

Abstract
Acute and chronic lung inflammation is associated with numerous important disease pathologies including asthma, chronic obstructive pulmonary disease and silicosis. Lung fibroblasts are a novel and important target of anti-inflammatory therapy, as they orchestrate, respond to, and amplify inflammatory cascades and are the key cell in the pathogenesis of lung fibrosis. Peroxisome proliferator-activated receptor gamma (PPARγ) ligands are small molecules that induce anti-inflammatory responses in a variety of tissues. Here, we report for the first time that PPARγ ligands have potent anti-inflammatory effects on human lung fibroblasts. 2-cyano-3, 12-dioxoolean-1, 9-dien-28-oic acid (CDDO) and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) inhibit production of the inflammatory mediators interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), COX-2, and prostaglandin (PG)E2 in primary human lung fibroblasts stimulated with either IL-1β or silica. The anti-inflammatory properties of these molecules are not blocked by the PPARγ antagonist GW9662 and thus are largely PPARγ independent. However, they are dependent on the presence of an electrophilic carbon. CDDO and 15d-PGJ2, but not rosiglitazone, inhibited NF-κB activity. These results demonstrate that CDDO and 15d-PGJ2 are potent attenuators of proinflammatory responses in lung fibroblasts and suggest that these molecules should be explored as the basis for novel, targeted anti-inflammatory therapies in the lung and other organs.
1. Introduction
Inflammation is associated with many diseases of the lung and can result from immunologic injury, infection, and inhalation of particulate matter. Diseases strongly associated with pulmonary inflammation include asthma, chronic obstructive pulmonary disease (COPD), and silicosis. Inflammation is also associated with an increased susceptibility to developing lung cancers and other malignancies [1–4]. Aside from glucocorticoids, few effective anti-inflammatory agents exist. In this regard, it is important to investigate new anti-inflammatory targets.
Peroxisome proliferator-activated receptor gamma (PPARγ) has emerged as an important potential anti-inflammatory target. PPARγ is a ligand-activated nuclear receptor that binds a variety of endogenous lipids and lipid-derived compounds. Upon ligand binding, PPARγ heterodimerizes with the retinoid X receptor (RXR), translocates to the nucleus, and regulates expression of genes containing PPARγ response elements (PPREs) [5]. PPARγ ligands regulate key cellular processes including differentiation, proliferation, adipogenesis, and insulin sensitization [6, 7]. PPARγ ligands have also been shown to attenuate inflammation in many tissues including skin, liver, kidney, and lung [8–11]. Several types of natural PPARγ ligands exist, including prostaglandins such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), and fatty acids such as lysophosphatidic acid and 15S-hydroxyeicosatetraenoic acid [12–15]. The thiazolidinedione (TZD) class of drugs, including rosiglitazone and pioglitazone, are synthetic PPARγ agonists that are used as insulin sensitizers for type II diabetes [16, 17]. 2-cyano-3, 12-dioxoolean-1, 9-dien-28-oic acid (CDDO) is a novel synthetic triterpenoid PPARγ ligand currently in clinical trials for treatment of several different forms of cancer [18, 19].
Rosiglitazone, CDDO and 15d-PGJ2 all tightly bind to PPARγ [12, 20, 21], activate PPARγ-dependent transcription [22, 23], and regulate adipogenesis via a PPARγ-dependent mechanism [12, 24, 25]. However, CDDO and 15d-PGJ2 also have effects which can be mediated through PPARγ-independent pathways [26–28]. 15d-PGJ2 and CDDO contain α/β-unsaturated ketone rings with electrophilic carbons susceptible to forming covalent bonds with cellular proteins through Michaels addition reactions [18, 29, 30]. Rosiglitazone and other synthetic TZDs lack these electrophilic centers. We have recently demonstrated the importance of these electrophilic carbons in preventing TGF-β-induced myofibroblast differentiation [26]. Understanding the molecular pathways modulated by PPARγ ligands will shed light on their potential therapeutic contribution(s) in the control of pulmonary inflammation.
In addition to their structural role, fibroblasts in the lung act as sentinel cells with significant effector roles in orchestrating and amplifying inflammatory cascades. They become activated when exposed to inflammatory stimuli and produce inflammatory mediators such as IL-6, monocyte chemoattractant protein-1 (MCP-1), cyclooxygenase-2 (COX-2), and PGE2 [31–34]. We hypothesized that PPARγ ligands would exhibit anti-inflammatory effects in human lung fibroblasts, and tested this hypothesis using IL-1β, a potent proinflammatory cytokines, and silica, an inhaled particulate with strong pro-inflammatory effects on lung fibroblasts [32, 33]. Here, we report for the first time that PPARγ ligands inhibit the inflammatory response of human lung fibroblasts, and do so via a largely PPARγ-independent pathway dependent on a strong electrophilic center.
2. Material and Methods
2.1. Cells and Cell Culture
Primary human lung fibroblasts were derived from tissue explants obtained from patients undergoing surgical resection for benign hamartoma. This is an abnormal but noncancerous growth within the lung, it is not an inflammatory or fibrotic disease. The tissue pieces used to obtain the fibroblasts were taken from a region of the resected tissue that was most distal to the hamartoma that was anatomically and histologically normal [35]. Patient samples were obtained with approval of the Institutional Review Board of the University of Rochester. These cells are morphologically consistent with fibroblasts [36]. They express collagen and vimentin, and they do not express CD45, factor VIII, or cytokeratin. Fibroblasts were cultured in minimum essential media (MEM, Life Technologies, Gaithersburg, Md, USA) supplemented with 10% fetal bovine serum (FBS, Sigma Aldrich, St. Louis, Mo, USA), 2 mM L-glutamine, penicillin (100 units/mL), streptomycin (100 μg/mL), and amphotericin (0.25 μg/mL) (Life Technologies) at 37°C in 7% CO2, as previously described [26]. Cells were used at passages 6–12.
2.2. Reagents
PPARγ agonists and related compounds rosiglitazone, 9,10-dihydro-15-deoxy-Δ12,14-PGJ2 (CAY10410), GW9662, and prostaglandin-A1 (PGA1) were from Cayman Chemical (Ann Arbor, MI). 2-cyano-3, 12-dioxoolean-1, 9-dien-28-oic acid (CDDO) was obtained from the NIH-RAID Program and Reata Pharmaceuticals (Dallas, Tex, USA) and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) was from Biomol (Plymouth Meeting, PA). These compounds were prepared as 10 mM stocks in DMSO and added to cell cultures to the final concentrations indicated. Control wells (media only) were supplemented with DMSO to the same final concentration (0.2%) as treated wells. One hour after pretreatment with PPARγ ligands, inflammatory stimulants interleukin-1β (IL-1β, R&D Systems, Minneapolis, Minn, USA) and amorphous silica (a generous gift of Dr. David R. Hemenway, University of Vermont) were added to the cell cultures at the final concentrations indicated for 24 hours. Amorphous silica was prepared by baking for 2 hours at 180°C prior to addition to MEM.
