

Abstract
The current drive for applications of biomass-derived compounds, for energy and advanced materials, has led to a resurgence of interest in the manipulation of plant polymers. The xyloglucans, a family of structurally complex plant polysaccharides, have attracted significant interest due to their intrinsic high affinity for cellulose, both in muro and in technical applications. Moreover, current cell wall models are limited by the lack of detailed structure–property relationships of xyloglucans, due to a lack of molecules with well-defined branching patterns. Here, we have developed a new, broad-specificity “xyloglucan glycosynthase”, selected from active-site mutants of a bacterial endoxyloglucanase, which catalyzed the synthesis of high molar mass polysaccharides, with complex side-chain structures, from suitable glycosyl fluoride donor substrates. The product range was further extended by combination with an Arabidopsis thaliana α(1→2)-fucosyltransferase to achieve the in vitro synthesis of fucosylated xyloglucans typical of dicot primary cell walls. These enzymes thus comprise a toolkit for the controlled enzymatic synthesis of xyloglucans that are otherwise impossible to obtain from native sources. Moreover, this study demonstrates the validity of a chemo-enzymatic approach to polysaccharide synthesis, in which the simplicity and economy of glycosynthase technology is harnessed together with the exquisite specificity of glycosyltransferases to control molecular complexity.
Introduction
The xyloglucans (XyGs) comprise a family of plant polysaccharides that are abundant in the primary cell walls of dicotyledons and nongraminaceous monocotyledons.(1) The basic structure of XyGs is a linear β-(1→4)-d-glucan chain, which is regularly branched with α-d-xylopyranosyl-(1→6) units. Both the density of xylose branching and the further substitution of these branches with other monosaccharides, such as galactopyranose, l-fucopyranose, and arabinofuranose, are determined by the plant species and tissue of origin.2−4 For example, fucogalactoxyloglucan (FucGalXyG), which has β-d-Galp-(1→2) and α-l-Fucp-(1→2)-β-d-Galp-(1→2) residues extending from a repeating Xyl3Glc4 oligosaccharide core, is a common structural glycan among land plants, including the model species Arabidopsis thaliana (Figure 1). Galactoxyloglucans (GalXyGs) are prevalent as seed storage polysaccharides in certain species, and a number of these have structures identical to that of FucGalXyGs, but lack terminal fucose residues. Because of their availability on the tonne-scale, GalXyGs have found widespread applications in the food, paper, textile, and pharmaceutical industries, where they are employed as rheology modifiers or cellulose cross-linking agents.5−9
Figure 1.

General structure of XXXG-type xyloglucans and structures of α-glycosyl fluoride substrates for xyloglucan glycosynthases. (a) Variable substitution of the Xyl3Glc4 core repeat in xyloglucans. The standard xyloglucan structural abbreviations(55) are given below the corresponding motifs, assuming full branch substitution. For example, the galactoxyloglucan from tamarind seeds is predominantly comprised of XXXG, XXLG, and XLLG units, with minor amounts of XLXG units.(56) In contrast, the fucogalactoxyloglucan from Arabidopsis thaliana primary cell walls contains primarily XXXG, XXFG, and XLFG units, with minor amounts of other structures.(48) (b) α-Glycosyl fluoride substrates for xyloglucan glycosynthases.
The composition of the branching pattern of XyGs is known to affect both solution properties and the interaction with cellulose.8,10−14 At present, however, the natural heterogeneity of XyGs, together with the inability of glycosidases to affect complete side chain removal,(10) poses a hurdle in the detailed structure–function analysis of these polysaccharides. Hence, there exists a need for (bio)synthetic strategies to produce bespoke xyloglucan oligo- and polysaccharides with well-defined branching patterns.
The glycosynthase technology introduced by Withers and co-workers(15) provides an efficient biocatalytic route for the synthesis of complex saccharides. Classical glycosynthases are anomeric-configuration-retaining β-glycoside hydrolases in which the catalytic nucleophile, a glutamate or aspartate, has been replaced by a shorter, essentially inert residue, such as glycine, alanine, or serine.(16) The resulting hydrolytically inactive variant is nonetheless able to catalyze glycosyl transfer to acceptor substrates using suitable α-glycosyl fluoride donors, which mimic the wild-type glycosyl-enzyme intermediate. Product yields are typically high, as glycosynthase reactions are not complicated by competing product hydrolysis incurred when wild-type glycosidases are employed to catalyze transglycosylation reactions.(17)
Glycosynthases have been successfully used to synthesize a diversity of oligo- and polysaccharides,17−22 including the use of glycosynthases derived from endo-glucanases (EC 3.2.1.4) and endo-xyloglucanases (EC 3.2.1.151) to produce well-defined xyloglucan structures.23−26 In particular, our group has recently demonstrated that the Humicola insolens endo-glucanase HiCel7B E197S nucleophile variant efficiently catalyzes the condensation of XXXGαF (Figure 1) to yield homogenously branched, nongalactosylated, (XXXG)n-type xyloglucans with molar masses up to 60 000 (n ≈ 55). These represent some of the largest polysaccharides produced so far by the glycosynthase technology.22,24
Unfortunately, there currently exists no comparable glycosynthase for the production of uniformly galactosylated (XLLG)n xyloglucans, or other such xyloglucans with extended branches, for structure–function comparisons. Remarkably, XLLGαF (Figure 1) was not a substrate for the HiCel7B E197A (ref (25)) and HiCel7B E197S glycosynthases,(24) the latter of which is otherwise extremely proficient in the homocondensation of the nongalactosylated homologue XXXGαF. In contrast, glycosynthases derived from GH16 xyloglucan endo-transglycosylases and xyloglucan endo-hydrolases are capable of condensing XLLGαF, but thus far degrees of condensation are limited to (XLLG)n products with n ≤ 8 (M ≈ 11 000).24,25
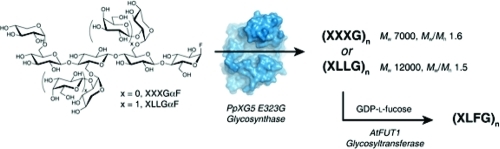
Motivated by the observation that Paenibacillus pabuli XG5 (PpXG5) is a highly specific endo-xyloglucanase (E.C. 3.2.1.151) with a particular selectivity for galactosylated side chains,(27) the E323A, E323S, and E323G nucleophile variants of this enzyme were generated in the present work to probe their capacity as glycosynthases for the polymerization of XLLGαF (Figure 2). Of these, the PpXG5 E323G glycosynthase was the most proficient, exhibiting slight selectivity for the digalactosylated donor over XXXGαF, and allowing the generation of (XLLG)n products with molar masses up to 60 000. Combination of this glycosynthase with the Arabidopsis thaliana xyloglucan-specific fucosyltransferase 1 (AtFUT1, glycosyltransferase family GT37, E.C. 2.4.1.69)28−30 allowed the production of fucogalactoxyloglucans of the type found in primary plant cell walls (Figure 2). This system comprises a toolkit for the enzymatic synthesis of well-defined xyloglucans, which are otherwise impossible to obtain from native sources.
