

Abstract
SHP-2 (PTPN11), a ubiquitously expressed protein tyrosine phosphatase, is critical for hematopoietic cell development and function owing to its essential role in growth factor/cytokine signaling. More importantly, germline and somatic mutations in this phosphatase are associated with Noonan syndrome, Leopard syndrome, and childhood hematologic malignancies. The molecular mechanisms by which SHP-2 mutations induce these diseases are not fully understood, as the biochemical bases of SHP-2 functions still remain elusive. Further understanding SHP-2 signaling activities and identification of its interacting proteins/substrates will shed light on the pathogenesis of PTPN11-associated hematologic malignancies, which, in turn, may lead to novel therapeutics for these diseases.
1. Introduction
SHP-2, encoded by PTPN11, is a ubiquitously expressed protein tyrosine phosphatase (PTP) that contains two tandem Src homology 2 (SH2) domains, a PTP domain, and a C-terminal tail with tyrosyl phosphorylation sites and a prolyl-rich motif [1–5]. It shares a similar overall structure and high homology with SHP-1, another SH2 domain-containing PTP that is predominantly expressed in hematopoietic cells [6, 7]. However, SHP-2 and SHP-1 play opposing roles in hematopoietic cell function [8–10]. Genetic analyses using the Xenopus model have revealed that both the SH2 domains and the PTP domains contribute to their signaling and thus functional specificities [11, 12]. The 2.0 Å X-ray crystal structure of SHP-2 reveals that the protein is self-inhibited by the binding of the N-terminal SH2 (N-SH2) domain to the PTP domain via hydrogen bonds [13, 14]. Upon growth factor/cytokine stimulation, binding of SHP-2 via its SH2 domains to phosphorylated tyrosine residues on growth factor receptors or docking proteins results in disruption of the autoinhibitory interaction, leading to exposure of the catalytic site and enzymatic activation [13, 14]. Remarkably, mutations in PTPN11 have been identified in several human diseases, such as the developmental disorders Noonan syndrome (NS) [15] and Leopard syndrome (LS) [16, 17], childhood hematologic malignancies [18–20], and sporadic solid tumors [21]. Most of these disease-associated mutations affect N-SH2 or PTP domain residues involved in the basal inhibition, resulting in either “activated mutants” or “inactivated mutants” of SHP-2. Although significant progress has been made in the past years, signaling mechanisms of SHP-2 and the molecular mechanisms by which SHP-2 mutations are associated with human diseases are still not fully understood. Emerging evidence has indicated that SHP-2 may also have important functions in the nucleus and the mitochondria [22–24]. Better understanding the role of SHP-2 in these organelles may provide new insights into the pathogenesis of SHP-2 mutation-associated human diseases. This paper focuses on the physiological and pathological roles of SHP-2 in hematopoietic cell development and leukemogenesis.
2. SHP-2 in Cytoplasmic Signal Transduction
SHP-2 is primarily localized to the cytosol and has been implicated in cell signaling initiated by growth factors, cytokines, and hormones, regulating cell survival/growth and differentiation [8, 25]. In addition, SHP-2 modulates cell adhesion molecules-induced signal transduction and has been found to play an important role in cell migration and motility [26–30]. This phosphatase plays complicated roles in cell signaling processes. It is involved in a variety of cell signaling cascades, such as the RAS-MAP kinase, JAK-STAT, PI3K-AKT, Rho, NF-κB, and NFAT pathways [8, 25]. Furthermore, it acts at multiple sites in individual pathways. SHP-2 appears to function in cell signaling in both catalytic-dependent and -independent manners. While its catalytic activity is important for cell signaling, SHP-2 can also function as an adaptor independent of catalytic activity [31–33]. Despite extensive studies over the past decade, the mechanisms of SHP-2 action are still poorly defined. SHP-2 interacts with a number of cell signaling components, such as growth factor/cytokine receptors, SIRPα/SHPS-1, PZR, Grb2, FRS, IRS-1, Gab1, Gab2, p85, STAT5/3/1, and Sprouty [8, 25]. Of these partners, some are the targets of SHP-2 enzymatic activity. However, none of the putative substrates identified to date can fully account for the signaling effects of SHP-2 on the many biological processes with which it has been implicated. PTPs were originally thought to be negative regulators owing to their opposite roles to protein-tyrosine kinases (PTKs). However, SHP-2 is a PTP that plays an overall