2.3. Cytotoxicity Assays
Cell viability was assessed by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay [37]. Fibroblasts were plated in triplicate at a density of 5,000 cells per well in 96 well plates and treated with TGF-β and PPARγ agonists for 24 h at the indicated concentrations. MTT was added for the final 4 hours. Production of the colored reaction product was measured at 560 nm, and the results were normalized to the negative control wells. LDH activity in culture medium was measured by a commercial assay (Sigma).
2.4. Prostaglandin and Cytokine Assays
Primary human lung fibroblasts (100,000 cells/well) were plated in six-well plates (Falcon/Becton Dickson, Franklin Lakes, Nj, USA), serum starved for 48 hours, and treated with IL-1β or silica and/or PPARγ agonists as described. PGE2 was measured in harvested supernatants using a commercially available competitive enzyme immunoassay (EIA) (Cayman Chemical) [38]. IL-6 and CCL2/MCP-1 in harvested supernatants were determined by ELISA according to the manufacturer's instructions (R&D Systems, Minneapolis, Minn, USA).
2.5. Western Blots for Cyclooxygenase-2 (COX-2)
Total cellular protein extracts were prepared from lung fibroblast cultures with 10% Nonidet P-40 (NP-40) lysis buffer supplemented with a protease inhibitor cocktail (Sigma). Lysates were clarified by centrifugation and proteins were quantitated by the bicinchoninic acid (Pierce, Rockford, IL). Typically, 5 μg of total solubilized cellular protein was separated by 10% SDS-PAGE under reducing conditions and transferred to nitrocellulose membranes. Blots were probed with a COX-2 specific monoclonal antibody (Cayman Chemical) using GAPDH (Abcam, Cambridge, Mass, USA) as a loading control. Proteins were visualized with enhanced chemiluminescence (Western Lightning, Perkin-Elmer, Wellesley, MA) [38] and densitometry of the resulting bands was performed using Kodak Molecular Imaging Software (Rochester, NY) normalized to the loading control.
2.6. COX-2 Immununfluorescence
Fibroblasts were cultured in eight-well chamber slides (5 × 104 cells/well) in serum-free MEM for 48 hours before treatment. Some cells were pretreated with PPARγ agonists for one hour prior to IL-1β (1 ng/mL) treatment for the indicated duration. Cells maintained in serum-free MEM were used as negative controls. Cells were washed in PBS and fixed and permeabilized for 15 minutes at room temperature in BD Cytofix/Cytoperm solution (BD Biosciences, San Diego, Calif, USA). The cells were then washed with BD Perm/Wash buffer (BD Biosciences) in this step and all future washes to maintain permeabilization. Nonspecific sites were blocked with 5% normal goat serum (Life Technologies) in BD Perm/Wash buffer for 15 minutes at room temperature or 4°C overnight. Monoclonal mouse anti-human COX-2 antibody was diluted in 1% normal goat serum in BD Perm/Wash buffer and incubated with the cells overnight at 4°C. Cells were washed and Alexa Fluor 488 (green-) tagged goat anti-mouse IgG (Invitrogen) diluted in 1% normal goat serum in BD Perm/Wash buffer was added to the cells for one hour at room temperature. Cells were washed a coverslipped with ProLong Antifade with DAPI (Invitrogen).
2.7. NF-κB Luciferase Assay
We generated human lung fibroblast strains that stably expressed a mammalian codon-optimized firefly luciferase reporter gene under the transcriptional control of NF-κB response elements using NF-κB Cignal Lenti Reporter lentiviral particles (SABiosciences Corporation, Frederick, Md, USA) at an MOI = 25. Two days post-infection, growth medium was removed and replaced with growth medium containing 1 μg/mL puromycin. Puromycin-resistant clones were identified and expanded for propagation. To investigate NF-κB activity, primary human lung fibroblasts expressing NF-κB-Luc (3,000 cells/well) were plated in 96-well plates, serum starved for 48 hours and treated with IL-1β and/or PPARγ agonists as described. Cells were lysed in 1x Passive Lysis Buffer and mixed with Luciferase Assay Reagent II as instructed by the manufacturer (Promega, Madison, Wis, USA). Equal protein quantities were used in luciferase assays; results were reported in relative light units.
2.8. Statistics
All data are expressed as mean ± SD. A one-way analysis of variance (ANOVA) with Tukey post-test were used to establish statistical significance. Results were considered significant if P < .05.
3. Results
3.1. PPARγ Ligands Inhibit IL-1β-Induced Inflammatory Cytokine Production in Human Lung Fibroblasts
To determine the efficacy of selected PPARγ ligands in inhibiting production of inflammatory mediators in lung fibroblasts, primary human lung fibroblasts were pretreated with rosiglitazone, CDDO, or 15d-PGJ2 for 1 hour and then co-treated with a powerful pro-inflammatory stimulus, IL-1β (1 ng/mL), for 24 hours. IL-1β strongly induced the pro-inflammatory mediators IL-6 and MCP-1 (Figure 1). Rosiglitazone, CDDO, and 15d-PGJ2 all show dose-dependent inhibition of cytokine production and significantly inhibited the release of these mediators. We also investigated an alternative inflammatory stimulus, crystalline silica, which we have previously reported is a potent pro-inflammatory stimulus in human lung fibroblasts [32]. Silica also strongly induced the production of IL-6 and MCP-1, which was inhibited by the PPARγ ligands in a dose-dependent manner (Figures 1(c) and 1(d)). Interestingly, all 3 ligands were 4-5-fold more effective at blocking MCP-1 than IL-6 (Figure 1(e)).
Figure 1.