Figure 2.

General scheme for the production of xyloglucans with defined, homogoenous side-chain structure using a combined glycosynthase/glycosyltransferase approach. Oligosaccharide nomenclature is identical to that shown in Figure 1.
Experimental Section
Production of PpXG5 Glycosynthases
Molecular Biology
All cloning steps were performed using standard molecular biology techniques. Nucleotides, buffers, and enzymes were purchased from Fermentas (Stockholm, Sweden). The oligonucleotide primers used in this study were obtained from Thermo Fisher Scientific (Ulm, D) and are given in Supporting Information Table S1. E. coli TOP10 cells, which were used for maintenance and propagation of all plasmids, were cultivated in LB-medium (yeast extract 5 g/L; tryptone 10 g/L; NaCl 10 g/L; pH 7.5) under appropriate selective conditions (ampicillin was added to a final concentration of 0.1 g/L). E. coli BL21DE3 cells were used for protein production in TBamp-medium (yeast extract 24 g/L; peptone from casein 12 g/L; glycerol 4 mL/L; KH2PO4-buffer 1 M, pH 7.5; ampicillin 0.1 g/L).
A synthetic gene of the PpXG5 E323S variant, codon-optimized for the expression in E. coli, was produced by GenScript (GenScript, NJ) and cloned into the vector pET28a+ using the restriction sites NcoI and XhoI. Site-directed mutagenesis was performed using the QuikChange II-E kit (Stratagene, Cedar Creek, TX) to obtain the variants PpXG5 E323G and PpXG5 E323A. The 1215 bp open reading frame of the PpXG5 sequence codes for a 395 amino acid protein; however, the mature protein comprises only 363 amino acids. The license-free software program SignalP predicted, with a probability of 99.6%, the first 32 amino acids to be a signal peptide, which is cleaved off post-translationally, resulting in a mature protein of an average molecular weight of 40.5 kDa. To avoid possible problems of this posttranslational modification in the heterologous host E. coli BL21DE3, we subcloned the genes PpXG5 E323G/A/S without the native signal sequence into the pET21a+ vector, providing a C-terminal His6-tag to facilitate purification. Full-length protein sequences encoded by the expression constructs are provided in Supporting Information Table S2.
The PCR reaction mix contained 2.5 Units Phusion DNA polymerase (Finnzymes; Espoo, Finland), ∼100 ng of plasmidic DNA, 5 pmol of each primer, 10 μM of each dNTP and 1× PCR buffer in a total volume of 50 μL. The mutagenic PCRs were done using the following conditions: 98 °C for 20 s; then 30 cycles of 98 °C for 10 s; 55 °C for 20 s; 72 °C for 1 min, followed by a final incubation at 72 °C for 7 min. After PCR, the methylated template-DNA was degraded by digestion with 10 Units DpnI at 37 °C for 3 h.(31) The remaining PCR products were separated by an agarose gel electrophoresis and purified using the QIAquick GelExtraction kit (QIAGEN; Sollentuna, SE). Five microliters of the PCR products were transformed into electro-competent E. coli TOP10 cells. Correct insertion of the PpXG5-encoding gene variants and the absence of undesired mutations were checked by DNA sequencing, using the primers T7fwd and T7rev (Supporting Information Table S1).
Gene Expression and Protein Purification
Cultures (1 L) of E. coli BL21DE3 transformants were grown in TBamp medium in baffled flasks at 37 °C and 220 rpm. Protein expression was induced at an OD600 of ∼0.5 by adding IPTG to a final concentration of 0.5 mM. After incubation at 25 °C for further 20 h, approximately 10 g of wet biomass per liter were harvested by centrifugation (4000g, 4 °C, 15 min) and resuspended in buffer A (NaH2PO4 50 mM; NaCl 500 mM; imidazole 20 mM; pH 7.5) containing the protease inhibitor PMSF (0.1% w/v). After disruption in a French Press (1200 psi), the crude cell extract was separated from cell debris by centrifugation (70 400g, 4 °C, 45 min) and used for protein purification by immobilized metal affinity chromatography (IMAC) with a 10 mL Ni-charged Sepharose 6 Fast Flow Resin (GE Healthcare, Uppsala, Sweden). Before the sample was loaded, the column was equilibrated with 10 column volumes (CV) of buffer A. After the protein sample was applied to the column, it was washed with 3 CV of the same buffer. Proteins were then eluted with a linear gradient of 5 CV of buffer B (NaH2PO4 50 mM; NaCl 500 mM; imidazole 1 M; pH 7.5). Appropriate fractions were combined, and imidazole was removed by ultrafiltration using an Amicon Ultra Centrifugal Filter Device (Millipore; Billerica, MA) with a 10 kDa cutoff membrane. The concentrated enzymes were washed three times with 10 mL of NaH2PO4 buffer (50 mM, pH 7.0) and finally diluted in the same buffer to a protein concentration of 20–30 g/L.
To check the purity of the glycosynthase preparations, SDS-PAGE analysis was done using precast 10% Bis-Tris gels (Invitrogen) and 1× MOPS-buffer (MOPS 10.46 g/L; EDTA 0.3 g/L; TrisBase 6.06 g/L; SDS 1.0 g/L; pH 7.7). Gels were run in the Novex MiniCell (Invitrogen) at 150 V for about 2 h. The protein mass standard used was the SeeBlue Plus 2 prestained standard (Invitrogen). Gels were stained with Coomassie blue. Electrospray ionization mass spectrometry (ESI MS) of the intact proteins was performed according to ref (32).
