

Abstract
Protein glycosylation commonly stabilizes proteins thereby increasing protein half-lives and protecting against denaturation or proteolytic degradation. While generally beneficial, such stabilization is potentially disadvantageous in the case of inhibitory serpins. These protease inhibitors are metastable and a conformational transition to a more stable form is key to their function. Instability is therefore essential for these inhibitory serpins and mutagenesis has demonstrated that substantial stabilization results in compromised function. We have used optical spectroscopy and hydrogen/deuterium exchange and mass spectrometry to investigate the effects of glycosylation on the human serpin alpha-1 antitrypsin (α1-AT). Previous studies found that unglycosylated recombinant α1-AT populates a molten globule at low denaturant and that the ability to populate this state is correlated with efficient protease inhibition. Further, a high degree of conformational flexibility was found in several important regions. Guanidine hydrochloride denaturation monitored by circular dichroism indicates that plasma α1-AT, which is glycosylated at 3 sites, is substantially stabilized relative to the unglycosylated form. However, hydrogen exchange reveals complete loss of protection in plasma α1-AT above 1 M GuHCl, similar to what is seen for the recombinant form. Sugars therefore appear to stabilize the compact denatured state of α1-AT without significant stabilization of the folded state. Native state hydrogen exchange reveals minor perturbations to native flexibility, but high flexibility in key regions such as the f helix is conserved. β-strand 1c is stabilized in plasma α1-AT, which may confer increased resistance to forming pathogenic polymers. Overall, our results indicate that glycosylation of inhibitory serpins does not interfere with either native state flexibility or the native instability that is required for efficient function, though it may confer resistance to degradation by proteases and thus extend the half-life of circulating serpins.
Keywords: Serpin, antitrypsin, glycosylation, hydrogen/deuterium exchange, protein stability
Introduction
Glycosylation is a very common form of post-translational modification in proteins and the majority of secreted proteins in eukaryotes undergo N-linked glycosylation during processing in the ER[1]. Glycosylation plays a number of roles, including modulating intermolecular interactions, helping prevent aggregation, increasing the lifetime of circulating proteins by conferring resistance to proteolytic degradation, and intrinsic stabilization of protein structure[1, 2]. In contrast to the vast number of biophysical studies on recombinant proteins expressed in E. coli, glycoproteins have received less attention. This is primarily due to the difficulty of obtaining homogeneous samples. While all N-linked glycoproteins contain a common core oligosaccharide, additional oligosaccharide units are usually added during processing, and the final product is generally not uniform even in copies of the same protein produced in a single cell[3]. Glycoproteins therefore generally exist as a distribution of multiple glycoforms, and it is often not possible to isolate one specific glycoform in sufficient amounts for biophysical studies. Nevertheless, the effects of glycosylation on the biophysical properties of proteins are of interest. Mass spectrometry is a premier tool for characterizing glycoproteins, and mass spectrometry coupled with hydrogen/deuterium exchange (HXMS) is a promising method for probing the local effects of glycosylation on protein structure, dynamics and stability.
α1-antitrypsin (α1-AT) is an important human glycoprotein that circulates in plasma at a concentration of 1–2 mg/ml and is the major inhibitor of neutrophil elastase[4]. α1-AT is the canonical member of the serpin class of protease inhibitors, sharing the common serpin tertiary structure consisting of three β-sheets (a, b and c) and 8–9 α helices with an extended reactive center loop (RCL) exposed at one end of the protein molecule to interact and bind with its cognate protease Figure 1A[5]. Inhibitory serpins employ a unique mechanism which involves a massive conformational change in which the flexible solvent exposed reactive center loop (RCL) inserts into β-sheet a, becoming a sixth strand[4]. This process occurs when the target protease is covalently bound to the RCL in an acylenzyme intermediate, and thus the protease is translocated nearly 70 Å from one end of the serpin molecule to the other (Figures 1B and 1C)[6, 7]. This conformational change is accompanied by a large increase in stability: serpins in the uncleaved metastable state unfold at ~60 °C while the cleaved serpins unfold at ≥ 120 °C[8]. Serpins are therefore highly unusual proteins in that they do not fold to their global free energy minimum. Rather, inhibitory serpins initially fold to a metastable state in which a considerable degree of “conformational strain” is present in the form of bad steric contacts and cavities[9]. During protease inhibition, serpins then convert to a stable conformation. Instability is therefore critical for serpin function. In fact, many serpins are marginally stable. The canonical serpin α1-AT populates a molten globule-like compact denatured state under very mildly denaturing conditions: in guanidine hydrochloride (GuHCl) the native→compact denatured transition occurs with a midpoint of 0.8 M[10, 11]. Studies have demonstrated that increasing the stability of the metastable form through mutation leads to compromised function[12]. It has further been shown that efficient inhibitory capacity is correlated with the ability to populate the compact denatured state at low concentrations of denaturant[12, 13]. In addition to global instability, a large degree of conformational flexibility is required in several specific parts of the structure in order to accomplish the inhibitory conformational change[14]. These include the conserved “breach” region which is the site of initial RCL insertion located at the top of the β-sheet a and the conserved “shutter” region which is believed to control the opening of β-sheet a during the inhibitory process and which consists of the middle of strands 3a, 5a and portion of strand 5b[14, 15]. Additionally, helix f lies directly in the path traversed by the RCL during the conformational change, and must be displaced if RCL insertion is to occur[16–18]. The majority of biophysical studies of α1-AT have employed a recombinant form that is produced in E. coli and is therefore unglycosylated. Glycosylation increases the global stability of proteins, and further, global stabilization is often accompanied by reduced flexibility in the native state[1]. It has been shown previously that glycosylation increases the stability of α1-AT against both thermal and chemical denaturation[19]. This raises the question: if glycosylation stabilizes α1-AT why does this stabilization not lead to compromised inhibitory efficiency? Here, we address this question by using HXMS and optical spectroscopy to characterize the global stability and local flexibility of glycosylated human plasma α1-AT (HPα1-AT) and comparing the results with those obtained for unglycosylated recombinant α1-AT (RCα1-AT)[20].