positive role in many cell-signaling processes, in particular, the RAS-ERK pathway, promoting cell growth and differentiation. The underlying mechanisms remain elusive. In most cases, SHP-2 is considered to regulate an upstream element necessary for RAS activation; yet, SHP-2 functions downstream of, or parallel to, RAS activation have also been demonstrated [34]. The PTP activity of SHP-2 has been shown to be required for full activation of RAS. Three possibilities have been proposed [35]. First, SHP-2 promotes RAS activation by dephosphorylating tyrosine-phosphorylated sites of receptor PTKs or docking proteins that bind p120 RAS-GTPase activating protein (RAS-GAP), a negative regulator of RAS activity; second, SHP-2 potentiates activation of Src family kinases (SFKs) by dephosphorylating Csk (a negative regulator of SFKs) binding protein (Cbp), thereby preventing access of Csk to SFKs. Enhanced activation of SFKs in turn leads to optimal RAS activation; finally, SHP-2 promotes RAS activation by dephosphorylating Sprouty, another negative regulator of RAS. Although in most circumstances, SHP-2 plays a positive role in transducing signals relayed from receptor PTKs, SHP-2 can also function as a negative regulator depending upon stimuli, binding partners, and interacting downstream signaling networks [36]. For example, it negatively regulates JAK/STAT signaling initiated by interferon-α and -γ [37]. Another prominent example is that SHP-2 negatively regulates gp130 signaling triggered by leukemia inhibitory factor, ciliary neurotrophic factor, and interleukin-6 (IL-6) [38, 39].
3. SHP-2 in the Nucleus and the Mitochondria
Emerging evidence shows that SHP-2 is distributed to the nucleus and the mitochondria, where it can also have important functions [22–24]. Prolactin stimulation of mammary cells leads to the nuclear translocation of SHP-2 as a complex with STAT5 which then binds to DNA and regulates transcription of milk protein genes [22]. Also, it has been demonstrated that nuclear SHP-2 dephosphorylates STAT1 and STAT3 at tyrosine and serine residues and thus inhibits their transcriptional activities [40, 41]. Recently, SHP-2 has been found to inhibit nuclear export of telomerase reverse transcriptase (TERT), the catalytic subunit of telomerase, in the nucleus by dephosphorylating Tyrosine707 of TERT, thereby enhancing nuclear telomerase activity [42]. Since telomerase plays an important role in maintaining telomere length and shortening of telomeres has been linked to chromosomal instability and cell aging, SHP-2 may thus be involved in the regulation of aging processes. Our previous studies showed that about 30%–40% of SHP-2 is localized to the nucleus in mouse embryonic fibroblasts and that SHP-2 plays an important role in DNA damage-induced cellular responses [23]. SHP-2 promotes DNA damage-induced apoptosis by enhancing the c-Abl kinase-mediated pathway [23]. It is also required for DNA damage-induced translocation of Cdc25C from the nucleus to the cytoplasm [43]. Loss of functional SHP-2 decreases DNA damage-induced apoptosis as well as the cell-cycle checkpoint (G2/M) response [23, 43]. Using “rescue” approaches, we have defined that SHP-2 functions in DNA damage-induced c-Abl activation, and thereby apoptosis, in a catalytic-dependent manner, whereas its role in the DNA damage-induced G2/M checkpoint does not require catalytic activity [44].
SHP-2 is also distributed to the mitochondria, specifically the intercristae/intermembrane space (IMS) [24, 45]. However, the role of SHP-2 in the mitochondria remains unclear. The mitochondrial oxidative phosphorylation (OxPhos) system provides the vast majority of cellular energy and produces reactive oxygen species (ROS). Recently, Lee et al. propose that OxPhos complexes might be direct or indirect targets of SHP-2 [46]. They have found that cytochrome c oxidase (CcO) activity and ROS levels are significantly increased, whereas mitochondrial membrane potential and ATP content are decreased in constitutively active SHP-2 mutant cells. CcO subunit II can be phosphorylated by Src kinase which is also localized to the IMS [47, 48]. As it has long been recognized that cytosolic SHP-2 and Src regulate each other, their observations raise the possibility that SHP-2 enhances CcO activity in the mitochondria by activating Src kinase. However, other signaling pathways may also be affected by mitochondrial SHP-2. Identification of mitochondrial substrate(s) and downstream target(s) of SHP-2 will shed light on the molecular mechanisms by which SHP-2 regulates mitochondrial function and mitochondria-dependent cellular activities.