Production of the α(1→2)-Fucosyltransferase from Arabidopsis thaliana, AtFUT1(28)
Protein Expression and Purification
A recombinant Pichia pastoris clone(33) expressing the Golgi-localized enzyme AtFUT1 (UniProt Q9SWH5, ref (34)) was kindly provided by Prof. Kenneth Keegstra (Michigan State University, MI). The P. pastoris clone was precultured in 250 mL of buffered glycerol-complex medium (BMGY) in a 2 L baffled flask at 30 °C overnight. The next day, OD600 was measured, an appropriate amount of culture was sampled, and cells were spun down and diluted in 1 L of buffered methanol-complex medium (BMMY) to an initial OD600 of 1.0. The culture was grown in special 2.5 L baffled flasks (Tunair; Sigma-Aldrich; Stockholm, Sweden) at 25 °C and 220 rpm for 120 h, and methanol (MeOH) was added to a final concentration of 0.5% every day. All media were prepared as described in the Invitrogen Pichia Expression Manual (Invitrogen, CA).
After 120 h of induction, cells were harvested by centrifugation (4000g, 4 °C, 15 min) and washed twice by resuspending in 200 mL of ultrapure H2O. After being washed, cells were resuspended in 100 mL of HEPES-KOH buffer (50 mM, pH 7.5) containing the complete-mini EDTA-free protease inhibitor (1 tablet per 10 mL of buffer; Roche, Basel, CH). Pichia cells were disrupted in a French Press (1200 psi), and cell debris was separated by subsequent centrifugation at 1000g at 4 °C for 5 min. The supernatant was then centrifuged at 10 000g at 4 °C for 15 min before a final centrifugation at 100 000g at 4 °C for 60 min in an Optima L-100 XP ultracentrifuge (Beckman Coulter, Stockholm, SE). The pellet, which represented the microsomal fraction, was then resuspended in 10 mL of HEPES-KOH buffer (50 mM, pH 7.5) containing the complete-mini EDTA-free protease inhibitor. A total protein concentration of 3.7 mg/mL was obtained in this preparation (Bradford assay). SDS-PAGE analysis (10 μL sample, 1 mg/mL) indicated a predominant band with an apparent mobility of 65 kDa in SDS-PAGE (Supporting Information Figure S1), which was consistent with the expected molar mass of 63.4 kDa (UniProt Q9SWH5(34)).
Glycosynthase Reactions
Substrates
The α-glycosyl fluorides of XXXG and XLLG, XXXGαF and XLLGαF, respectively (Figure 1), were prepared as previously described.(25) A pure XLLG standard was produced by digestion of tamarind seed xyloglucan (Megazyme International Ireland Ltd., Bray), per-O-acetylation, flash chromatography, and deprotection as previously described.(35)
Analytical Instruments
Fluoride Ion Electrode
The rate of fluoride ion release from α-glycosyl fluoride substrates was measured using an Orion ionplus combination fluoride electrode (model 96-09) coupled to a Vernier Electrode Amplifier and Lab Pro interface connected to a Windows-based computer (Vernier Software and Technology, Beaverton, OR). Data were captured, and initial kinetic rates were extracted using the Vernier Logger Pro 3 software program, as in previous studies.24,25 The reaction temperature was kept constant at 30 °C by a jacketed glass vessel and water circulation from a thermostat-controlled bath.
HPAEC-PAD
High-performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) was performed using a Dionex ICS-3000 system and a Dionex PA-200 column. A ternary solvent system was used: solvent A, ultrapure water; solvent B, 1.0 M NaOH; solvent C, 1.0 M NaOAc; flow rate, 0.5 mL/min. The gradient program was as follows: 0–4 min, 100 mM NaOH, 60 mM NaOAc; 4–17 min, linear gradient from 60 to 250 mM NaOAc; 17–18 min, gradient up to 500 mM NaOH and 500 mM NaOAc, then initial conditions for 4 min. Ten microliters of suitably diluted samples were injected.
HPSEC-ELS
High-performance size-exclusion chromatography with evaporative light-scattering detection (HPSEC-ELS) was performed with a Gynkotek M480G pump (Optimize Technologies, OR), PL-ELS 1000 ELS detector (Polymer Laboratories, CA), and two 300 × 7.5 mm PLgel 10 μm MIXED-B columns (Polymer Laboratories) coupled in series with a matching 50 mm guard column; the columns were maintained at 70 °C. The eluent was 100% DMSO (1 mL/min), and the injection volume was 100 μL. Polymer Laboratories pullulan standards were used for molar mass calibration (Mw 738–1 660 000).
MALDI-TOF-MS
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) was performed using a Biflex IV workstation (Bruker Daltonics, Bremen, Germany) equipped with a nitrogen laser (337 nm) and operated in positive mode. After a delayed extraction time of 200 ns, the ions were accelerated to a kinetic energy of 19 kV and subsequently detected in reflector mode. 0.5 μL of sample solution were mixed on a MALDI-TOF-plate (Bruker Daltonics) with 1 μL of matrix solution containing 10 mg/mL 2,5-dihydroxybenzoic acid (Sigma, Stockholm, Sweden) in acetone, followed by drying under a stream of air.
Initial Rate Enzyme Kinetics
Protein concentrations of all glycosynthase variants were determined at 595 nm by the Bradford assay(36) using the BioRad Protein Assay Kit with BSA as standard; all subsequent enzyme specific activity calculations assume 100% enzyme activity.
For the initial comparison of the three glycosynthase variants, PpXG5 E323G, E323A, and E323S, the specific activity of each in the homocondensation of XLLGαF and XLLGαF was determined from the rate of fluoride ion release at 30 °C, using substrate concentrations of 2 mM in 50 mM sodium phosphate buffer (pH 7.5) in independent experiments.
The pH optimum for the homocondensation of XLLGαF (2.0 mM) by PpXG5 E323G (11.7 μM) was determined using the following buffer systems: 50 mM sodium citrate (pKa1 = 3.06, pKa2 = 4.74, pKa3 = 5.40) was used in the pH range 2.1–6.4; 50 mM sodium phosphate (pKa1 = 7.21, pKa2 = 12.32) was used in the pH range 6.2–8.0; and 50 mM Tris (pKa = 8.06) was used in the pH range 7.5–9.0.
Subsequent determination of the apparent macroscopic kinetic parameters kcat and Km for PpXG5 E323G was performed by measuring the initial rate of fluoride ion release using XXXGαF and XLLGαF independently over the concentration range 1 μM to 6 mM in phosphate buffer (50 mM, pH 7.5). 11.7 μM PpXG5 E323G was used in each assay. The standard Michaelis–Menten equation was fit to the data using the Origin software program, v.8 (OriginLab Corp., Northampton, MA).