Figure 1. Serpin Structure and Mechanism.
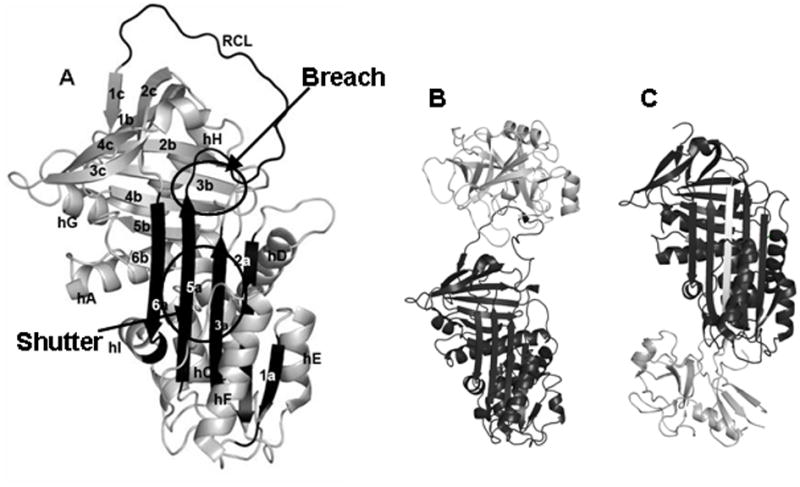
A. The structure of α1-antitrypsin (1QLP). Secondary structure elements are labeled. B. The Michaelis complex between a serpin (black) and target protease (white) (1K90). C. The covalently linked serpin-protease complex after the inhibitory conformational change (1EZX). In addition to the protease, the serpin’s inserted reactive center loop is also shown in white.
Material and Methods
Purification of wild-type and human plasma α1-antitrypsin and activity assay
Unglycosylated recombinant α1-AT (RCα1-AT) was expressed and purified as described[20]. Human plasma α1-AT (HPα1-AT) purchased from Sigma and was further purified as follows. The protein was solublized in 10 mM sodium phosphate (pH 6.5), 1 mM EDTA, 0.2 mM PMSF, and 1 mM β-mercaptoethanol (β-ME) (Buffer A) and loaded onto Hiprep 16/10 DEAE FF column (Amersham). Proteins were eluted with a linear gradient of buffer A containing Buffer B (1M NaCl, 10 mM sodium phosphate (pH 6.5), 1 mM EDTA, 0.2 mM PMSF, and 1 mM BME). Fractions containing HPα1-AT were pooled, and buffer-exchanged into 20 mM bistris (pH 6.5), 1 mM EDTA, and 1 mM β-ME (buffer C) with Amicon Ultra-15 (Millipore). This sample was further loaded onto MonoQ 4.6/100 PE (Amersham), and HPα1-AT was eluted with a linear gradient of buffer C containing 1M NaCl. Fractions containing HPα1-AT were pooled and buffer-exchanged into 10 mM sodium phosphate (pH 7.5) and 50 mM NaCl. The purified protein concentrations were determined in 6 M GuHCl using A1cm1% = 4.3 at 280 nm as described previously[20]. The activity assays on the two proteins were performed as described earlier[20]. For all the samples, the stoichiometry of inhibition was determined to be 1.0.
Peptide mapping by HPLC-tandem mass spectrometry
A total of 10 μg of purified HPα1-AT in 120 μl of 10 mM sodium phosphate, pH 7.5 and 50 mM NaCl was mixed with 115 μl of 100 mM NaH2PO4, pH 2.4 followed by the addition of 5 μg of porcine pepsin (1 μg/μl), dissolved in 0.05% TFA and H2O for pepsin digestion. HPα1-AT was digested for 5 min on ice. The final concentration of HPα1-AT was 940 nM. The digested sample was injected into a micropeptide trap (Michrom Bioresources) connected to a C18 HPLC column (5 cm × 1 mm, Alltech) coupled to a ThermoElectron LTQ ion-trap mass spectrometer (ThermoElectron). Peptic fragments were eluted using a gradient of acetonitrile (Burdick and Jackson) at a flow rate of 50 μl/min for a tandem mass spectrometry experiment to sequence each of the peptic fragments. Peptic fragments were identified by using the search algorithm SEQUEST (ThermoElectron) and manual inspection.
