 Hz), 7.62 (1H, d, J = 7.6 Hz), 7.40 (1H, t, J = 7.2 Hz), 7.35 (2H, d, J = 8.4 Hz), 7.27 (1H, t, J = 7.6 Hz), 7.14 (1H, s), 4.52 (2H, d, J = 17.2 Hz), 4.36 (2H, d, J = 17.2 Hz), 3.22 (3H, s), 2.35 (3H, s). 13C NMR (400 MHz, acetone-d6, δ ppm): 169.3, 146.1, 140.0, 136.6, 131.1, 130.6, 128.0, 126.0, 124.2, 122.9, 122.2, 115.5, 65.7, 55.0, 50.4, 32.3, 23.3, 21.5, 14.4. HRMS: calcd: 426.1057 (M)+, found: 449.0948 (M + Na)+.
2-(hydroxymethyl)phenylethynyl MIDA boronate (5)
Prepared according to General Procedure A using 2-iodobenzyl alcohol (0.834 g, 3.56 mmol), ethynyl MIDA boronate (0.588 g, 3.24 mmol), CuI (0.054 g, 0.28 mmol), PdCl2(PPh3)2 (0.114 g, 0.162 mmol), DMF (15 mL) and Et3N (1.32 mL). The reaction mixture was allowed to stir for 6 h. The crude product was purified by flash column chromatography (SiO2; gradient: ethyl acetate → 9:1 ethyl acetate/ethanol) to afford 5 as an off-white solid (0.277 g, 27 %). 1H NMR (400 MHz, acetone-d6, δ ppm): 7.57 (1H, d, J = 7.6 Hz), 7.46 (1H, d, J = 8.0 Hz), 7.39 (1H, t, J = 7.2 Hz), 7.25 (1H, t, J = 7.6 Hz), 4.80 (2H, s), 4.34 (2H, d, J = 16.8 Hz), 4.18 (2H, d, J = 16.8 Hz), 3.34 (3H, s), 2.88 (1H, s). 13C NMR (400 MHz, acetone-d6, δ ppm): 168.6, 145.2, 132.9, 129.6, 127.5, 127.3, 121.4, 63.0, 62.4, 48.6. HRMS: calcd: 287.0965 (M)+, found: 310.0859 (M + Na)+.
Isobenzofuran-1(3H)-ylidenemethyl MIDA boronate (6)
Prepared according to General Procedure B using compound 5 (25.8 mg, 0.09 mmol), ([Ph3PAu)3O]BF4 (4 mg, 0.00269 mmol) and anhydrous THF (2.5 mL). The reaction was stirred at 65 ºC for 7 h. The crude product was purified by flash column chromatography (SiO2; gradient: 49:1 → 19:1 → 9:1 ethyl acetate/ethanol) to afford 6 as an off-white solid (18.3 mg, 71 %). 1H NMR (400 MHz, acetone-d6, δ ppm): 7.58 (1H, d, J = 6.4 Hz), 7.31–7.41 (3H), 5.33 (2H, s), 4.81 (1H, s), 3.91 (2H, d, J = 16.4 Hz), 3.84 (2H, d, J = 16.0 Hz), 2.86 (3H, s). 13C NMR (400 MHz, acetone-d6, δ ppm): 168.3, 166.8, 140.7, 134.1, 130.1, 128.6, 121.9, 121.6, 74.7, 62.9, 47.3. HRMS: calcd: 287.0965 (M)+, found: 288.1038 (M + H)+.
2-(4′-tert-butylphenyl)benzofuran (7a)
Prepared according to General Procedure C using Pd(OAc)2 (0.9 mg, 0.0045 mmol, 5 mol %), SPhos (3.1 mg, 0.0090 mmol, 10 mol %), 3.0 M aqueous K3PO4 (0.23 mL, 7.5 equiv of base), and 1-bromo-4-tert-butylbenzene (0.014 mL, 0.090 mmol, 1 equiv). The mixture was stirred under nitrogen at 60 °C for 4 h. The crude product was purified by flash column chromatography (SiO2; hexanes) to afford 7a as an off-white solid (14.4 mg, 64 % over two steps). 1H NMR (400 MHz, CDCl3, δ ppm): 7.86 (2H, d, J = 6.4 Hz), 7.63 (1H, d, J = 6.8 Hz), 7.57 (1H, d, J = 8.4 Hz), 7.53 (2H, d, J = 6.4 Hz), 7.30 (2H, m), 7.03 (1H, s), 1.42 (9H, s). 13C NMR (400 MHz, CDCl3, δ ppm): 156.3, 155.0, 152.0, 129.5, 127.9, 125.9, 124.9, 124.2, 123.0, 120.9, 111.3, 100.9, 35.0, 31.4. HRMS: calcd: 250.1358 (M)+, found: 250.1362.