Preparative Scale Synthesis of Xyloglucan Homopolymers
PpXG5 E323G (11.7 μM) was incubated at 30 °C with either 2 mM XXXGαF or XLLGαF in 1 mL of phosphate buffer (50 mM, pH 7.5), with constant shaking at 150 rpm overnight. The next day, the enzyme was denatured by heating the reaction mixture at 70 °C for 15 min, and the precipitated protein was spun down (13 000 rpm, 4 °C, 15 min). The supernatant was analyzed directly, after preparing a suitable dilution in ultrapure water, by HPAEC-PAD. For HPSEC-ELS analysis, the supernatant (0.5 mL) was frozen in liquid nitrogen, lyophilized, redissolved in 0.5 mL of dimethyl sulfoxide (DMSO), and filtered through a 0.45 μm nylon centrifuge filter.
Fucosylation of XLLGαF and Glycosynthetic (XLLG)nαF Polymers with AtFUT1
Reactions were performed in 40 mM HEPES-KOH (pH 6.8) containing 0.4 mM DTT, 0.16 M sucrose, 2 mM MgCl2, 1% Triton X-100, 2 mM GDP-fucose, and acceptor substrate. In the case of XLLGαF, the acceptor concentration was 2 mM. (XLLG)nαF products were fucosylated directly from glycosynthase reactions originally containing 2 mM XLLGαF after inactivation of the glycosynthase by heating to 70 °C for 15 min. Reactions were started by addition of the microsomal preparation of AtFUT1 (total protein concentration of 3.7 mg/mL) in an amount comprising 10% (v/v) of the final reaction volume (500 μL). The reaction mixture was incubated at room temperature for different time intervals (0 min, 6 h, 17 h), and the glycosyltransferase reaction was stopped by heating at 70 °C for 15 min. For product analyses, the polysaccharides were treated with 1.5 units of Chrysosporium lucknowense endo-xyloglucanase Xgl (Xgl74A)(37) (70 680 U/25 g; Dyadic, Wageningen, NL) at room temperature overnight.
Results and Discussion
PpXG5 Glycosynthase
Protein Production
The E323G, E323A, and E323S glycosynthase variants of PpXG5 were produced in good yield from 1 L shake-flask cultivations of E. coli and subsequent purification by IMAC; ca. 50 mg of each protein was obtained (Supporting Information Table S3). SDS-PAGE analysis indicated that all three variants were obtained in high purity (Supporting Information Figure S2). ESI MS analysis (Supporting Information Figure S3) of the intact, pure proteins confirmed that each had an Mr value within one unit of that expected for the properly translated primary sequence (Supporting Information Table S3), with cleavage of the N-terminal methionine (Supporting Information Table S2).
Specific Activity
The specific activity of the three different glycosynthase variants PpXG5 E323G, E323A, and E323S in the homocondensation of both XXXGαF and XLLGαF at 2 mM was determined for comparison with values obtained for previous xyloglucan glycosynthases.24,25 As shown in Table 1, all of the PpXG5 glycosynthase variants showed significantly higher specific activities in the homocondensation of the substrate XLLGαF than the PttXET16-34 E85A or TmNXG1 E94A glycosynthases. These two latter enzymes, which were derived from a GH16 xyloglucan endo-transglycosylase (EC 2.4.1.207) and a GH16 xyloglucan endo-hydrolase (endo-xyloglucanase, EC 3.2.1.151), respectively, were the first two, and this far only, glycosynthases with significant activity toward XLLGαF reported (although neither was capable of producing high molar mass polysaccharides, vide infra). Notably, each PpXG5 glycosynthase variant exhibited a higher specific activity toward XLLGαF than XXXGαF at 2 mM, which mirrors the selectivity of the wild-type enzyme in the hydrolysis of aryl glycosides of these oligosaccharides.(27) The PpXG5 E323G variant, in particular, exhibited the highest specific activity toward XLLGαF (2–3-fold higher than the E323A and E323S variants). Moreover, the specific activity of PpXG5 E323G toward XXXGαF was only one-half of that of the proficient HiCel7B, which was previously shown to generate high molar mass nongalactosylated xyloglucan.(24) Hence, the PpXG5 E323G variant and its reaction products were analyzed in more detail.
Table 1. Specific Activity of Glycosynthases toward α-Xyloglucosyl Fluoride Substrates.
Initial Rate Kinetics
The pH optimum of PpXG5 E323G was 7.0 using 2.0 mM XLLGαF (Supporting Information Figure S4); all subsequent analyses were performed in 50 mM sodium phosphate buffer, pH 7.5, to ensure sufficient buffering capacity for HF released by glycosynthase reactions.
The enzymatic homocondensations of XXXGαF and XLLGαF by PpXG5 E323G were each measured across a range of substrate concentrations (Figure 3), and apparent macroscopic kinetic parameters were determined (Table 2). In the polymerization of XXXGαF, PpXG5 E323G had a ca. 2-fold lower catalytic efficiency than HiCel7B E197S, which is the fastest glycosynthase on this substrate reported so far.(24) Notably, this difference is primarily due to the higher kcat value of HiCel7B E197S (120 vs 70 min–1 for PpXG5 E323G), because these enzymes have similar Km values (ca. 10 mM, Table 2). PpXG5 E323G was, however, much more specific for XLLGαF (Table 2). Although the kcat value for this substrate was apparently lower than that for XXXGαF, a significantly lower Km value for XLLGαF yielded a 5-fold higher specificity value (kcat/Km) for the digalactosylated substrate. Consistent with specific activity measurements (Table 1), this detailed kinetic analysis highlights that the PpXG5 E323G glycosynthase has maintained the substrate selectivity of the wild-type hydrolase.(27)
Figure 3.

PpXG5 E323G activity on α-xyloglucosyl fluorides. (A) Initial rate kinetics of PpXG5 E323G with XXXGαF; (B) initial rate kinetics of PpXG5 E323G with XLLGαF. Error bars represent standard deviations of duplicate measurements.