Hydrogen/deuterium exchange
HPα1-AT sample containing 10 μg of active protein in 10 mM sodium phosphate, pH 7.5 and 50 mM NaCl was diluted 24-fold with 10 mM sodium phosphate, pD 7.8 and 50 mM NaCl dissolved in D2O (Cambridge Isotope Laboratories) at 25 °C to label the sample with deuterium. The deuteration reaction was finally quenched at different time points by adding equal volume of 100 mM NaH2PO4, pH 2.4, which dropped the pH of the sample to 2.5, and quickly frozen in a dry ice-ethanol bath. Samples were stored at −80 °C until use. The final concentration of HPα1-AT was 940 nM.
Isotope analysis by HPLC-electrospray ionization mass spectrometry
The frozen sample was quickly thawed and digested with 5 μg of pepsin (1 μg/μl, dissolved in 0.05% TFA and H2O) on ice for 5 min followed by immediate injection into a micropeptide trap connected to a C18 HPLC column (5 cm × 1 mm, Alltech) coupled to a ThermoElectron LTQ ion-trap mass spectrometer. Peptic peptides were eluted in 12 min using a gradient of 10–45% acetinitrile at a flow rate of 50μl/min. The micropeptide trap as well as the C18 HPLC column was immersed in ice during the entire run to minimize the back exchange. The amount of deuterium incorporation for each fragment was calculated using the following equation:
where m is the mass of deuterated peptic fragment, m0% and m100% are the mass of the unlabeled and fully deuterated peptic fragments, respectively, N is the total number of exchangeable amide hydrogen atoms in each peptic fragment, and D is the number of amide hydrogen atoms incorporated in each peptic fragment.
Equilibrium unfolding in GuHCl monitored by Circular dichroism and fluorescence spectroscopy
CD spectra were obtained using Aviv CD spectrometer Model 215 at 25°C with 1 nm/10 sec signal averaging from 210 to 250 nm using a 1 mm path-length cuvette. RCα1-AT was treated as described previously (10). 11μM HPα1-AT was incubated in 10 mM sodium phosphate (pH 7.5), 50 mM NaCl containing different concentrations of optical grade GuHCl (Pierce) for 3 hr at 25°C. The equilibrium unfolding curves were determined from the signal at 222 nm as a function of the denaturant concentration. The data were fitted to two and three state equations.
The intrinsic tryptophan fluorescence spectra of HPα1-AT were measured at different concentrations of GuHCl to monitor the unfolding of the glycosylated protein. Fluorescence spectra were obtained using a FluoroMax-3 (HoribaJobinYvon) fluorescence spectrophotometer with an excitation wavelength of 295 nm and the emission spectra were recorded from 310–430 nm and having the slit width of 5 nm. For equilibrium unfolding of HPα1-AT in presence of GuHCl, the purified protein was incubated in 10 mM sodium phosphate (pH 7.5) and 50 mM NaCl, containing various concentrations of GuHCl at 25 °C. Samples were allowed to equilibrate for 3 hr. The protein concentration for unfolding transition was 10 μg/ml.
Equilibrium unfolding of HPα1-AT by hydrogen/deuterium exchange
HPα1-AT sample containing 10 μg of active protein in was incubated 10 mM sodium phosphate, pH 7.5 and 50 mM NaCl in presence of 1.2 and 4 M GuHCl separately for 3 hr at 25 °C to achieve the equilibrium. Then the sample was diluted 24-fold with 10 mM sodium phosphate, pD 7.8 and 50 mM NaCl containing the same concentration of GuDCl dissolved in D2O at 25 °C for 10 sec to label the sample with deuterium. The deuteration reaction was finally quenched by adding equal volume of 100 mM NaH2PO4, pH 2.4, which dropped the pH of the sample to 2.5, and quickly frozen in a dry ice-ethanol bath. Samples were stored at −80 °C freezer until use. Finally the samples were digested with pepsin and analyzed by mass spectrometry as described above.
Results
Peptide Mapping and Coverage
Tandem mass spectrometry identified peptides covering 68.18% of the HPα1-At sequence. We did not identify any of the peptide fragments containing the glycosylation sites Asn46, Asn83 and Asn247. The 20 peptide fragments were chosen and analyzed in H/D-MS experiments are well distributed throughout the protein molecule. Sequence differences between plasma AT and the specific recombinant form used in Tsutsui et al[20], as well as possible interference by the bulky glycans altered the digestion pattern slightly. An addition to several missing peptides, two new peptides were identified which were not previously analyzed in RCα1-AT.
Equilibrium Unfolding Monitored by Circular Dichroism and Fluorescence spectroscopy
GuHCl induced unfolding of plasma AT was monitored by CD spectroscopy. Results are shown in Figure 2 together with the unfolding curve previously measured for RCα1-AT[10]. The signal at 222 nm reveals a single transition with a midpoint at ~2.3 M. The transition was well fit to a 2-state unfolding model. Fitting yields a ΔG of unfolding of 3 kcal/mol, a value that is smaller than the ΔG of unfolding that has been determined previously for recombinant AT[21]. This puzzling result is due to the fact that the unfolding of plasma AT is only apparently 2-state (see below).
Figure 2. Unfolding Monitored by CD Spectroscopy.