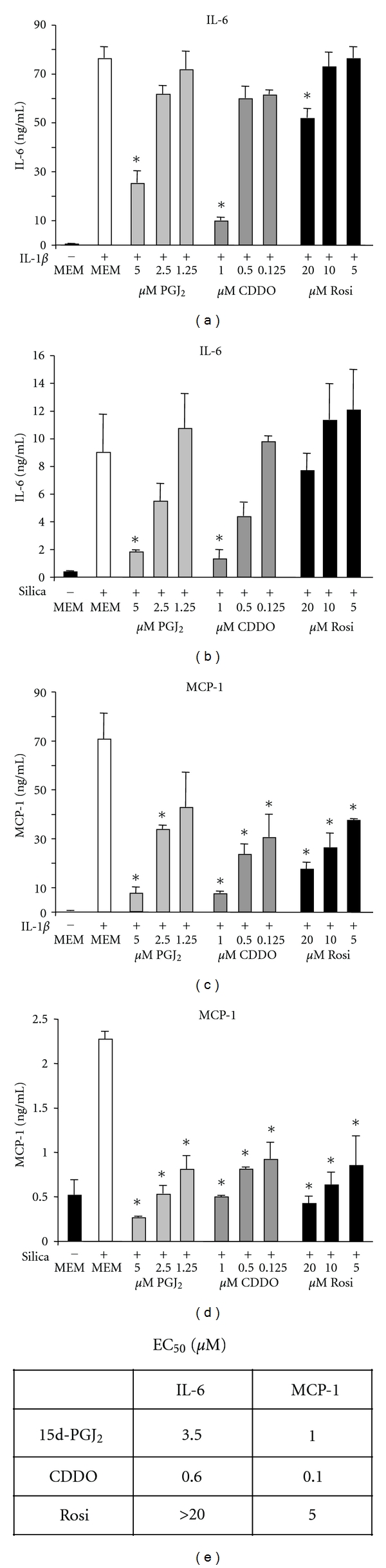
PPARγ ligands attenuate IL-6 and MCP-1 production by human lung fibroblasts induced by IL-1β or crystalline silica. Primary human lung fibroblasts were pretreated with the indicated concentrations of rosiglitazone (Rosi), CDDO or 15d-PGJ2 (PGJ2) for 1 hour and then cotreated with 1 ng/mL IL-1β (a, b) or 10 μg/mL crystalline silica (c, d) for 24 hours. Supernatants were harvested and cytokine concentrations were determined by ELISA. Results are mean ± standard error for quadruplicate wells and are representative of 2 independent experiments that yielded similar results. (e) The EC50 was determined from the given data and averaged for each cytokine. Data were analyzed by one-way ANOVA. * = P < .05 compared to stimulus alone (MEM).
It should be noted that the maximum dose used for each compound is the highest dose that does not cause overt cyotoxicity (Figure 2 and data not shown). Rosiglitazone is at least 10-fold less effective than CDDO or 15d-PGJ2 and is a very poor inhibitor of IL-6 release even at the maximum tolerable dose of 20 μM.
Figure 2.
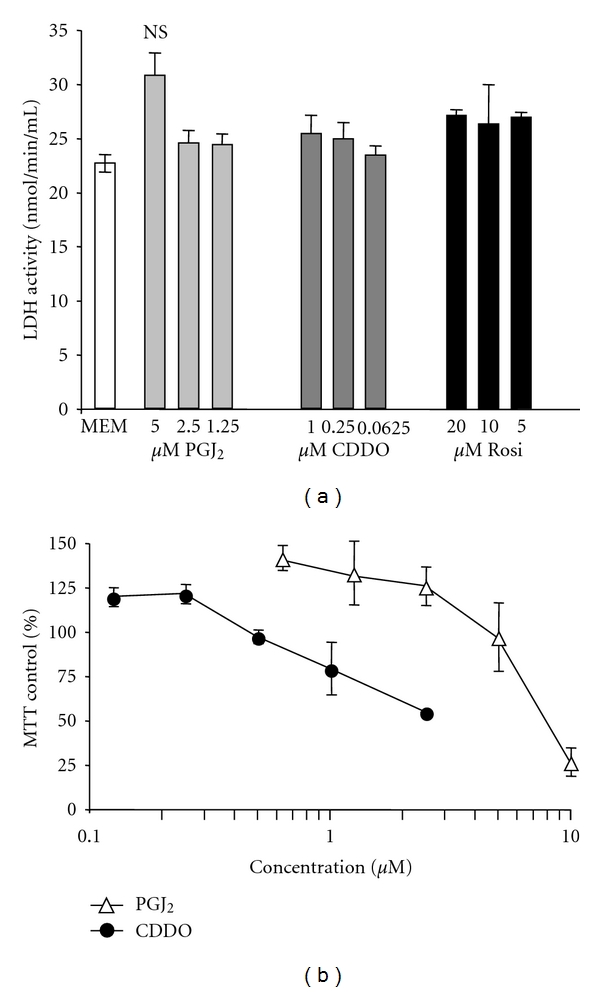
The PPARγ ligands are not overtly cytotoxic at the doses used. (a) Primary human lung fibroblasts were treated for 24 hours with the indicated concentrations of CDDO, or 15d-PGJ2 (PGJ2), and LDH released into the media was measured by commercial LDH activity assay. There were no significant differences between any of the treatment groups compared to MEM control. Results are mean ± standard deviation for triplicate wells and are representative of 2 independent experiments that yielded similar results. (b) Primary human lung fibroblasts were plated in a 96-well plate and treated with the indicated concentrations of CDDO or 15d-PGJ2 (PGJ2). Cell viability was determined after 24 hours by MTT assay. The results shown are the mean ± standard deviation of quadruplicate wells and are normalized to untreated control wells.
3.2. PPARγ Ligands Inhibit IL-1β-Induced Upregulation of COX-2 and PGE2
To further evaluate the potential antiinflammatory actions of PPARγ ligands, lung fibroblasts were treated with IL-1β and PPARγ ligands, and expression of COX-2 was determined by Western blot. As expected, IL-1β strongly induces COX-2 (Figure 3). CDDO and 15d-PGJ2 significantly inhibited IL-1β-induced COX-2 production. However, rosiglitazone failed to suppress COX-2 induction in human lung fibroblasts.
Figure 3.

CDDO and 15d-PGJ2 attenuate IL-1β-induced COX-2 production in human lung fibroblasts. Primary human lung fibroblasts were pretreated with 20 μM rosiglitazone (Rosi), 1 μM CDDO, or 5 μM 15d-PGJ2 (PGJ2) for 1 hour and then cotreated with 1 ng/mL of IL-1β for 24 hours. Protein lysates were harvested and Western blot analysis was performed by probing for protein expression of COX-2 and GAPDH (for normalization). (a) Representative samples are shown. (b) Quadruplicate samples were analyzed by densitometry and normalized to GAPDH. COX-2 expression was significantly reduced in CDDO and 15d-PGJ2 treated cells compared to IL-1β alone (*P < .001). CDDO and 15d-PGJ2 were significantly more potent than rosiglitazone († P < .001). Results are mean ± standard deviation for quadruplicate wells and are representative of 3 independent experiments that yielded similar results. Data were analyzed by ANOVA.