2-phenylbenzofuran (7b)
Prepared according to General Procedure C using Pd(OAc)2 (0.9 mg, 0.0045 mmol, 5 mol %), SPhos (3.1 mg, 0.0090 mmol, 10 mol %), 3.0 M aqueous K3PO4 (0.23 mL, 7.5 equiv of base), and chlorobenzene (9 μL, 0.090 mmol, 1 equiv). The mixture was stirred under nitrogen at 60 °C for 4.5 h. The crude product was purified by flash column chromatography (SiO2; hexanes) to afford 7b as a white solid (12 mg, 69 % over two steps). 1H NMR (400 MHz, CDCl3, δ ppm): 7.89 (2H, d, J = 7.0 Hz), 7.61 (1H, d, J = 7.8 Hz), 7.55 (1H, d, J = 8.2 Hz), 7.48 (2H, t, J = 7.3 Hz), 7.38 (1H, t, J = 7.5 Hz), 7.23–7.33 (2H, m), 7.06 (1H, s). 13C NMR (400 MHz, CDCl3, δ ppm): 156.1, 155.1, 147.9, 130.6, 129.4, 129.0, 128.7, 125.3, 124.4, 123.5, 121.6, 121.1, 111.5, 103.9, 101.5. HRMS: calcd: 194.0732 (M)+, found: 194.0731.
2-(4′-trifluoromethylphenyl)benzofuran (7c)
Prepared according to General Procedure C using Pd(OAc)2 (0.9 mg, 0.0045 mmol, 5 mol %), SPhos (3.1 mg, 0.0090 mmol, 10 mol %), 3.0 M aqueous K3PO4 (0.23 mL, 7.5 equiv of base), and 4-chlorobenzotrifluoride (0.012 mL, 0.090 mmol, 1 equiv). The mixture was stirred under nitrogen at 60 °C for 5 h. The crude product was purified by flash column chromatography (SiO2; hexanes) to afford 7c as a white solid (11.3 mg, 48 % over two steps). 1H NMR (400 MHz, CDCl3, δ ppm): 8.00 (2H, d, J = 8.4 Hz), 7.72 (2H, d, J = 8.0 Hz), 7.64 (1H, d, J = 7.2 Hz), 7.56 (1H, d, J = 8.0 Hz), 7.27 (1H, t, J = 7.6 Hz), 7.20 (1H, s). 13C NMR (400 MHz, CDCl3, δ ppm): 129.4, 128.9, 126.3, 125.7, 125.5, 124.0, 123.8, 122.1, 121.9, 121.1, 111.8, 103.9. HRMS: calcd: 262.0605 (M)+, found: 262.0607.
2-(4′-tert-butylphenyl)benzofuran (7d)
Prepared in the same way as 7a starting with 39.5 mg of the precursor phenol, but Catalyst C was used instead of Catalyst A. Upon purification, 7d was afforded as a white solid (13.7 mg, 38 % over two steps). Characterization data was identical to 7a (i.e. same compound).