Table 2. Apparent Kinetic Constants for Different Glycosynthases on α-Xyloglucosyl Fluoride Substrates.
| XXXGαF |
XLLGαF |
|||||
|---|---|---|---|---|---|---|
| enzyme | KM(app) (mM) | kcat(app) (min–1) | kcat/KM (mM–1 min–1) | KM(app) (mM) | kcat(app)(min–1) | kcat/KM (mM–1 min–1) |
| HiCel7B E197Sa | 8.2 ± 0.8 | 120 ± 10 | 15 | |||
| PttXET16-34 E85Ab | 1.6 ± 0.4 | 1.1 ± 0.1 | 0.66 | 3.8 ± 0.2 | 1.4 ± 0.04 | 0.38 |
| TmNXG1 E94Aa | 1.2 ± 0.2 | 2.6 ± 0.1 | 2.2 | 1.4 ± 0.2 | 8.2 ± 0.8 | 1.4 |
| PpXG5 E323G | 9.5 ± 2.7 | 73 ± 14 | 7.7 | 0.8 ± 0.09 | 28.9 ± 1.2 | 37 |
When compared to other XLLGαF-polymerizing glycosynthases, PpXG5 E323G is the fastest thus far (Table 2). The kcat value of PpXG5 E323G is 3.5-fold higher and the Km value is 2-fold lower as compared to the TmNXG1 E94A variant,(24) which was notably faster than the PttXET16-34 E85A variant(25) (Table 2). These glycosynthase activity differences qualitatively mirror the wild-type activities of these enzymes. PttXET16-34 is a strict xyloglucan endo-transglycosylase (EC 2.4.1.207), and it might be assumed that this protein scaffold is well-adapted to condense two xyloglucan oligosaccharides. However, wild-type PttXET16-34 has a turnover number (apparent kcat value) of ca. 5 min–1 (ref (38)), which is of the same magnitude as the PttXET16-34 E85A glycosynthase reaction(25) (Table 2). Similarly, TmNXG1 is a predominant endo-xyloglucanase with some transglycosylation activity, which is involved in the digestion of storage polysaccharide in germinating nasturtium (Tropaeolum majus) seeds.(38) Here again, kcat values for hydrolysis and transglycosylation by the wild-type are <5 min–1, which are in the same range as the best TmNXG1 glycosynthase variant, E94A (ref (24)) (Table 2). The bacterial PpXG5 is a significantly faster hydrolase than TmNXG1 (by approximately 3 orders of magnitude on tamarind xyloglucan or XLLG-2-chloro-4-nitrophenyl glycoside(39)); however, it is clear that not all of this catalytic potential is directly translated to glycosynthase efficiency. The reasons for this are unresolved, but may involve nonoptimized binding in the positive enzyme subsites. Indeed, wild-type PpXG5 does not appear to catalyze transglycosylation reactions using the activated substrate XLLG-2-chloro-4-nitrophenyl glycoside, which causes accumulation of the glycosyl-enzyme intermediate.(27)
Preparative Scale Synthesis of (XXXG)n and (XLLG)n Homopolymers
Encouraged by the high apparent glycosynthase kinetics of PpXG5 E323G, the ability of the enzyme to produce high molar mass xyloglucan homopolymers was tested. Thus, PpXG5 E323G was incubated with either 2 mM XXXGαF or XLLGαF at room temperature overnight followed by product analysis with HPAEC-PAD and HPSEC-ELS. HPAEC-PAD indicated that multiple (XXXG)nαF and (XLLG)nαF coupling products were observed for both substrates, with n ranging from 2 to over 10 in each case. As HPAEC-PAD suffers from decreasing sensitivity with increasing degrees of polymerization, and because of product mixtures resulting from the decomposition of the α-glycosyl fluorides under the high pH chromatography conditions,24,25 the upper limit of n was difficult to estimate for both products (Supporting Information Figure S5). For more reliable product analysis, the overnight glycosynthase reactions with 2 mM XXXGαF and XLLGαF were repeated at 30 °C, followed by HPSEC-ELS versus pullulan molar mass standards (Figure 4).
Figure 4.

HPSEC-ELS analysis of the PpXG5 E323G glycosynthase reactions with XXXGαF or XLLGαF as substrates. Dashed line, XXXGαF products formed after overnight incubation at 30 °C; solid line, XLLGαF products formed after overnight incubation at 30 °C. The eluent was DMSO, and molar mass calibration was performed with pullulan standards.
Both pullulans and native xyloglucans behave as random coils in aqueous solution.40,41 A good correlation of molar mass values between the standards and samples was found in the low molar mass range by HPSEC using DMSO as an eluent (Figure 4): XXXGαF (calcd 1065, obs 1100), XLLGαF (calcd 1389, obs 1280), and dimer XXXGXXXGαF (calcd 2110, obs 2250). Extrapolating this correlation, the (XXXG)nαF products had an Mw value of 7000 with a polydispersity index (PDI, Mw/Mn) of 1.6; the highest molar masses observed (<1% of the products) approached 30 000. These values correspond to values of n = 7 at the peak of the chromatogram and n = 29 for the longest XXXG-based products, assuming a molar mass of the repeating unit of 1045.
No product precipitation was apparent in these reactions, in contrast to previous observations with the HiCel7B E197S glycosynthase working under identical substrate concentration and buffer composition. HiCel7B E197S produced (XXXG)nαF products with molar masses up to 60 000 (n = 57), but due to limited solubility of the nongalactosylated polymer, products with molar masses larger than ca. 20 000 (n = 20) precipitated from aqueous solution (but could be dissolved in DMSO for HPSEC-ELS analysis).(24) Clearly, the precipitation of longer polysaccharides depends both on glycosynthase kinetics and the total amount of high mass products formed18,22,42−44 as well as on the branching pattern (vide infra).
The XLLGαF reaction contained notably lower amounts of starting material and dimers than was observed with XXXGαF, consistent with the faster initial rate kinetics observed for the former substrate (Figure 3, Table 2). Indeed, the (XLLG)nαF products had a higher Mw value of 12 000 (PDI, Mw/Mn 1.45), and polymers were observed with molar masses approaching 60 000 (<1% of the products, Figure 4). These values correspond to values of n = 9 at the chromatographic peak and n = 44 for the longest XLLG-based products (repeat molar mass 1369). The largest (XLLG)nαF products previously obtained, produced using the TmNXG1 E94A glycosynthase, were limited to oligo- and polysaccharides with n ≤ 8.(24) Thus, the PpXG5 E323G variant is the first glycosynthase capable of producing significant amounts of homogenously digalactosylated xyloglucan polysaccharides with high molar masses.