We also performed immunofluorescence to confirm the induction of COX-2 protein and to localize the expression of COX-2 in IL-1β-treated lung fibroblasts. COX-2-specific staining demonstrated that untreated fibroblasts expressed minimal COX-2 at baseline (Figure 4(a)). Upon treatment with IL-1β, cytoplasmic COX-2 production was markedly upregulated (Figure 4(b)). Rosiglitazone failed to suppress IL-1β-induced COX-2 upregulation (Figure 4(c)). In contrast, however, both CDDO and 15d-PGJ2 suppressed cytoplasmic expression of COX-2 in IL-1β-treated fibroblasts (Figures 4(d) and 4(e)). CAY10410, a structural analogue of 15d-PGJ2, did not suppress IL-1β-induced COX-2 upregulation (Figure 4(f)), suggesting structural differences between the ligands account for their anti-inflammatory actions.
Figure 4.

Immunofluorescence of human lung fibroblasts demonstrates CDDO and 15d-PGJ2 attenuate IL-1β-induced COX-2 expression. Fibroblasts were pretreated with PPARγ agonists for 1 hour prior to IL-1β (1 ng/mL) treatment for 24 hours. Cells were fixed and permeabilized and probed for COX-2 protein. COX-2 was visualized with Alexa Fluor 488 (green) and cell nuclei with DAPI (blue). (a) MEM control; (b) IL-1β; (c) IL-1β + 20 μM rosiglitazone; (d) IL-1β + 1 μM CDDO; (e) IL-1β + 5 μM 15d-PGJ2, (f) IL-1β + 5 μM CAY10410. IL-1β-induced COX-2 was inhibited by CDDO (d) and 15d-PGJ2 (e).
COX-2 mediates the first step in the conversion of arachidonic acid to prostaglandins. The immunomodulatory prostaglandin PGE2, a product of this reaction, was measured in lung fibroblast culture supernatants following treatment with PPARγ ligands and IL-1β. Consistent with the COX-2 results, CDDO and 15d-PGJ2 inhibited IL-1β-induced production of PGE2 by greater than 90% compared to controls treated with IL-1β alone (Figure 5). Rosiglitazone also inhibited IL-1β-induced production of PGE2, but was less effective than CDDO and 15d-PGJ2.
Figure 5.
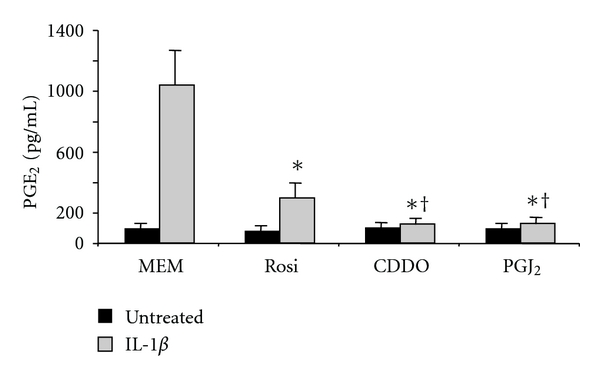
PPARγ ligands attenuate IL-1β-induced PGE2 production in human lung fibroblasts. Primary human lung fibroblasts were pretreated with 20 μM rosiglitazone (Rosi), 1 μM CDDO, or 5 μM 15d-PGJ2 (PGJ2) and then cotreated with IL-1β for 24 hours as previously described. Supernatants were harvested, and PGE2 was determined by EIA. PGE2 production is significantly reduced in PPARγ ligand-treated fibroblasts compared to IL-1β alone (*P < .001). CDDO and 15d-PGJ2 were significantly more potent than rosiglitazone († P < .05). Results are mean ± standard deviation for quadruplicate wells and are representative of 3 independent experiments that yielded similar results. Data were analyzed by ANOVA.
3.3. Suppression of Inflammatory Mediators by PPARγ Ligands in Human Lung Fibroblasts Occurs via a PPARγ-Independent Mechanism
We used a pharmacological approach to determine whether the anti-inflammatory actions of PPARγ ligands are dependent on or independent of PPARγ. GW9662 is an irreversible PPARγ antagonist that covalently binds to a cysteine residue in the ligand binding site of PPARγ [39]. GW9662 inhibits PPARγ agonist-driven adipogenesis, which is a completely PPARγ-dependent process [24]. Primary human lung fibroblasts were pretreated for 4 hours with GW9662 and one hour with CDDO or 15d-PGJ2, followed by IL-1β. IL-1β strongly induced IL-6, MCP-1 and PGE2 compared to MEM control (Figures 6(a)–6(c)). As previously shown, CDDO and 15d-PGJ2 significantly inhibited the IL-1β-induced production of these inflammatory mediators. GW9662 did not reverse the suppressive effects of CDDO and 15d-PGJ2 ligands on cytokine and PGE2 production (Figures 6(a)–6(c)). This indicates that PPARγ is not essential for the anti-inflammatory effects of these ligands, and that PPARγ independent pathways are therefore likely important.
Figure 6.

The PPARγ antagonist GW9662 does not inhibit the anti-inflammatory effects of CDDO and 15d-PGJ2. Primary human lung fibroblasts were pretreated with 1 μM GW9662 for 3 hours, then PPARγ ligands were added for 1 hour, then 1 ng/mL IL-1β was added for 24 hours. Supernatants were harvested and proinflammatory mediators were measured by ELISA or EIA. (a) IL-6, (b) MCP-1, (c) PGE2. Pretreatment with GW9662 did not significantly alter the attenuation of pro-inflammatory mediator production by CDDO or 15d-PGJ2 alone. Results are mean ± standard deviation for quadruplicate wells and are representative of 2 independent experiments that yielded similar results.
3.4. A Strong Electrophilic Center Is Important for PPARγ Ligand-Mediated Suppression of Inflammation in Human Lung Fibroblasts
CDDO and 15d-PGJ2 contain α/β-unsaturated ketone rings with electrophilic carbons that can form covalent bonds with free sulfhydryls in cellular proteins [40, 41]. CAY10410 (9,10-dihydro-15-deoxy-Δ12,14-PGJ2) is a structural analog of 15d-PGJ2 that lacks the unsaturated ketone containing the electrophilic carbon. To investigate the importance of the electrophilic center in suppressing inflammatory endpoints, we compared the ability of 15d-PGJ2 and CAY10410 to inhibit the pro-inflammatory effects of IL-1β on human lung fibroblasts. CAY10410 treatment resulted in a small reduction in IL-1β-induced IL-6 production that was not statistically significant, and a 60% reduction in MCP-1 production compared to 98% inhibition by 15d-PGJ2 (Figure 7).