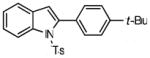
1-tosyl-2-(4′-tert-butylphenyl)indole (8a)
Prepared according to General Procedure C using Pd(OAc)2 (0.9 mg, 0.0045 mmol, 5 mol %), SPhos (3.1 mg, 0.0090 mmol, 10 mol %), 3.0 M aqueous K3PO4 (0.23 mL, 7.5 equiv of base), and 1-bromo-4-tert-butylbenzene (0.014 mL, 0.090 mmol, 1 equiv). The mixture was stirred under nitrogen at 60 °C for 5 h. The crude product was purified by flash column chromatography (SiO2; gradient: 1:3 → 2:3 → 1:1 dichloromethane/hexanes) to afford 8a as a white solid (19.3 mg, 58 % over two steps). 1H NMR (400 MHz, CD2Cl2, δ ppm): 8.23 (1H, d, J = 8.4 Hz), 7.25–7.50 (5H), 7.08 (2H, d, J = 8.4 Hz), 2.29 (3H, s), 1.40 (9H, s). 13C NMR (400 MHz, CD2Cl2, δ ppm): 152.3, 145.4, 130.4, 129.8, 128.9, 127.2, 125.2, 125.0, 124.8, 124.5, 121.1, 117.0, 114.0, 35.2, 31.6, 21.8. HRMS: calcd: 403.1606 (M)+, found: 403.1612.
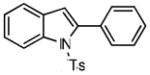
1-tosyl-2-phenylindole (8b)
Prepared according to General Procedure C using Pd(OAc)2 (0.9 mg, 0.0045 mmol, 5 mol %), SPhos (3.1 mg, 0.0090 mmol, 10 mol %), 3.0 M aqueous K3PO4 (0.23 mL, 7.5 equiv of base), and chlorobenzene (9.2 μL, 0.090 mmol, 1 equiv). The mixture was stirred under nitrogen at 60 °C for 5 h. The crude product was purified by flash column chromatography (SiO2; gradient: 1:3 → 3:7 → 2:3 dichloromethane/hexanes) to afford 8b as a white tacky solid (21.9 mg, 70 % over two steps). 1H NMR (400 MHz, CD2Cl2, δ ppm): 7.97 (1H, d, J = 8.4 Hz), 7.76 (2H, d, J = 8.4 Hz), 7.50–7.60 (3H, m), 7.20–7.32 (7H, m), 6.69 (1H, s), 2.33 (3H, s). 13C NMR (400 MHz, CD2Cl2, δ ppm): 145.9, 135.6, 135.3, 131.3, 130.4, 127.3, 126.9, 125.0, 123.8, 121.9, 113.9, 109.6, 21.8. HRMS: calcd: 347.0980 (M)+, found: 347.0986.
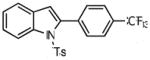
1-tosyl-2-(4′-trifluoromethylphenyl)indole (8c)
Prepared according to General Procedure C using Pd(OAc)2 (0.9 mg, 0.0045 mmol, 5 mol %), SPhos (3.1 mg, 0.0090 mmol, 10 mol %), 3.0 M aqueous K3PO4 (0.23 mL, 7.5 equiv of base), and 4-chlorobenzotrifluoride (0.012 mL, 0.090 mmol, 1 equiv). The mixture was stirred under nitrogen at 60 °C for 15 h. The crude product was purified by flash column chromatography (SiO2; gradient: 1:3 → 3:7 → 2:3 dichloromethane/hexanes) to afford 8c as a white solid (21 mg, 56 % over two steps). 1H NMR (400 MHz, acetone-d6, δ ppm): 8.02 (1H, d, J = 7.6 Hz), 7.87 (2H, d, J = 8.4 Hz), 7.71 (1H, s), 7.58 (2H, d, J = 8.0 Hz), 7.35 (5H, m), 7.23 (1H, t, J = 7.2 Hz), 6.79 (1H, s), 2.33 (3H, s). 13C NMR (400 MHz, acetone-d6, δ ppm): 146.9, 136.5, 136.2, 132.4, 132.1, 131.4, 129.1, 128.3, 128.0, 127.5, 125.9, 125.6, 124.7, 124.6, 110.6, 101.4, 21.9. 19F NMR (400 MHz, acetone-d6, δ ppm): −62.16. HRMS: calcd: 415.0854 (M)+, found: 415.0855.