4. SHP-2 in Normal Hematopoiesis and the Immune System
Hematopoiesis is the cellular process in which all types of blood cells including erythroid, myeloid, and lymphoid cells are produced from pluripotent hematopoietic stem cells (HSCs). It is tightly controlled by environmental cues, such as cytokines and growth factors. Dysregulation of cytokine/growth factor signaling can result in blood disorders, including hematologic malignancies. SHP-2 is highly expressed in hematopoietic cells. Our previous studies have demonstrated that in contrast to the highly related SHP-1 phosphatase which negatively regulates hematopoietic cell development [49, 50], SHP-2 plays an overall positive role in hematopoiesis [51–53]. N-SH2 deletion generates a loss of function mutation in SHP-2. This mutation severely suppresses the development of erythroid and myeloid progenitors in homozygous mutant embryos [51]. Consistent with this result, neither erythroid nor myeloid progenitors derived from mutant embryonic stem (ES) cells with the N-SH2 deletion mutation in SHP-2 were detectable in the fetal liver or bone marrow of the chimeric animals generated from these ES cells [52]. In addition, SHP-2 is required for lymphopoiesis [53]. In Rag2-deficient blastocyst rescue experiments, differentiation of lymphoid lineages from ES cells with the N-SH2 deletion mutation of SHP-2 was blocked before pro-T- and pro-B-cell stages [53]. These data suggest that SHP-2 is required for the development of all blood cell lineages. Recent studies have demonstrated that SHP-2 plays a critical role in the survival and maintenance of HSCs. Even heterozygous N-SH2 deletion mutation in SHP-2 causes the HSC pool to reside in a less quiescent (G0) state. HSC repopulating capacity and self-renewal are markedly reduced by this mutation [54]. Furthermore, depletion of SHP-2 from hematopoietic cells in SHP-2 conditional knockout mice results in rapid loss of HSCs and immature progenitors of all hematopoietic lineages in a gene dosage-dependent and cell-autonomous manner [55].
The signaling mechanisms by which SHP-2 regulates hematopoietic cell development and function are not well understood. SHP-2 participates in the signal transduction of many hematopoietic cytokines, such as IL-3, IL-5, IL-6, IL-9, IL-11, EPO, SCF, GM-CSF, M-CSF, and Flt3-ligand [9]. SHP-2 appears to promote hematopoietic cell development by positively regulating critical signaling processes of hematopoietic growth factors/cytokines. We have demonstrated that SHP-2 is required for the signal transduction of IL-3, a cytokine involved in hematopoietic cell survival, proliferation, and differentiation [33]. IL-3-induced activation of JAK/STAT, ERK, and PI3K pathways in hematopoietic cells with N-SH2 deletion mutation of SHP-2 was impaired [33]. However, in catalytically deficient SHP-2 C459S-overexpressing cells, IL-3-induced PI3K activation remained unaltered, while activation of JAK2 and ERK was reduced [33]. These observations suggest that SHP-2 plays multiple roles in IL-3 signal transduction, acting in both catalytic-dependent and -independent manners in the JAK/STAT, ERK, and PI3K pathways (Figure 1). Interestingly, SHP-2 also has a negative effect on hematopoietic cell survival. Overexpression of wild-type SHP-2 in hematopoietic cells compromised their hematopoietic activities and enhanced cytokine deprivation-induced apoptosis by dephosphorylating STAT5 [56].
Figure 1.
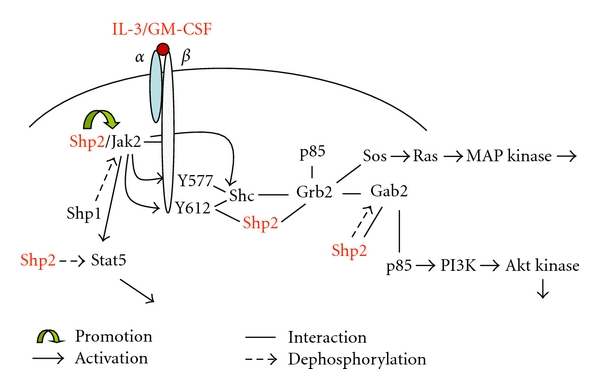
Catalytic-dependent and -independent roles of SHP-2 in IL-3/GM-CSF signaling.