Figure 7.

A strong electrophilic carbon is necessary for PPARγ ligand-mediated attenuation of inflammation in IL-1β-treated human lung fibroblasts. Primary human lung fibroblasts were pretreated with 1 μM CDDO, 5 μM 15d-PGJ2 (PGJ2), 5 μM CAY10410, (CAY) or 15 μM PGA1 for 1 hour and then co-treated with 1 ng/mL of IL-1β for 24 hours. Supernatants were harvested, and cytokine concentrations were measured by ELISA. (a) CDDO, 15d-PGJ2, and PGA1, but not CAY10410, significantly inhibited production of IL-6 (*P < .01). (b) CDDO, 15d-PGJ2, PGA1, and CAY10410 all significantly reduced MCP-1 production (*P < .01). CDDO, 15d-PGJ2, and PGA1 were significantly more potent than CAY10410 († P < .01). Results are mean ± standard deviation for quadruplicate wells and are representative of 3 independent experiments.
To further investigate the importance of the electrophilic center, we tested another prostaglandin that is also a potent electrophile, PGA1. PGA1 was partially effective at inhibiting IL-1β-induced production of IL-6 and completely effective in blocking MCP-1 production (Figure 7).
3.5. A Strong Electrophilic Center Is Important for PPARγ Ligand-Mediated Suppression of NF-κB in Human Lung Fibroblasts
To better understand the mechanism involved in PPARγ ligand-mediated immune suppression, we investigated the effect of PPARγ ligands on the activation of NF-κB, a transcription factor that regulates the expression of numerous pro-inflammatory mediators. Primary human lung fibroblasts were transfected with an NF-κB luciferase reporter construct, and treated with PPARγ ligands and IL-1β. CDDO, 15d-PGJ2, and PGA1, but not CAY10410 or rosiglitazone, significantly decreased IL-1β-induced NF-κB luciferase activity (Figure 8).
Figure 8.

CDDO and PGA1 inhibit IL-1β-induced NF-κB transcriptional activity in human lung fibroblasts. Primary human lung fibroblasts were transfected with a luciferase reporter, then pretreated with 20 μM rosiglitazone (Rosi), 1 μM CDDO, 5 μM 15d-PGJ2 (PGJ2), 5 μM CAY10410 (CAY), or 15 μM PGA1 and cotreated with of IL-1β for 24 hours as described. NF-κB-dependent luciferase activity was measured in lysates as described, normalized to protein concentration, and expressed as relative light units (RLU). NF-κB activation is significantly reduced in PPARγ ligand-treated fibroblasts compared to IL-1β alone (*P < .01). Results are mean ± standard deviation for quadruplicate wells and are representative of 2 independent experiments that yielded similar results.
4. Discussion
Inflammation is associated with many diseases of the lung including asthma, chronic obstructive pulmonary disease (COPD), silicosis, and lung cancer [1–4]. Aside from glucocorticoids, there are few effective anti-inflammatory therapies; therefore, the development of novel therapies that have the potential to alleviate pulmonary diseases associated with inflammatory etiologies is a priority.
PPARγ ligands are receiving increasing attention as potential anti-inflammatory therapeutics because of their anti-inflammatory properties in a variety of tissues in vivo and cells in vitro [42]. The anti-inflammatory effects of PPARγ ligands have not previously been reported in human lung fibroblasts, a sentinel cell of inflammatory cascades in the lung [31, 34, 43, 44]. Here, we report that PPARγ ligands have potent anti-inflammatory effects in human lung fibroblasts exposed to divergent inflammatory stimuli, and that the mechanism is largely PPARγ-independent.
To induce a pro-inflammatory response in human lung fibroblasts, we used two different inflammatory stimuli. IL-1β is an acute phase inflammatory cytokine, while silica is a particulate that has potent proinflammatory effects when inhaled and is capable of causing both acute and chronic inflammatory lung disease [32, 33]. Both IL-1β and silica induced the inflammatory mediators IL-6 and MCP-1, which were inhibited by CDDO, rosiglitazone, and 15d-PGJ2 (Figure 1). Interestingly, rosiglitazone was much less effective at inhibiting IL-6 and MCP-1, with an EC50 5–10-fold higher than 15d-PGJ2 and at least 30-fold higher than CDDO. CDDO and 15d-PGJ2, but not rosiglitazone, also blocked upregulation of COX-2 and PGE2 (Figures 3 and 4). This is in agreement with our previous finding that rosiglitazone is less effective than CDDO or 15d-PGJ2 at inhibiting the pro-fibrotic effects of TGF-β in lung fibroblasts [26], and suggests that there are significant differences in the mechanism of action between rosiglitazone and CDDO and 15d-PGJ2.
Rosiglitazone, CDDO and 15d-PGJ2 all tightly bind the PPARγ receptor [12, 20, 21], activate PPARγ-dependent transcription [22, 23], and promote adipogenesis via a solely PPARγ-dependent mechanism [12, 25]. However, in addition to stimulating PPARγ-dependent transcriptional changes, CDDO and 15d-PGJ2 are reported to have effects that are mediated through PPARγ-independent pathways [26, 28, 45]. To determine whether CDDO, and 15d-PGJ2 might be acting via a PPARγ-independent mechanism, we used a pharmacological approach to block PPARγ. GW9662 is an irreversible competitive PPARγ antagonist that covalently binds to a cysteine residue in the ligand binding domain of PPARγ [46]. GW9662 is a highly effective inhibitor of PPARγ-dependent processes including differentiation of osteoclasts and activation of hepatic stellate cells [47, 48]. We have previously reported that rosiglitazone, CDDO and 15d-PGJ2 drive the differentiation of fibroblasts to adipocytes. GW9662 at 1 μM completely inhibits this effect, demonstrating that this compound is effective at blocking the PPARγ-dependent actions of these PPARγ ligands [24]. Here, GW9662 did not reverse the anti-inflammatory effects of CDDO and 15d-PGJ2 (Figure 6), indicating that the anti-inflammatory effects of CDDO and 15d-PGJ2 on human lung fibroblasts are largely independent of the PPARγ-dependent transcriptional pathway. Rosiglitazone was such a poor inhibitor of the inflammatory effects of IL-1β that it was not possible to show a reversal of inhibition by GW9662, which would be expected if rosiglitazone acted by a purely PPARγ-dependent mechanism.