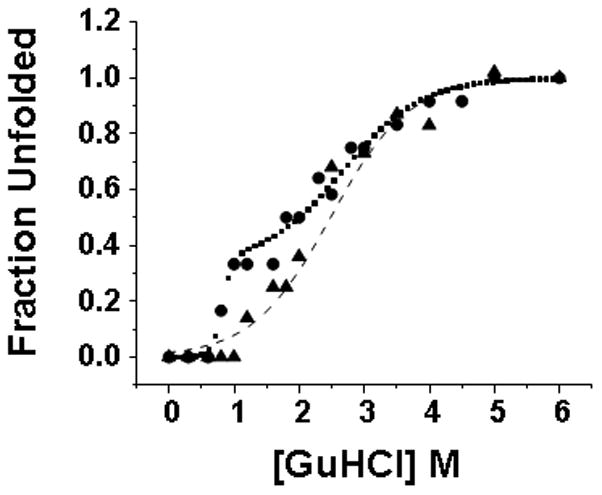
Unfolding of recombinant (filled circles) and plasma (filled triangles) α1-AT. Recombinant data was fit to a 3-state unfolding model (dotted line) while plasma was fit to a 2-state model (dashed line).
HPα-1AT contains two tryptophan residues Trp194 and Trp238 which were used to study the conformational change within the protein at different GuHCl. A small red shift is apparent from 1 to 1.8 M GuHCl, followed by a much larger red shift from 1.8 through 4.5 M GuHCl: above 4.5 M, the emission maximum remains essentially constant (Figure 3). The transition indicated by the larger red shift has a midpoint of 2.5 M GuHCl, which is close to the midpoint of 2.3 M for the single transition seen by CD.
Figure 3. Unfolding Monitored by Fluorescence.
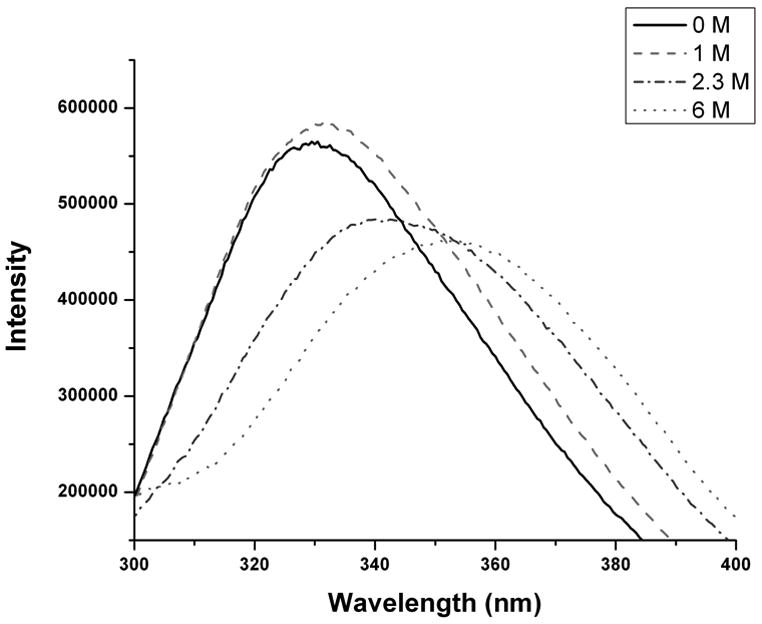
Fluorescence emission spectra for HPα1-AT at increasing concentrations of GuHCl. Excitation wavelength was 295 nm.
Equilibrium unfolding and pulse labeling H/D exchange
When combined with equilibrium chemical denaturation, pulse labeling HXMS can detect double isotopic envelopes which reveal the relative populations of folded and unfolded species[22]. This type of analysis can uncover details of the unfolding mechanism that are not apparent to more conventional spectroscopic methods. Pulse labeling of plasma AT was performed under conditions similar to those used in [10] for recombinant AT in order to facilitate comparison. In Figure 4, it can be seen that two populations are discernable at 1 M GuHCl, with the folded and unfolded populations being approximately equal. By 1.2 M GuHCl, only the unfolded population is detectable. Similar results were obtained for all peptides, indicating a cooperative transition to the unfolded state involving the entire protein. The results of pulse labeling HXMS are at variance with those obtained by CD spectroscopy, which indicates that AT is almost fully native at 1 M GuHCl and only 15% unfolded at 1.2 M.
Figure 4. Unfolding Monitored by HXMS.

Mass spectra for 8 peptides from diverse regions of HPα1-AT pulse labeled for 10 s in D2O/GuDCl after equilibration at increasing concentrations of GuHCl.
Native State Hydrogen/Deuterium Exchange
Hydrogen/Deuterium (H/D) exchange of glycosylated HPα-1AT was performed at pD 7.8 and 25 °C followed by HPLC-MS to quantify the mass of each peptic fragment. The corrected levels of deuterium incorporation by different peptic peptides at different time points were calculated from the H/D exchange experiments and are given in Table 1 and normalized deuterium uptake vs time curves for HPα1-AT and RCα1-AT are shown in Figure 5. As with recombinant α1-AT, the regions containing significant α-helical and β-sheet content exchange more slowly than regions consisting largely of turns or loops. Residues 62-77 (helix c), 100-118 (helix d and strand 2a), 227-240 (strands 1b, 2b and part of 3b), 318-329, 325-338 (strand 5a) and 374-384 (strands 4b and 5b) and 266-275 (helix h) all show relatively slow exchange. Also as expected the RCL and other surface loops show rapid exchange. While H/D exchange patterns in recombinant and plasma α-1AT are similar overall, there are several differences. Peptides 188-205 containing a loop and a portion of strand 4c and 318-329 containing a loop between helix i and strand 5a both show increased exchange in HPα1-AT. Peptides 208-227, 227-240 and 353-372 show reduced exchange in HPα1-AT relative to RCα1-AT. Two peptides in this study were not previously reported in other HXMS studies of RCα1-AT. Residues 100-118 include part of helix d and strand 2a and shows significant protection against exchange, indicating that these secondary structure elements are stable in both HPα1-AT and RCα1-AT. Residues 374-384 include parts of strands 4 and 5 b and a linker between them. These residues also show substantial protection against exchange, as expected since β-sheet b forms part of the stable core of α1-AT.