1-(4′-methoxybenzylidene)-1,3-dihydroisobenzofuran (9)
Prepared according to General Procedure C using Pd(OAc)2 (1.8 mg, 0.009 mmol, 5 mol %), SPhos (6.2 mg, 0.018 mmol, 10 mol %), 3.0 M aqueous K3PO4 (0.46 mL, 7.5 equiv of base), and 4-bromoanisole (0.023 mL, 0.090 mmol, 1 equiv). The mixture was stirred under nitrogen at 60 °C for 15 h. The crude product was purified by flash column chromatography (SiO2; gradient: hexanes → 1:4 → 3:7 → 2:3 dichloromethane/hexanes) to afford 9a as a white solid (11.3 mg, 53 % over two steps). 1H NMR (400 MHz, CD2Cl2, δ ppm): 7.67 (2H, d, J = 8.8 Hz), 7.56 (1H, d, J = 6.8 Hz), 7.36 (3H, m), 6.88 (2H, d, J = 6.8 Hz), 5.92 (1H, s), 5.51 (2H, s), 3.81 (3H, s). 13C NMR (400 MHz, CD2Cl2, δ ppm): 157.2, 154.5, 138.8, 134.5, 128.9, 128.5, 128.1, 127.7, 127.2, 120.9, 119.2, 113.8, 113.4, 95.2, 74.5, 54.8. HRMS: calcd: 238.0994 (M)+, found: 238.0993.
Acknowledgments
We gratefully acknowledge NIHGMS (RO1 GM073932) and Amgen for financial support. G.W.A. thanks the National Council for Scientific and Technological Development (CNPq)-Brazil for a postdoctoral fellowship. We also thank Professor Martin D. Burke and his group at the University of Illinois, Urbana-Champaign for their generous gift of ethynyl MIDA boronate and Johnson Matthey for a donation of AuCl3. Gold catalysts B and C were provided by Dr. William E. Brenzovich.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.a) De Luca L, Nieddu G, Porcheddu A, Giacomelli G. Curr Med Chem. 2009;16:1–20. doi: 10.2174/092986709787002826. [DOI] [PubMed] [Google Scholar]; b) Bräse S, Gil C, Knepper K. Bioorg Med Chem. 2002;10:2415–2437. doi: 10.1016/s0968-0896(02)00025-1. [DOI] [PubMed] [Google Scholar]; c) Mor M, Spadoni G, Di Giacomo B, Diamantini G, Bedini A, Tarzia G, Plazzi PV, Rivara S, Nonno R, Lucini V, Pannacci M, Fraschini F, Stankov BM. Bioorg Med Chem. 2001;10:1045–1057. doi: 10.1016/s0968-0896(00)00322-9. [DOI] [PubMed] [Google Scholar]; d) Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; e) Engler TA, LaTessa KO, Iyengar R, Chai W, Agrios K. Bioorg Med Chem. 1996;4:1755–1769. doi: 10.1016/0968-0896(96)00192-7. [DOI] [PubMed] [Google Scholar]; f) Dai JR, Hallock YF, Cardellina JH, Boyd M. J Nat Prod. 1998;61:351–353. doi: 10.1021/np970519h. [DOI] [PubMed] [Google Scholar]; g) Diyasena MNC, Sotheeswaran S, Surendrakumar S, Balasubramanian S, Bokel M, Kraus W. J Chem Soc Perkin Trans. 1985;1:1807–1809. [Google Scholar]; h) Dean FM. In: The Total Synthesis of Natural Products. ApSimon J, editor. Vol. 1 Wiley; New York: 1973. [Google Scholar]
- 2.a) Katritzky AR, Li J, Stevens CV. J Org Chem. 1995;60:3401–3404. [Google Scholar]; b) Kraus GA, Guo H. Org Lett. 2008;10:3061–3063. doi: 10.1021/ol801034x. [DOI] [PubMed] [Google Scholar]; c) Taber DF, Tian W. J Am Chem Soc. 2006;128:1058–1059. doi: 10.1021/ja058026j. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Fang YQ, Lautens M. Org Lett. 2005;7:3549–3552. doi: 10.1021/ol051286l. [DOI] [PubMed] [Google Scholar]; e) Gribble GW. J Chem Soc Perkin Trans 1. 2000;7:1045–1075. [Google Scholar]; f) Yue D, Yao T, Larock RC. J Org Chem. 2006;71:62–69. doi: 10.1021/jo051549p. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Kumar MP, Liu RS. J Org Chem. 2006;71:4951–4955. doi: 10.1021/jo0606711. [DOI] [PubMed] [Google Scholar]
- 3.For examples of boron and silicon containing substrates in gold-catalyzed cycloisomerization reactions see: Park S, Lee D. J Am Chem Soc. 2006;128:10664–10665. doi: 10.1021/ja062560p.Horino W, Luzung MR, Toste FD. J Am Chem Soc. 2006;128:11364–11365. doi: 10.1021/ja0636800.Matsuda T, Kadowaki S, Yamaguchi Y, Murakami M. Chem Commun. 2008:2744–2746. doi: 10.1039/b804721a.Lee JCH, Hall DG. Tetrahedron Lett. 2011;52:321–324.