Comparing the chemical structures of rosiglitazone, CDDO, and 15d-PGJ2, it is notable that CDDO and 15d-PGJ2 have strong electrophilic carbons, whereas rosiglitazone does not. 15d-PGJ2 has one α/β-unsaturated ketone ring with an electrophilic carbon capable of forming covalent bonds through Michael addition reactions [49], whereas CDDO has two [18, 30]. We have recently demonstrated the importance of these electrophilic carbons in preventing TGF-β-inducedmyofibroblast differentiation [26, 50]. We hypothesize that the electrophilic carbons of CDDO and 15d-PGJ2 are also important for their anti-inflammatory effects. To test this hypothesis, we used CAY10410, a structural analog of 15d-PGJ2 that lacks the α/β-unsaturated ketone, and PGA1, another electrophilic prostaglandin. In lung fibroblasts stimulated with IL-1β, CAY10410 did not inhibit COX-2 upregulation or IL-6 production and was half as effective as 15d-PGJ2 at blocking MCP-1 production (Figures 4 and 7). On the other hand, PGA1 significantly attenuated IL-6 and completely blocked production of MCP-1 (Figure 7). Because CAY10410 has an identical structure to 15d-PGJ2 except for the electrophilic carbon, the fact that CAY10410 lacks the effects of 15d-PGJ2 strongly suggests that the electrophilic centers present in CDDO and 15d-PGJ2 are critical for mediating their maximal anti-inflammatory therapeutic potential. CDDO and 15d-PGJ2, but not rosiglitazone or CAY10410, significantly inhibited IL-1β-induced NF-κB activity (Figure 8).
The molecular targets of CDDO and 15d-PGJ2 in inflammation are not completely known. 15d-PGJ2 can bind to the NF-κB components IκB and p65 [50]. Another candidate is the transcription factor Nrf2, which regulates anti-oxidant and anti-inflammatory pathways. CDDO and 15d-PGJ2 activate Nrf2 in mouse cells and human cancer cells [51, 52]. However, these compounds do not activate Nrf2 in human lung fibroblasts [27, 53]. We have previously reported that CDDO activates AP-1 transcriptional activity in human lung fibroblasts [27]. However, AP-1 is a promoter, rather than an inhibitor of inflammation, and AP-1 activation leads to upregulation of IL-6 via NF-κB [54]. We hypothesize that these electrophilic compounds suppress inflammation and activate AP-1 via different pathways, and that the anti-inflammatory effects are stronger and override the potentially proinflammatory effects of AP-1 activation.
In addition to PPARγ-independent effects, PPARγ ligands have anti-inflammatory effects that are moderated via a PPARγ-dependent mechanism. This PPARγ-dependent mechanism can be accessed by TZDs such as rosiglitazone and pioglitazone [9, 55–57], and indeed, rosiglitazone has limited anti-inflammatory properties in this report. However, while TZDs are currently used clinically as insulin sensitizers in type 2 diabetes, they have a complex sideeffect profile including edema, weight gain, bone weakness, and potentially an increased risk of cardiovascular disease [58–60], that may limit their widespread use as anti-inflammatory therapies. Although TZDs have high binding affinity for PPARγ they lack electrophilic centers and are thus unable to access PPARγ-independent anti-inflammatory pathways that use this mechanism [27, 49, 61, 62]. We suggest that additional research on the PPARγ-independent anti-inflammatory activities of CDDO and 15d-PGJ2, including identification of additional targets beyond NF-κB, should lead to development of novel compounds with greater specificity for the anti-inflammatory targets of PPARγ ligands but decreased binding of PPARγ itself, with fewer resulting side-effects. As CDDO is orally active, has a long half-life, and is currently in clinical trials as an anticancer therapy, it may be a useful platform for derivatization and further study. Further development of small compounds with strong electrophilic centers is warranted as these drugs may be effective anti-inflammatory treatments for human lung diseases.
Acknowledgments
This work was supported in part by the National Institutes of Health Clinical and Translational Sciences Award and Grants nos.HL75432-0451, HL75432-0452, DE011390, ES01247, HL095402, EY017123, EY015836, T32-HL066988, T32-HL007152, and ES07026. C. M. Hagon was supported by Award no. 5TL1RR024135 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or National Institutes of Health.
References
- 1.Barnes PJ. Future advances in COPD therapy. Respiration. 2001;68(5):441–448. doi: 10.1159/000050547. [DOI] [PubMed] [Google Scholar]
- 2.Bowden DH, Adamson IYR. The role of cell injury and the continuing inflammatory response in the generation of silicotic pulmonary fibrosis. Journal of Pathology. 1984;144(3):149–161. doi: 10.1002/path.1711440302. [DOI] [PubMed] [Google Scholar]
- 3.Brody JS, Spira A. Chronic obstructive pulmonary disease, inflammation, and lung cancer. Proceedings of the American Thoracic Society. 2006;3(6):535–537. doi: 10.1513/pats.200603-089MS. [DOI] [PubMed] [Google Scholar]
- 4.Engels EA. Inflammation in the development of lung cancer: epidemiological evidence. Expert Review of Anticancer Therapy. 2008;8(4):605–615. doi: 10.1586/14737140.8.4.605. [DOI] [PubMed] [Google Scholar]
- 5.Issemann I, Prince RA, Tugwood JD, Green S. The peroxisome proliferator-activated receptor:retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. Journal of Molecular Endocrinology. 1993;11(1):37–47. doi: 10.1677/jme.0.0110037. [DOI] [PubMed] [Google Scholar]
- 6.Cabrero A, Laguna JC, Vázquez M. Peroxisome proliferator-activated receptors and the control of inflammation. Curr Drug Targets Inflamm Allergy. 2002;1(3):243–248. doi: 10.2174/1568010023344616. [DOI] [PubMed] [Google Scholar]
- 7.Tan NS, Michalik L, Desvergne B, Wahli W. Multiple expression control mechanisms of peroxisome proliferator-activated receptors and their target genes. Journal of Steroid Biochemistry and Molecular Biology. 2005;93(2–5):99–105. doi: 10.1016/j.jsbmb.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 8.Belvisi MG, Hele DJ, Birrell MA. Peroxisome proliferator-activated receptor gamma agonists as therapy for chronic airway inflammation. European Journal of Pharmacology. 2006;533(1–3):101–109. doi: 10.1016/j.ejphar.2005.12.048. [DOI] [PubMed] [Google Scholar]