TABLE 1.
Corrected level of deuterium incorporation after H/D exchange experiments at different time points
| Residues | Peptic peptides | Amide hydrogens (N) | Corrected level of deuterium uptake (D) |
||||
|---|---|---|---|---|---|---|---|
| 10 sec | 50 sec | 100 sec | 500 sec | 1000 sec | |||
| 62-77 | AMLSLGTKADTHDEIL | 15 | 4.2 | 4.8 | 5.1 | 5.5 | 6.0 |
| 100-118 | LRTLNQPDSQLQLTTGNGL | 17 | 3.8 | 6.1 | 6.2 | 7.7 | 8.6 |
| 120-130 | LSEGLKLVDKF | 10 | 5.3 | 6.1 | 6.9 | 7.5 | 7.7 |
| 131-142 | LEDVKKLYHSEA | 11 | 2.9 | 4.9 | 5.8 | 7.3 | 7.9 |
| 143-159 | FTVNFGDTEEAKKQIND | 16 | 7.2 | 8.4 | 9.0 | 11.0 | 12.8 |
| 160-171 | YVEKGTQGKIVDL | 11 | 7.0 | 9.1 | 9.5 | 9.9 | 10.0 |
| 172-182 | LVKELDRDTVF | 10 | 7.3 | 8.0 | 8.6 | 9.0 | 9.2 |
| 188-205 | IFFKGKWERPFEVKDTEE | 16 | 8.1 | 10.1 | 11.3 | 12.1 | 12.4 |
| 208-227 | FHVDQVTTVKVPMMKRLGMF | 18 | 6.3 | 7.3 | 7.6 | 8.8 | 9.2 |
| 227-240 | FNIQHCKKLSSWVL | 13 | 3.2 | 3.7 | 4.0 | 4.1 | 4.3 |
| 251-266 | IFFLPDEGKLQHLENE | 14 | 3.9 | 4.5 | 4.7 | 6.1 | 6.2 |
| 266-275 | ELTHDIITKF | 9 | 1.1 | 2.1 | 2.4 | 3.2 | 3.2 |
| 286-303 | LHLPKLSITGTYDLKSVL | 16 | 8.2 | 8.3 | 8.5 | 9.2 | 9.4 |
| 299-317 | KSVLGQLGITKVFSNGAD | 18 | 9.6 | 13.2 | 13.8 | 16.0 | 17.7 |
| 318-329 | LSGVTEEAPLKL | 10 | 2.6 | 3.8 | 4.6 | 5.2 | 5.8 |
| 325-338 | APLKLSKAVHKAVL | 12 | 3.1 | 4.2 | 4.5 | 4.9 | 5.0 |
| 339-352 | TIDEKGTEAAGAMF | 13 | 11.8 | 12.3 | 12.4 | 12.6 | 12.7 |
| 353-372 | LEAIPMSIPPEVKFNKPFVF | 15 | 7.5 | 7.5 | 8.0 | 8.2 | 8.2 |
| 374-384 | MIEQNTKSPLF | 9 | 0.9 | 2.1 | 2.5 | 3.1 | 4.3 |
| 385-394 | MGKVVNPTQK | 8 | 3.5 | 3.8 | 3.8 | 3.9 | 4.0 |
Figure 5. Effects of glycosylation on native H/D exchange.

Normalized deuterium uptake vs time curves for 20 peptides from HPα1-AT (squares) and RCα1-AT (diamonds).
Discussion
In this study we have focused on the HPα1-AT which is normally fully glycosylated at three different Asn residues (Asn46, Asn83 and Asn247) with a mixture of bi- and tri-antennary complex glycans[23]. The glycan moieties of different glycoproteins play crucial role in stabilizing the glycoprotein from proteolytic degradation and denaturation. In this study, we have analyzed the distribution of the conformational flexibility in the metastable HPα1-AT as well as the equilibrium unfolding study to compare with the non-glycosylated form RCα1-AT.
Numerous prior studies of recombinant α1-AT, including our own, have identified a 3-state unfolding mechanism during GuHCl or urea induced denaturation[11, 12, 16]. CD spectroscopy reveals a transition to an unfolding intermediate with a midpoint of ~0.8 M GuHCl[11]. This intermediate retains approximately 70% of its native ellipticity at 222 nm and is more compact than the fully unfolded state, but lacks protection against H/D exchange[10]. Studies on stabilized mutants of recombinant α1-AT suggest that the ability to populate this compact denatured state at modest denaturant concentrations correlates with inhibitory efficiency. Additionally, transient unfolding during protease translocation and RCL insertion was identified by HXMS[24]. All of these results suggest that α1-AT must transiently populate a partially unfolded state during the conformational change that accompanies protease inhibition.