Production of High Molar Mass Fucosylated Xyloglucans Using the α(1→2)-Fucosyltransferase from Arabidopsis thaliana (AtFUT1)
The glycosyltransferase family GT37 α(1→2)-fucosyltransferase from Arabidopsis thaliana (AtFUT1, UniProt Q9SWH5) catalyzes the regiospecific addition of an l-fucosyl moiety to XXLG and XLLG oligosaccharides in xyloglucans to produce XXFG and XLFG units, respectively, using GDP-l-fucose as a donor substrate(45) (see Figure 1 for oligosaccharide structures). Building upon the demonstrated ability of PpXG5 E323G to assemble high mass (XLLG)n xyloglucans, we turned our attention toward extending the glycosynthase reaction to build homogenously fucosylated xyloglucans with a repeating (XLFG)n structure. We originally envisioned a route involving the fucosylation of (XLLG)αF (Figure 1) using AtFUT1 to generate (XLFG)αF building blocks for the subsequent assembly of homogenously branched fucogalactoxyloglucans by the glycosynthase. However, preliminary experiments indicated that a microsomal preparation of AtFUT1 was essentially inactive on (XLLG)αF (data not shown). These results were consistent with previous results on the homologue PsFT1 (62% sequence identity) from pea (Pisum sativum), which exhibited maximum activity on seed galactoxyloglucans, significantly lower activity on Glc12-based xyloglucan oligosaccharides, and essentially no activity on Glc4-based xyloglucan oligosaccharides.(34)
Consequently, the ability of AtFUT1 to directly fucosylate the high molar mass (XLLG)nαF glycosynthase products was explored. AtFUT1 was incubated with the polysaccharide and GDP-l-Fuc, and, following heat inactivation and removal of the glycosyltransferase, the product was digested with the glycoside hydrolase family GH74 endo-xyloglucanase Xgl from Chrysosporium lucknowense (ClXgl74A). This enzyme cleaves xyloglucans specifically to produce Glc4-based oligosaccharides with an unbranched Glc residue (G, Figure 1) at their reducing ends,(37) thus allowing relative quantitation of component oligosaccharides to be obtained by MALDI-TOF MS analysis.(46) As shown in Figure 5, ClXgl74A treatment of the original (XLLG)nαF glycosynthase products yielded only XLLG, whereas after 6 and 17 h incubation, increasing amounts of XLFG units were obtained in the polysaccharide, approaching ca. 75% of the oligosaccharide repeats present (relative abundance based on MALDI-TOF MS peak heights, assuming identical ionization efficiencies, as previously described(46)). We assume that the distribution of fucosylation sites along the polysaccharide is random, although, due to technical limitations in polysaccharide sequencing, this is essentially impossible to examine at present.
Figure 5.

Fucosylation of (XLLG)n-based xyloglucans by AtFUT1. MALDI-TOF MS of pure XLLG standard (A) and endo-xyloglucanase limit digests of (XLLG)nαF (Mw 12 000, Mw/Mn 1.45) incubated with AtFUT1 and GDP-l-Fuc for 0 min (B), 6 h (C), and 17 h (D).
These results highlight the current challenges and limitations of using glycosyltransferases in the synthesis of complex oligo- and polysaccharides. Although kinetic parameters for AtFUT1 are presently lacking, the Km value for GDP-l-Fuc of the homologue PsFT1 is ca. 30 μM (ref (34)), and indeed Km values in the 10 μM range are typical of NDP-sugars for other glycosyltransferases (e.g., ref (47)). Considering the acceptor substrate, PsFT1 has a Km value of 0.4 μM (0.46 g/L) for tamarind xyloglucan,(34) which is equivalent to a galactose-equivalent Km value of 300 μM (0.3 mM), assuming that ca. 75% of the oligosaccharides are XXLG and XLLG(35) (fucosylation site underlined(45)). Thus, in the conditions used herein for the synthesis of fucosylated xyloglucans (2 mM donor and acceptor), the donor substrate would likely have been maintained in saturating conditions at the ca. 75% conversion obtained after 17 h (Figure 5), while the concentration of acceptor galactosyl units would have been approximately equivalent to the Km value at this time (i.e., the rate would have only dropped to ca. 50% of Vmax). It is presently unclear, due to the lack of specific kinetic parameters for (XXLG)n and (XLLG)n polysaccharides, how acceptor substrate structure might affect the efficiency of AtFUT1, although the potential of PpXG5 E323G to make xyloglucan with homogenously monogalactosylated repeats could allow more in-depth analysis of this and related glycosyltransferases.
This analysis highlights that the inability to produce completely fucosylated (XLFG)n xyloglucans (as found, e.g., in wild-type Arabidopsis thaliana(48)) was due to the limited activity of the AtFUT1 preparation. In the present study, AtFUT1 was produced recombinantly in the yeast Pichia pastoris and extracted as a crude microsomal preparation that had limited storage stability (data not shown). Previously, AtFUT1 has been obtained in low levels from the technically more complex COS(28) and 293T(29) mammalian cell expression systems, which further underscores the difficulty in obtaining plant glycosyltransferases for preparative work.(49) Moreover, the cost of the donor substrate is also a significant concern: The largest 0.5 mL reactions run in this study required ca. 0.5 mg of GDP-l-Fuc at a current price of 150 Euro/mg to produce ca. 1.5 mg of polysaccharide. Although continuing developments in the area of chemo-enzymatic nucleotide sugar synthesis will likely reduce the cost of GDP-l-Fuc to some extent in the future,(50) there is a clear opportunity for the further development of glycosynthases (“α-l-fucosynthases”51,52) for the regiospecific addition of fucose to well-defined xyloglucan oligo- and polysaccharides.