- 4.a) Gillis EP, Burke MD. Aldrichimica Acta. 2009;42:17–27. [PMC free article] [PubMed] [Google Scholar]; b) Lee SJ, Anderson TM, Burke MD. Angew Chem Int Ed. 2010;49:8860–8863. doi: 10.1002/anie.201004911. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Woerly EM, Cherney AH, Davis EK, Burke MD. J Am Chem Soc. 2010;132:6941–6943. doi: 10.1021/ja102721p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dick GR, Knapp DM, Gillis EP, Burke MD. Org Lett. 2010;12:2314–2317. doi: 10.1021/ol100671v. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Struble JR, Lee SJ, Burke MD. Tetrahedron. 2010;66:4710–4718. [Google Scholar]
- 6.For recent relevant reviews on gold-catalysis see: Bandini M. Chem Soc Rev. 2011;40:1358–1367. doi: 10.1039/c0cs00041h.Corma A, Leya-Perez A, Sbater MA. Chem Rev. 2011;111:1657–1712. doi: 10.1021/cr100414u.de Mendoza P, Echavarren AM. Pure Appl Chem. 2010;82:801.Shapiro ND, Toste FD. Synlett. 2010:675–691. doi: 10.1055/s-0029-1219369.Abu Sohel AM, Liu R-S. Chem Soc Rev. 2009;38:2269–2281. doi: 10.1039/b807499m.Kirsch SF. Synthesis. 2008:3183–3204.Patil NT, Yamamoto Y. Chem Rev. 2008;108:3395–3442. doi: 10.1021/cr050041j.
- 7.For related tandem cyclization/coupling processes, see: Ishikura M, Takahashi N, Yamada K, Yanada R. Tetrahedron. 2006;62:11580–11591.Zhu G, Tong X, Cheng J, Sun Y, Li D, Zhang Z. J Org Chem. 2005;70:1712–1717. doi: 10.1021/jo048238j.Zhu G, Zhang Z. Org Lett. 2004;6:4041–4044. doi: 10.1021/ol048301r.Cheung WS, Patch RJ, Player MR. J Org Chem. 2005;70:3741–3744. doi: 10.1021/jo050016d.Thielges S, Meddah E, Bisseret P, Eustache J. Tetrahedron Lett. 2004;45:907–910.