Unfolding of glycosylated α1-AT monitored by CD spectroscopy suggests the disappearance of the compact denatured state that was seen during the unfolding of recombinant AT, as there is no indication of an unfolding transition below 1 M. However, when unfolding is monitored by pulse labeling HXMS, it is clear that α1-AT loses its protection against exchange by 1.2 M, similar to recombinant. In addition to the loss of protection from H/D exchange, the small changes in the fluorescence spectrum at low concentrations of GuHCl also suggest destabilization of the native structure. From 1 to 1.8 M GuHCl, a small red shift accompanied by an increase in emission intensity is observed. It has been reported before that Trp residues are strongly quenched in the native structure of α1-AT, and the increased emission intensity likely reflects decreased quenching due to disruption of the native structure[21]. It therefore appears that glycosylation stabilizes the compact denatured form of α1-AT, but not the native state. Further, glycosylation appears to alter the properties of the compact denatured state of α1-AT. Unlike recombinant, which loses ~30% of its native ellipticity above 1M GuHCl, plasma α1-AT retains near native levels of ellipicity. It has been argued that compaction of the polypeptide chain induces secondary structure[25], and our results suggest that plasma α1-AT at 1.2 M GuHCl is more compact than recombinant α1-AT under the same conditions, although direct comparison of hydrodynamic radii or other measures of chain dimensions is complicated by the presence of glycans in plasma α1-AT. Despite the increased secondary structure content of plasma AT at low GuHCl concentrations, it retains virtually no protection against H/D exchange under these conditions, indicating that whatever secondary structure is present must be marginally stable and subject to frequent and significant fluctuations.
α1-AT is thus highly unusual in that glycosylation does not appear to stabilize the native state. In the case of inhibitory serpins, this peculiar behavior may reflect functional requirements since significant stabilization of the native metastable form of α1-AT could lead to compromised inhibitory activity. Previous studies on the effects of glycosylation on protein folding and stability have found that the primary effect of glycosylation is to destabilize the denatured state[26]. In the case of α1-AT, the effects of glycosylation on the unfolded state appear to be decoupled from the stability of the native state. In a protein with a simple 2-state folding mechanism, such destabilization would naturally drive the protein to the native state. The equilibrium folding/unfolding mechanism of α1-AT, however, is not 2-state. Instead, AT unfolds through a complex mechanism involving a molten globule intermediate. Our results suggest that glycosylation does destabilize the unfolded state of α1-AT at mild GuHCl concentrations, but that this drives α1-AT into the compact denatured state rather than to the native state.
Regarding native state dynamics, glycosylation increases flexibility in some regions and decreases it in others (Figure 6). Peptides 208-227 and 227-240 include β-strands 3c and 1b as well as portions of β-strands 4c and 2b and both show decreased H/D exchange in plasma α1-AT. Peptide 208-227 contains residues that are spatially adjacent to the glycan at position 46, and might be stabilized by direct interactions with the sugar. Such stabilization could propagate to neighboring residues in β-sheets b and c. Stabilization of residues in peptide 227-240 by interactions with the glycan at position 83 are also possible. There is no obvious structural explanation for the destabilization seen at the bottom of strand 5a, as it is not close to either a site of glycosylation or a region that is affected by glycosylation. However, a previous study found that interactions can propagate allosterically through the structure of α1-AT to distant regions[13].
Figure 6. Perturbed Flexibility Mapped on the Structure of α1-AT.
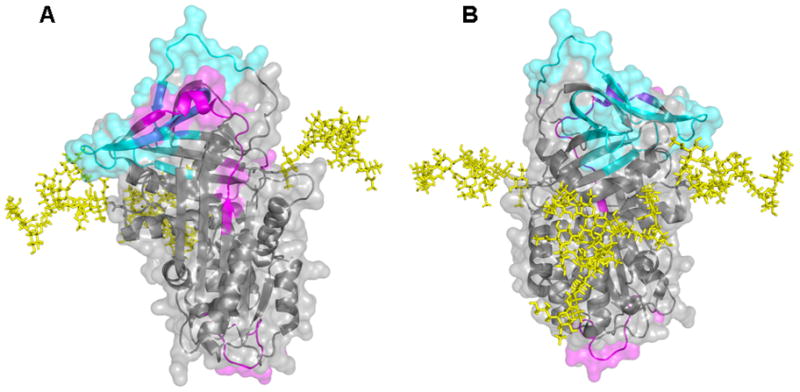
A. Front and B. Back of α1-AT (1QLP). The structure is displayed as both a ribbon diagram and as solvent accessible surface. Regions where normalized deuterium levels at 1000 s for HPα1-AT are greater or less than RCα1-AT by 10% or more are shown in magenta and cyan, respectively. The glycans have been modeled in to the structure using the GlyProt web server[29] and are shown as yellow sticks.