Conclusion
The glycosynthase variant PpXG5 E323G from Paenibacillus pabuli has been shown to possess a uniquely high catalytic activity toward (XLLG)αF, and consequently is the first glycosynthase capable of producing digalactosylated xyloglucans with molar mass values up to 60 000 (Mw 12 000). From these homogenously branched polysaccharides, it was subsequently possible to produce fucosylated xyloglucans with a XLFG to XLLG ratio of 3:1 utilizing the GT37 Arabidopsis thaliana α(1→2)-fucosyltransferase, AtFUT1. Together with the HiCel7B E197S glycosynthase,(24) these enzymes comprise a toolkit for the synthesis of XyG variants with well-defined branching patterns. An inherent limitation of the method, however, is that such products are not monodisperse, but rather a mixture of polysaccharides of various backbone lengths, which are practically challenging to fractionate. Nonetheless, the ability to produce regularly substituted polysaccharides, which is otherwise difficult to achieve, will facilitate future structure–function studies of xyloglucans in the context of their interaction with cellulose and other biomolecules.13,53,54
Acknowledgments
The Knut and Alice Wallenberg Foundation is thanked for funding via the Wallenberg Wood Science Center (http://wwsc.se/) to O.S., C.X., and H.B. H.B. is a Special Research Fellow (Rådsforskare) of the Swedish Research Council (Vetenskapsrådet); project funding from Vetenskapsrådet, Formas (via CarboMat, http://www.biotech.kth.se/glycoscience/CarboMat), and the Swedish Foundation for Strategic Research (via Biomime, http://www.biomime.org/) is also gratefully acknowledged. Work in York was supported by the Biotechnology and Biological Sciences Research Council (BBSRC); G.J.D. is a Royal Society/Wolfson Research Merit Award recipient. We are grateful to Prof. Kenneth Keegstra (Michigan State University, MI) for providing the Pichia pastoris AtFUT1 expression clone.
Supporting Information Available
Complete author list for ref (55); three tables and five figures of supporting data: Table S1, oligonucleotide primer sequences; Table S2, primary structures of the PpXG5 glycosynthase variants; Table S3, yield and Mr values of the PpXG5 E323G, E323A, and E323S glycosynthase variants; Figure S1, SDS-PAGE analysis of the microsomal preparation from a recombinant P. pastoris clone expressing the α(1→2)-fucosyltransferase from Arabidopsis thaliana, AtFUT1; Figure S2, SDS-PAGE analysis of recombinant PpXG5 E323G, E323A, and E323S variants produced in Escherichia coli; Figure S3, electrospray mass spectra of PpXG5 glycosynthase variants; Figure S4, pH rate profile of PpXG5 E323G; and Figure S5, HPAEC-PAD product analysis of PpXG5 E323G glycosynthase reactions. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ Petersburg Nuclear Physics Institute, Russian Academy of Science, Molecular and Radiation Biology Division, Gatchina, St. Petersburg 188300, Russia.
Supplementary Material
References
- Vogel J. Curr. Opin. Plant Biol. 2008, 11, 301–307. [DOI] [PubMed] [Google Scholar]
- Peña M. J.; Darvill A. G.; Eberhard S.; York W. S.; O’Neill M. A. Glycobiology 2008, 18, 891–904. [DOI] [PubMed] [Google Scholar]
- Hoffman M.; Jia Z. H.; Pena M. J.; Cash M.; Harper A.; Blackburn A. R.; Darvill A.; York W. S. Carbohydr. Res. 2005, 340, 1826–1840. [DOI] [PubMed] [Google Scholar]
- Hsieh Y. S. Y.; Harris P. J. Mol. Plant 2009, 2, 943–965. [DOI] [PubMed] [Google Scholar]
- Brumer H.Enzymatic functionalization of cellulosic fibres: Xyloglucan as a molecular anchor In Advances in Textile Biotechnology; Nierstrasz V., Cavaco-Paulo A., Eds.; Woodhead Publishing: Cambridge, 2010; pp 266–287. [Google Scholar]
- Zhou Q.; Rutland M. W.; Teeri T. T.; Brumer H. Cellulose 2007, 14, 625–641. [Google Scholar]
- Mishra A.; Malhotra A. V. J. Mater. Chem. 2009, 19, 8528–8536. [Google Scholar]
- Kochumalayil J.; Sehaqui H.; Zhou Q.; Berglund L. A. J. Mater. Chem. 2010, 20, 4321–4327. [Google Scholar]
- Yamatoya K.; Shirakawa M. Curr. Trends Polym. Sci. 2003, 8, 27–72. [Google Scholar]
- Shirakawa M.; Yamatoya K.; Nishinari K. Food Hydrocolloids 1998, 12, 25–28. [Google Scholar]
- de Lima D. U.; Buckeridge M. S. Carbohydr. Polym. 2001, 46, 157–163. [Google Scholar]
- Lima D. U.; Loh W.; Buckeridge M. S. Plant Physiol. Biochem. 2004, 42, 389–394. [DOI] [PubMed] [Google Scholar]
- Lopez M.; Bizot H.; Chambat G.; Marais M. F.; Zykwinska A.; Ralet M. C.; Driguez H.; Buleon A. Biomacromolecules 2010, 11, 1417–1428. [DOI] [PubMed] [Google Scholar]
- Hanus J.; Mazeau K. Biopolymers 2006, 82, 59–73. [DOI] [PubMed] [Google Scholar]
- Mackenzie L. F.; Wang Q. P.; Warren R. A. J.; Withers S. G. J. Am. Chem. Soc. 1998, 120, 5583–5584. [Google Scholar]
- Perugino G.; Trincone A.; Rossi M.; Moracci M. Trends Biotechnol. 2004, 22, 31–37. [DOI] [PubMed] [Google Scholar]
- Wang L. X.; Huang W. Curr. Opin. Chem. Biol. 2009, 13, 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faijes M.; Planas A. Carbohydr. Res. 2007, 342, 1581–94. [DOI] [PubMed] [Google Scholar]
- Ben-David A.; Bravman T.; Balazs Y. S.; Czjzek M.; Schomburg D.; Shoham G.; Shoham Y. ChemBioChem 2007, 8, 2145–2151. [DOI] [PubMed] [Google Scholar]
- Hancock S. M.; Vaughan M. D.; Withers S. G. Curr. Opin. Chem. Biol. 2006, 10, 509–19. [DOI] [PubMed] [Google Scholar]