- 8.Kadieva MG, Oganesyan ET. Chem Heterocycl Compd. 1997;33:1245–1258. [Google Scholar]
- 9.Gribble GW. Contemp Org Synth. 1994;1:145–172. [Google Scholar]
- 10.a) Praveen C, Iyyappan C, Perumal PT. Tetrahedron Lett. 2010;51:4767–4771. [Google Scholar]; b) Gabriele B, Salerno G, Fazio A, Pitelli R. Tetrahedron. 2003;59:6251–6259. [Google Scholar]; c) Hiroya K, Jouka R, Kameda M, Yasuhara A, Sakamoto T. Tetrahedron. 2001;57:9697–9710. [Google Scholar]
- 11.a) Solé D, Serrano O. J Org Chem. 2010;75:6267–6270. doi: 10.1021/jo101054j. [DOI] [PubMed] [Google Scholar]; b) Sun Q, Zhou X, Islam K, Kyle DJ. Tetrahedron Lett. 2001;42:6495–6497. [Google Scholar]
- 12.a) Mancuso R, Mehta S, Gabriele B, Salerno G, Jenks WS, Larock RC. J Org Chem. 2010;75:897–901. doi: 10.1021/jo902333y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kusama H, Sawada T, Okita A, Shiozawa F, Iwasawa N. Org Lett. 2006;8:1077–1079. doi: 10.1021/ol052951t. [DOI] [PubMed] [Google Scholar]; c) Wang F, Miao Z, Chen R. Org Biomol Chem. 2009;7:2848–2850. doi: 10.1039/b908313h. [DOI] [PubMed] [Google Scholar]; d) Yue D, Della Ca N, Larock RC. J Org Chem. 2006;71:3381–3388. doi: 10.1021/jo0524573. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Butin AV, Abaev VT, Mel’chin VV, Dmitriev AS. Tetrahedron Lett. 2005;46:8439–8441. [Google Scholar]; f) Villeneuve K, Tam W. Organometallics. 2007;26:6082–6090. [Google Scholar]
- 13.a) Movassaghi M, Hill MD. Org Lett. 2008;10:3485–3488. doi: 10.1021/ol801264u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gilmore CD, Allan KM, Stoltz BM. J Am Chem Soc. 2008;130:1558–1559. doi: 10.1021/ja0780582. [DOI] [PubMed] [Google Scholar]; c) Wang B, Lu B, Jiang Y, Zhang Y, Ma D. Org Lett. 2008;10:2761–2763. doi: 10.1021/ol800900a. [DOI] [PubMed] [Google Scholar]; d) Hui BWQ, Chiba S. Org Lett. 2009;11:729–732. doi: 10.1021/ol802816k. [DOI] [PubMed] [Google Scholar]; e) Niu YN, Yan ZY, Gao GL, Wang HL, Shu XZ, Ji KG, Liang YM. J Org Chem. 2009;74:2893–2896. doi: 10.1021/jo900010m. [DOI] [PubMed] [Google Scholar]; f) Fischer D, Tomeba H, Pahadi NK, Patil NT, Huo Z, Yamamoto Y. J Am Chem Soc. 2008;130:15720–15725. doi: 10.1021/ja805326f. [DOI] [PubMed] [Google Scholar]; g) Yang YY, Shou WG, Chen ZB, Hong D, Wang YG. J Org Chem. 2008;73:3928–3930. doi: 10.1021/jo8003259. [DOI] [PubMed] [Google Scholar]
- 14.a) Kotera A, Uenishi J, Uemura M. Tetrahedron Lett. 2010;51:1166–1169. [Google Scholar]; b) Hellal M, Bourguignon JJ, Bihel FJJ. Tetrahedron Lett. 2008;49:62–65. [Google Scholar]; c) Patil NT, Yamamoto Y. J Org Chem. 2004;69:5139–5142. doi: 10.1021/jo049416b. [DOI] [PubMed] [Google Scholar]
- 15.For examples of the use of this complex, see: Sherry BD, Toste FD. J Am Chem Soc. 2004;126:15978–15979. doi: 10.1021/ja044602k.Sherry BD, Maus L, Lafortezza BN, Toste FD. J Am Chem Soc. 2006;128:8132–8133. doi: 10.1021/ja061344d.LaLonde RL, Brenzovich WE, Jr, Benitez D, Tkatchouk E, Kelley K, Goddard WA, III, Toste FD. Chem Sci. 2010;1:226–233. doi: 10.1039/C0SC00255K.
- 16.For discussions of ligand effects on gold catalyzed reactions see: Raubenheimer HG, Schmidbaur HS. Afr J Sci. 2011 doi: 10.4102/sajs/v10713/4.459.Benitez D, Shapiro ND, Tkatchouk E, Wang Y, Goddard WA, III, Toste FD. Nat Chem. 2009;1:482–486. doi: 10.1038/nchem.331.Gorin DJ, Sherry BD, Toste FD. Chem Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g.
- 17.a) Nesmeyanov AN, Perevalova EG, Struchkov YT, Antipin MY, Grandberg KI, Dyadchenko VP. J Organomet Chem. 1980;201:343–349. [Google Scholar]; b) Yang Y, Ramamoorthy V, Sharp PR. Inorg Chem. 1993;32:1946–1950. [Google Scholar]
- 18.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]