- 9.Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Frontiers in Bioscience. 2008;13(5):1813–1826. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehrke M, Lazar MA. The many faces of PPARγ . Cell. 2005;123(6):993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 11.Rizzo G, Fiorucci S. PPARs and other nuclear receptors in inflammation. Current Opinion in Pharmacology. 2006;6(4):421–427. doi: 10.1016/j.coph.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83(5):813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 13.McIntyre TM, Pontsler AV, Silva AR, et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(1):131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schopfer FJ, Lin Y, Baker PRS, et al. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor γ ligand. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shappell SB, Gupta RA, Manning S, et al. 15S-hydroxyeicosatetraenoic acid activates peroxisome proliferator-activated receptor γ and inhibits proliferation in PC3 prostate carcinoma cells. Cancer Research. 2001;61(2):497–503. [PubMed] [Google Scholar]
- 16.Elte JWF, Blicklé JF. Thiazolidinediones for the treatment of type 2 diabetes. European Journal of Internal Medicine. 2007;18(1):18–25. doi: 10.1016/j.ejim.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 17.Zieleniak A, Wójcik M, Woźniak LA. Structure and physiological functions of the human peroxisome proliferator-activated receptor γ . Archivum Immunologiae et Therapiae Experimentalis. 2008;56(5):331–345. doi: 10.1007/s00005-008-0037-y. [DOI] [PubMed] [Google Scholar]
- 18.Chintharlapalli S, Papineni S, Konopleva M, Andreef M, Samudio I, Safe S. 2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid and related compounds inhibit growth of colon cancer cells through peroxisome proliferator-activated receptor γ-dependent and -independent pathways. Molecular Pharmacology. 2005;68(1):119–128. doi: 10.1124/mol.105.011437. [DOI] [PubMed] [Google Scholar]
- 19.Ray DM, Morse KM, Hilchey SP, et al. The novel triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) induces apoptosis of human diffuse large B-cell lymphoma cells through a peroxisome proliferator-activated receptor gamma-independent pathway. Experimental Hematology. 2006;34(9):1202–1211. doi: 10.1016/j.exphem.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ) The Journal of Biological Chemistry. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Porter WW, Suh N, et al. A synthetic triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), is a ligand for the peroxisome proliferator-activated receptor γ . Molecular Endocrinology. 2000;14(10):1550–1556. doi: 10.1210/mend.14.10.0545. [DOI] [PubMed] [Google Scholar]
- 22.Burgess HA, Daugherty LE, Thatcher TH, et al. PPARγ agonists inhibit TGF-β induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. American Journal of Physiology. 2005;288(6):L1146–L1153. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]
- 23.Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP, Sime PJ. PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS ONE. 2011;6(1, article e15909) doi: 10.1371/journal.pone.0015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feldon SE, O’Loughlin CW, Ray DM, Landskroner-Eiger S, Seweryniak KE, Phipps RP. Activated human T lymphocytes express cyclooxygenase-2 and produce proadipogenic prostaglandins that drive human orbital fibroblast differentiation to adipocytes. American Journal of Pathology. 2006;169(4):1183–1193. doi: 10.2353/ajpath.2006.060434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-deoxy-Δ, -prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ . Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 26.Ferguson HE, Kulkarni A, Lehmann GM, et al. Electrophilic peroxisome proliferator-activated receptor-γ ligands have potent antifibrotic effects in human lung fibroblasts. American Journal of Respiratory Cell and Molecular Biology. 2009;41(6):722–730. doi: 10.1165/rcmb.2009-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferguson HE, Thatcher TH, Olsen KC, et al. Peroxisome proliferator-activated receptor-γ ligands induce heme oxygenase-1 in lung fibroblasts by a PPARγ-independent, glutathione-dependent mechanism. American Journal of Physiology. 2009;297(5):L912–L919. doi: 10.1152/ajplung.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melichar B, Konopleva M, Hu W, Melicharova K, Andreeff M, Freedman RS. Growth-inhibitory effect of a novel synthetic triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, on ovarian carcinoma cell lines not dependent on peroxisome proliferator-activated receptor-γ expression. Gynecologic Oncology. 2004;93(1):149–154. doi: 10.1016/j.ygyno.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 29.Shi B, Greaney MF. Reversible Michael addition of thiols as a new tool for dynamic combinatorial chemistry. Chemical Communications. 2005;(7):886–888. doi: 10.1039/b414300k. [DOI] [PubMed] [Google Scholar]
- 30.Suh N, Wang Y, Honda T, et al. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9- dien-28-oic acid, with potent differentiating, antiproliferative, and anti- inflammatory activity. Cancer Research. 1999;59(2):336–341. [PubMed] [Google Scholar]
- 31.Jordana M, Sarnstrand B, Sime PJ, Ramis I. Immune-inflammatory functions of fibroblasts. European Respiratory Journal. 1994;7(12):2212–2222. doi: 10.1183/09031936.94.07122212. [DOI] [PubMed] [Google Scholar]
- 32.O’Reilly KMA, Phipps RP, Thatcher TH, Graf BA, Van Kirk J, Sime PJ. Crystalline and amorphous silica differentially regulate the cyclooxygenase-prostaglandin pathway in pulmonary fibroblasts: Implications for pulmonary fibrosis. American Journal of Physiology. 2005;288(6):L1010–L1016. doi: 10.1152/ajplung.00024.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olman MA, White KE, Ware LB, et al. Pulmonary edema fluid from patients with early lung injury stimulates fibroblast proliferation through IL-1β-induced IL-6 expression. Journal of Immunology. 2004;172(4):2668–2677. doi: 10.4049/jimmunol.172.4.2668. [DOI] [PubMed] [Google Scholar]
- 34.Smith RS, Smith TJ, Blieden TM, Phipps RP. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. The American Journal of Pathology. 1997;151(2):317–322. [PMC free article] [PubMed] [Google Scholar]
- 35.Fries KM, Sempowski GD, Gaspari AA, Blieden T, Looney RJ, Phipps RP. CD40 expression by human fibroblasts. Clinical Immunology and Immunopathology. 1995;77(1):42–51. doi: 10.1016/0090-1229(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 36.Baglole CJ, Reddy SY, Pollock SJ, et al. Isolation and phenotypic characterization of lung fibroblasts. Methods in Molecular Medicine. 2005;117:115–127. doi: 10.1385/1-59259-940-0:115. [DOI] [PubMed] [Google Scholar]