- Perugino G.; Cobucci-Ponzano B.; Rossi M.; Moracci M. Adv. Synth. Catal. 2005, 347, 941–950. [Google Scholar]
- Pérez X.; Faijes M.; Planas A. Biomacromolecules 2011, 12, 494–501. [DOI] [PubMed] [Google Scholar]
- Faure R.; Saura-Valls M.; Brumer H.; Planas A.; Cottaz S.; Driguez H. J. Org. Chem. 2006, 71, 5151–5161. [DOI] [PubMed] [Google Scholar]
- Gullfot F.; Ibatullin F. M.; Sundqvist G.; Davies G. J.; Brumer H. Biomacromolecules 2009, 10, 1782–1788. [DOI] [PubMed] [Google Scholar]
- Piens K.; Henriksson A. M.; Gullfot F.; Lopez M.; Faure R.; Ibatullin F. M.; Teeri T. T.; Driguez H.; Brumer H. Org. Biomol. Chem. 2007, 5, 3971–3978. [DOI] [PubMed] [Google Scholar]
- Faure R.; Cavalier D.; Keegstra K.; Cottaz S.; Driguez H. Eur. J. Org. Chem. 2007, 26, 4313–4319. [Google Scholar]
- Gloster T. M.; Ibatullin F. M.; Macauley K.; Eklof J. M.; Roberts S.; Turkenburg J. P.; Bjornvad M. E.; Jorgensen P. L.; Danielsen S.; Johansen K. S.; Borchert T. V.; Wilson K. S.; Brumer H.; Davies G. J. J. Biol. Chem. 2007, 282, 19177–89. [DOI] [PubMed] [Google Scholar]
- Perrin R. M.; DeRocher A. E.; Bar-Peled M.; Zeng W.; Norambuena L.; Orellana A.; Raikhel N. V.; Keegstra K. Science 1999, 284, 1976–9. [DOI] [PubMed] [Google Scholar]
- Vanzin G. F.; Madson M.; Carpita N. C.; Raikhel N. V.; Keegstra K.; Reiter W. D. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 3340–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel B. L.; Coutinho P. M.; Rancurel C.; Bernard T.; Lombard V.; Henrissat B. Nucleic Acids Res. 2009, 37, D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. L.; Wilkinson M. F. BioTechniques 1997, 23, 588–590. [DOI] [PubMed] [Google Scholar]
- Sundqvist G.; Stenvall M.; Berglund H.; Ottosson J.; Brumer H. J. Chromatogr., B 2007, 852, 188–194. [DOI] [PubMed] [Google Scholar]
- Davis J.; Brandizzi F.; Liepman A. H.; Keegstra K. Plant J. 2010, 64, 1028–1037. [DOI] [PubMed] [Google Scholar]
- Faik A.; Bar-Peled M.; DeRocher A. E.; Zeng W.; Perrin R. M.; Wilkerson C.; Raikhel N. V.; Keegstra K. J. Biol. Chem. 2000, 275, 15082–9. [DOI] [PubMed] [Google Scholar]
- Greffe L.; Bessueille L.; Bulone V.; Brumer H. Glycobiology 2005, 15, 437–445. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. Anal. Biochem. 1976, 72, 248–54. [DOI] [PubMed] [Google Scholar]
- Grishutin S. G.; Gusakov A. V.; Markov A. V.; Ustinov B. B.; Semenova M. V.; Sinitsyn A. P. Biochim. Biophys. Acta, Gen. Subj. 2004, 1674, 268–281. [DOI] [PubMed] [Google Scholar]
- Baumann M. J.; Eklöf J. M.; Michel G.; Kallas Å. M.; Teeri T. T.; Czjzek M.; Brumer H. Plant Cell 2007, 19, 1947–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibatullin F. M.; Baumann M. J.; Greffe L.; Brumer H. Biochemistry 2008, 47, 7762–7769. [DOI] [PubMed] [Google Scholar]
- Nishinari K.; Kohyama K.; Williams P. A.; Phillips G. O.; Burchard W.; Ogino K. Macromolecules 1991, 24, 5590–5593. [Google Scholar]
- Ren Y. L.; Picout D. R.; Ellis P. R.; Ross-Murphy S. B. Biomacromolecules 2004, 5, 2384–2391. [DOI] [PubMed] [Google Scholar]
- Faijes M.; Ima T.; Bulone V.; Planas A. Biochem. J. 2004, 380, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort S.; Boyer V.; Greffe L.; Davies G.; Moroz O.; Christiansen L.; Schulein M.; Cottaz S.; Driguez H. J. Am. Chem. Soc. 2000, 122, 5429–5437. [Google Scholar]
- Hrmova M.; Imai T.; Rutten S. J.; Fairweather J. K.; Pelosi L.; Bulone V.; Driguez H.; Fincher G. B. J. Biol. Chem. 2002, 277, 30102–30111. [DOI] [PubMed] [Google Scholar]
- Perrin R. M.; Jia Z. H.; Wagner T. A.; O’Neill M. A.; Sarria R.; York W. S.; Raikhel N. V.; Keegstra K. Plant Physiol. 2003, 132, 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerouxel O.; Choo T. S.; Seveno M.; Usadel B.; Faye L.; Lerouge P.; Pauly M. Plant Physiol. 2002, 130, 1754–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly H. D.; Lougheed B.; Wakarchuk W. W.; Withers S. G. Biochemistry 2002, 41, 5075–5085. [DOI] [PubMed] [Google Scholar]
- Pauly M.; Eberhard S.; Albersheim P.; Darvill A.; York W. S. Planta 2001, 214, 67–74. [DOI] [PubMed] [Google Scholar]
- Petersen B. L.; Egelund J.; Damager I.; Faber K.; Jensen J. K.; Yang Z.; Bennett E. P.; Scheller H. V.; Ulvskov P. Glycoconjugate J. 2009, 26, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G. H.; Guan W. Y.; Cai L.; Wang P. G. Nat. Protoc. 2010, 5, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobucci-Ponzano B.; Conte F.; Bedini E.; Corsaro M. M.; Parrilli M.; Sulzenbacher G.; Lipski A.; Dal Piaz F.; Lepore L.; Rossi M.; Moracci M. Chem. Biol. 2009, 16, 1097–1108. [DOI] [PubMed] [Google Scholar]
- Wada J.; Honda Y.; Nagae M.; Kato R.; Wakatsuki S.; Katayama T.; Taniguchi H.; Kumagai H.; Kitaoka M.; Yamamoto K. FEBS Lett. 2008, 582, 3739–3743. [DOI] [PubMed] [Google Scholar]
- Nordgren N.; Eklof J.; Zhou Q.; Brumer H.; Rutland M. W. Biomacromolecules 2008, 9, 942–948. [DOI] [PubMed] [Google Scholar]
- Stiernstedt J.; Brumer H.; Zhou Q.; Teeri T. T.; Rutland M. W. Biomacromolecules 2006, 7, 2147–2153. [DOI] [PubMed] [Google Scholar]
- Fry S. C.; et al. Physiol. Plant. 1993, 89, 1–3. [Google Scholar]
- Marry M.; Cavalier D. M.; Schnurr J. K.; Netland J.; Yang Z. Y.; Pezeshk V.; York W. S.; Pauly M.; White A. R. Carbohydr. Polym. 2003, 51, 347–356. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.