It is notable that H/D exchange rates in regions that play central roles in the metastable→stable transition are not affected by glycosylation. Helix f lies across the face of β-sheet a, and directly blocks the path that the RCL must traverse during inhibition. Previously it was found that in recombinant α1-AT the top of helix f, which packs against sheet a, is extremely flexible, showing negligible protection against H/D exchange[20]. It was proposed that this high degree of flexibility would allow helix f to be readily displaced from its position by the RCL during translocation. The top of helix f retains its flexibility in glycosylated α1-AT (peptide 160-171). The “breach” and “shutter” regions, which play major parts in the RCL insertion process were found to be stable in recombinant α1-AT, suggesting that the flexibility required during the native→cleaved transition must be induced by RCL cleavage. In those portions where we have coverage, rigidity in the breach and shutter regions seems to be neither significantly increased or decreased in HPα1-AT, but is retained at approximately the same level as in RCα1-AT (peptides 325-338, 339-352). A degree of increased exchange is seen in residues 188-205, which include two residues in strand 3a. However, this peptide also includes helix h and a portion of strand 4c and it is unclear which residues account for the increased exchange. Since helix h and strand 4c are adjacent to other regions affected by glycosylation while strand 3a is not, the former two regions most likely contain the destabilized residues. The present study did identify one difference in the native state dynamics of recombinant and glycosylated α1-AT that has potential functional significance. It has been demonstrated that the release of β-strand 1c from the rest of β-sheet c is required for the formation of pathological serpin polymers[27]. In recombinant α1-AT, strand c shows substantial protection from H/D exchange indicating a stable interaction with the rest of sheet c. This interaction would clearly deter polymer formation. In glycosylated α1-AT, stand 1c shows even greater protection from H/D exchange. Previous work has shown that HPα1-AT is more resistant to aggregation/polymerization than RCα1-AT[19]. Our results suggest that increased stability of strand 1c is one factor contributing to increased resistance to the formation of pathological polymers.
Like other proteins, α1-AT is not homogeneously glycosylated, instead it is produced as a mixture of glycoforms representing different combinations of bi- and tri-antennary complex glycans. Proteomic studies have identified two major glycoforms comprising ~74% of total α1-AT in human plasma together with several minor glycoforms[23]. Multiple glycoforms were present in the material used in this study (supplementary Figure 1), and yet this heterogeneity was not evident in the H/D exchange behavior. If a region of the α1-AT structure, covered by a specific pepsin derived peptide, exchanged hydrogen with deuterium at different rates in the different glycoforms, then peptides derived from the more flexible species should shift to higher m/z at a faster rate then peptides derived from the more rigid species. This should result in an unusually broad peak, at least at some of the labeling times. We did not observe such behavior. Peak widths for HPα1-AT were not significantly different from those for RCα1-AT, even though RCα1-AT represents a single pure species while HPα1-AT does not. This is consistent with previous observations on the effects of glycosylation on the global stability of proteins. It was found that the major effects on stability were caused by the core trisaccharide, while further additions to the oligosaccharide chain made only minor contributions[28]. Since all species of HPα1-AT contain the core trisaccharide, they therefore display indistinguishable local and global stability.
In summary, we have shown how the serpin α1-AT, which requires metastability for its function, deals with the stabilization conferred by glycosylation: stabilization is conferred largely on the compact denatured form, rather than on the native state. We have additionally demonstrated the utility of HXMS for detecting “hidden” unfolding intermediates that are not easily detected by common spectroscopic methods such as CD. Finally, this work provides a further demonstration that HXMS is a powerful and convenient method for profiling the biophysical properties of glycoproteins.
Supplementary Material
Acknowledgments
This work is supported by NIH R01HL085469.
Abbreviations
- α1-AT
α1-antitrypsin
- HPα1-AT
human plasma α1-antitrypsin
- RCα1-AT
recombinant α1-antitrypsin
- RCL
reactive center loop
- GuHCl
guanidine hydrochloride
- HXMS
hydrogen/deuterium exchange mass spectrometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shental-Bechor D, Levy Y. Folding of glycoproteins: toward understanding the biophysics of the glycosylation code. Curr Op Struct Biol. 2009 Oct;19(5):524–33. doi: 10.1016/j.sbi.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Varki A. Biological roles of oligosaccharides - all of the theories are correct. Glycobiology. 1993 Apr;3(2):97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudd PM, Dwek RA. Glycosylation: Heterogeneity and the 3D structure of proteins. Critical Reviews in Biochemistry and Molecular Biology. 1997;32(1):1–100. doi: 10.3109/10409239709085144. [DOI] [PubMed] [Google Scholar]