- 37.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods. 1983;65(1-2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 38.Koumas L, Smith TJ, Phipps RP. Fibroblast subsets in the human orbit: thy-1 and Thy-1 subpopulations exhibit distinct phenotypes. European Journal of Immunology. 2002;32(2):477–485. doi: 10.1002/1521-4141(200202)32:2<477::AID-IMMU477>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 39.Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-δ12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3-kinase-akt-NF-κB-p300 pathway independent of peroxisome proliferator-activated receptor γ . Journal of Immunology. 2004;173(8):5196–5208. doi: 10.4049/jimmunol.173.8.5196. [DOI] [PubMed] [Google Scholar]
- 40.Atsmon J, Sweetman BJ, Baertschi SW, Harris TM, Roberts LJ. Formation of thiol conjugates of 9-deoxy-delta 9,delta 12(E)-prostaglandin D2 and delta 12(E)-prostaglandin D2. Biochemistry. 1990;29:3760–3765. doi: 10.1021/bi00467a023. [DOI] [PubMed] [Google Scholar]
- 41.Shibata T, Yamada T, Ishii T, et al. Thioredoxin as a molecular target of cyclopentenone prostaglandins. The Journal of Biological Chemistry. 2003;278(28):26046–26054. doi: 10.1074/jbc.M303690200. [DOI] [PubMed] [Google Scholar]
- 42.Asada K, Sasaki S, Suda T, Chida K, Nakamura H. Antiinflammatory roles of peroxisome proliferator-activated receptor γ in human alveolar macrophages. American Journal of Respiratory and Critical Care Medicine. 2004;169(2):195–200. doi: 10.1164/rccm.200207-740OC. [DOI] [PubMed] [Google Scholar]
- 43.Kaufman J, Graf BA, Leung EC, et al. Fibroblasts as sentinel cells: role of the CD40-CD40 ligand system in fibroblast activation and lung inflammation and fibrosis. Chest. 2001;120(1, supplement):S53–S55. doi: 10.1378/chest.120.1_suppl.s53. [DOI] [PubMed] [Google Scholar]
- 44.Koumas L, King AE, Critchley HOD, Kelly RW, Phipps RP. Fibroblast heterogeneity: existence of functionally distinct Thy 1+ and Thy 1− human female reproductive tract fibroblasts. American Journal of Pathology. 2001;159(3):925–935. doi: 10.1016/S0002-9440(10)61768-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Derlacz RA, Hyc K, Usarek M, Jagielski AK, Drozak J, Jarzyna R. PPAR-γ-independent inhibitory effect of rosiglitazone on glucose synthesis in primary cultured rabbit kidney-cortex tubules. Biochemistry and Cell Biology. 2008;86(5):396–404. doi: 10.1139/o08-105. [DOI] [PubMed] [Google Scholar]
- 46.Leesnitzer LM, Parks DJ, Bledsoe RK, et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41(21):6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- 47.Bendixen AC, Shevde NK, Dienger KM, Willson TM, Funk CD, Pike JW. IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator-activated receptor γ1. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2443–2448. doi: 10.1073/pnas.041493198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyahara T, Schrum L, Rippe R, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. The Journal of Biological Chemistry. 2000;275(46):35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 49.Scher JU, Pillinger MH. The anti-inflammatory effects of prostaglandins. Journal of Investigative Medicine. 2009;57(6):703–708. doi: 10.2310/JIM.0b013e31819aaa76. [DOI] [PubMed] [Google Scholar]
- 50.Straus DS, Pascual G, Li M, et al. 15-Deoxy-Δ-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(9):4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim HR, Kim S, Kim EJ, et al. Suppression of Nrf2-driven heme oxygenase-1 enhances the chemosensitivity of lung cancer A549 cells toward cisplatin. Lung Cancer. 2008;60(1):47–56. doi: 10.1016/j.lungcan.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 52.Sussan TE, Rangasamy T, Blake DJ, et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(1):250–255. doi: 10.1073/pnas.0804333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baglole CJ, Sime PJ, Phipps RP. Cigarette smoke-induced expression of heme oxygenase-1 in human lung fibroblasts is regulated by intracellular glutathione. American Journal of Physiology. 2008;295(4):L624–L636. doi: 10.1152/ajplung.90215.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viedt C, Vogel J, Athanasiou T, et al. Monocyte chemoattractant protein-1 induces proliferation and interleukin-6 production in human smooth muscle cells by differential activation of nuclear factor-κB and activator protein-1. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(6):914–920. doi: 10.1161/01.atv.0000019009.73586.7f. [DOI] [PubMed] [Google Scholar]
- 55.Jacob A, Wu R, Zhou M, Wang P. Mechanism of the anti-inflammatory effect of curcumin: PPAR-γ activation. PPAR Research. 2007 doi: 10.1155/2007/89369. Article ID 89369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakajima A, Wada K, Miki H, et al. Endogenous PPARγ mediates anti-inflammatory activity in murine ischemia-reperfusion injury. Gastroenterology. 2001;120(2):460–469. doi: 10.1053/gast.2001.21191. [DOI] [PubMed] [Google Scholar]
- 57.Rollins MD, Sudarshan S, Firpo MA, et al. Anti-inflammatory effects of PPAR-γ agonists directly correlate with PPAR-γ expression during acute pancreatitis. Journal of Gastrointestinal Surgery. 2006;10(8):1120–1130. doi: 10.1016/j.gassur.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 58.Chaggar PS, Shaw SM, Williams SG. Review article: thiazolidinediones and heart failure. Diabetes & Vascular Disease Research. 2009;6(3):146–152. doi: 10.1177/1479164109338772. [DOI] [PubMed] [Google Scholar]
- 59.Rizos CV, Elisaf MS, Mikhailidis DP, Liberopoulos EN. How safe is the use of thiazolidinediones in clinical practice? Expert Opinion on Drug Safety. 2009;8(1):15–32. doi: 10.1517/14740330802597821. [DOI] [PubMed] [Google Scholar]
- 60.Shah P, Mudaliar S. Pioglitazone: side effect and safety profile. Expert Opinion on Drug Safety. 2010;9(2):347–354. doi: 10.1517/14740331003623218. [DOI] [PubMed] [Google Scholar]
- 61.Giaginis C, Giagini A, Theocharis S. Peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands as potential therapeutic agents to treat arthritis. Pharmacological Research. 2009;60(3):160–169. doi: 10.1016/j.phrs.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 62.Jung WK, Park IS, Park SJ, et al. The 15-Deoxy-Δ-prostaglandin J2 inhibits LPS-stimulated AKT and NF-κB activation and suppresses interleukin-6 in osteoblast-like cells MC3T3E-1. Life Sciences. 2009;85(1-2):46–53. doi: 10.1016/j.lfs.2009.04.010. [DOI] [PubMed] [Google Scholar]