- 4.Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002 Dec;102(12):4751–804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 5.Elliott PR, Pei XY, Dafforn TR, Lomas DA. Topography of a 2.0 A structure of alpha1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci. 2000 Jul;9(7):1274–81. doi: 10.1110/ps.9.7.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000 Oct 19;407(6806):923–6. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 7.Ye S, Cech AL, Belmares R, et al. The structure of a Michaelis serpin-protease complex. Nat Struct Biol. 2001 Nov;8(11):979–83. doi: 10.1038/nsb1101-979. [DOI] [PubMed] [Google Scholar]
- 8.Kaslik G, Kardos J, Szabo E, et al. Effects of serpin binding on the target proteinase: global stabilization, localized increased structural flexibility, and conserved hydrogen bonding at the active site. Biochemistry. 1997 May 6;36(18):5455–64. doi: 10.1021/bi962931m. [DOI] [PubMed] [Google Scholar]
- 9.Seo EJ, Im H, Maeng JS, et al. Distribution of the native strain in human alpha 1-antitrypsin and its association with protease inhibitor function. J Biol Chem. 2000 Jun 2;275(22):16904–9. doi: 10.1074/jbc.M001006200. [DOI] [PubMed] [Google Scholar]
- 10.Tsutsui Y, Wintrode PL. Cooperative unfolding of a metastable serpin to a molten globule suggests a link between functional and folding energy landscapes. J Mol Biol. 2007 Aug;371(1):245–55. doi: 10.1016/j.jmb.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 11.James EL, Whisstock JC, Gore MG, Bottomley SP. Probing the unfolding pathway of alpha(1)-antitrypsin. J Biol Chem. 1999 Apr;274(14):9482–8. doi: 10.1074/jbc.274.14.9482. [DOI] [PubMed] [Google Scholar]
- 12.Seo EJ, Lee C, Yu MH. Concerted regulation of inhibitory activity of alpha(1)-antitrypsin by the native strain distributed throughout the molecule. J Biol Chem. 2002 Apr;277(16):14216–20. doi: 10.1074/jbc.M110272200. [DOI] [PubMed] [Google Scholar]
- 13.Sengupta T, Tsutsui Y, Wintrode PL. Local and Global Effects of a Cavity Filling Mutation in a Metastable Serpin. Biochemistry. 2009 Sep;48(34):8233–40. doi: 10.1021/bi900342d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whisstock JC, Skinner R, Carrell RW, Lesk AM. Conformational changes in serpins: I. The native and cleaved conformations of alpha(1)-antitrypsin. J Mol Biol. 2000 Jan 21;295(3):651–65. doi: 10.1006/jmbi.1999.3375. [DOI] [PubMed] [Google Scholar]
- 15.Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 2000 Dec;10(12):1845–64. doi: 10.1101/gr.gr-1478r. [DOI] [PubMed] [Google Scholar]
- 16.Cabrita LD, Whisstock JC, Bottomley SP. Probing the role of the F-helix in serpin stability through a single tryptophan substitution. Biochemistry. 2002 Apr;41(14):4575–81. doi: 10.1021/bi0158932. [DOI] [PubMed] [Google Scholar]
- 17.Cabrita LD, Dai W, Bottomley SP. Different conformational changes within the F-helix occur during serpin folding, polymerization, and proteinase inhibition. Biochemistry. 2004 Aug 3;43(30):9834–9. doi: 10.1021/bi0491346. [DOI] [PubMed] [Google Scholar]
- 18.Gettins PG. The F-helix of serpins plays an essential, active role in the proteinase inhibition mechanism. FEBS Lett. 2002 Jul 17;523(1–3):2–6. doi: 10.1016/s0014-5793(02)02924-1. [DOI] [PubMed] [Google Scholar]
- 19.Kwon KS, Yu MH. Effect of glycosylation on the stability of alpha(1)-antitrypsin toward urea denaturation and thermal deactivation. Biochimica Et Biophysica Acta. 1997 Jun;1335(3):265–72. doi: 10.1016/s0304-4165(96)00143-2. [DOI] [PubMed] [Google Scholar]
- 20.Tsutsui Y, Liu L, Gershenson A, Wintrode PL. The conformational dynamics of a metastable serpin studied by hydrogen exchange and mass spectrometry. Biochemistry. 2006 May;45(21):6561–9. doi: 10.1021/bi060431f. [DOI] [PubMed] [Google Scholar]
- 21.James EL, Whisstock JC, Gore MG, Bottomley SP. Probing the unfolding pathway of alpha1-antitrypsin. J Biol Chem. 1999 Apr 2;274(14):9482–8. doi: 10.1074/jbc.274.14.9482. [DOI] [PubMed] [Google Scholar]
- 22.Deng YZ, Zhang ZQ, Smith DL. Comparison of continuous and pulsed labeling amide hydrogen exchange/mass spectrometry for studies of protein dynamics. J Am Soc Mass Spectrom. 1999 Aug;10(8):675–84. doi: 10.1016/S1044-0305(99)00038-0. [DOI] [PubMed] [Google Scholar]
- 23.Mills K, Mills PB, Clayton PT, et al. Identification of alpha(1)-antitrypsin variants in plasma with the use of proteomic technology. Clinical Chem. 2001 Nov;47(11):2012–22. [PubMed] [Google Scholar]
- 24.Baek JH, Yang WS, Lee C, Yu MH. Functional Unfolding of alpha(1)-Antitrypsin Probed by Hydrogen-Deuterium Exchange Coupled with Mass Spectrometry. Mol Cell Proteom. 2009 May;8(5):1072–81. doi: 10.1074/mcp.M800365-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yee DP, Chan HS, Havel TF, Dill KA. Does compactness induce secondary structure in proteins - a study of poly-alanine chains computed by distance geometry. J Mol Biol. 1994 Aug;241(4):557–73. doi: 10.1006/jmbi.1994.1531. [DOI] [PubMed] [Google Scholar]
- 26.Shental-Bechor D, Levy Y. Effect of glycosylation on protein folding: A dose book at thermodynamic stabilization. Proc Natl Acad USA. 2008 Jun;105(24):8256–61. doi: 10.1073/pnas.0801340105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang WSW, Whisstock JC, Hopkins PCR, et al. Importance of the release of strand 1C to the polymerization mechanism of inhibitory serpins. Protein Science. 1997;6(1):89–98. doi: 10.1002/pro.5560060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanson SR, Culyba EK, Hsu TL, et al. The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proc Natl Acad Sci USA. 2009 Mar;106(9):3131–6. doi: 10.1073/pnas.0810318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohne-Lang A, von der Lieth CW. GlyProt: in silico glycosylation of proteins. Nucleic Acids Res. 2005;33:W214–W9. doi: 10.1093/nar/gki385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.