SHP-2 also plays an important role in the regulation of the immune system through its effects on cytokine and inhibitory receptor signaling pathways [57]. In addition to its well-established role in cytokine responses, SHP-2 has been implicated as an important mediator of inhibitory receptor signaling. Inhibitory receptors contain one or more immunoreceptor tyrosine-based inhibitory motifs (ITIMs) within their cytoplasmic domains essential for generation and transduction of inhibitory signals. Tyrosine phosphorylation of the ITIM allows it to bind and activate phosphatases containing SH2 domains. SHP-2 has been found to be recruited to several inhibitory receptors, including the T-lymphocyte-associated antigen 4 (CTLA-4), programmed death-1 (PD-1), B- and T-lymphocyte attenuator (BTLA), killer cell Ig-like receptors (KIRs), CD31 in lymphocytes, and ITIM-containing receptors in granulocytes and platelets [57]. The role of SHP-2 in T cells is still controversial. Frearson and Alexander [58] showed that SHP-2 played a positive role in T cell receptor (TCR) signaling. Expression of catalytically deficient SHP-2 C459S significantly inhibited TCR-induced activation of ERK, but had no effect on TCR-zeta chain tyrosine phosphorylation or TCR-elicited Ca2+ transients. However, another study implicates SHP-2 as a negative regulator in TCR signaling [59]. Overexpression of Gab2, a critical downstream substrate/target of SHP-2, in T cells resulted in downregulation of TCR-mediated NFAT activation and IL-2 production [59]. Gab2 mutants lacking SHP-2-binding sites abrogated the inhibitory activity of Gab2, but its inhibitory function was restored by fusing to active SHP-2 as a chimeric protein, suggesting that the inhibitory function of Gab2 is largely dependent on SHP-2 [59]. Additionally, SHP-2 might be involved in the downregulation of T-cell adhesion processes [60]. TCR-induced ROS production selectively inhibits SHP-2-mediated dephosphorylation of Vav1 and ADAP associated with SLP-76, which limits TCR-induced adhesion and integrin clustering [60]. In consistent, overexpression of SHP-2 C459S enhanced TCR-induced LFA-1 clustering and the adhesion of Jurkat T cells to fibronectin [60].
Recent mouse studies have provided new insights into the role of SHP-2 in immune responses. Transgenic mice with expression of dominant-negative SHP-2 C459S in T cells have normal T cell development but display increased T cell activation in vivo, and aged mice have elevated serum antibodies [61]. In addition, phosphorylation of the adaptor LAT (linker for activation of T cells) downstream of TCR stimulation is defective in SHP-2 C459S T cells, whereas SHP-2 C459S-expression does not affect the majority of TCR-induced tyrosine phosphorylation, ERK activation, and TCR-driven proliferation [61]. These results suggest that wild-type SHP-2 plays an important role in suppressing the differentiation of T cells to a Th2 phenotype. Considering that overall TCR response is unchanged in SHP-2 C459S-expressing mice, it is likely that the phenotypes observed in the SHP-2 C459S transgenic mice may not be due to direct effects of SHP-2 on TCR-mediated signaling but rather on signaling pathways downstream of other receptors. However, this conclusion was challenged by more recent studies with selective SHP-2 depletion in thymocytes [62]. Lck-Cre-mediated deletion of SHP-2 in the thymus resulted in a significant block in thymocyte differentiation/proliferation and reduced expansion of CD4+ T cells. Furthermore, SHP-2-depleted mature T cells showed decreased TCR signaling in vitro. These data supports that SHP-2 is a common signal transducer for pre-TCR and TCR in promoting T cell maturation and proliferation.
5. PTPN11 Mutations in Hematologic Malignancies
Notably, germline mutations in PTPN11 (SHP-2) are found in ∼50% of the cases with the developmental disorder NS [15] and nearly all patients of the developmental disorder LS that shares certain features with NS [16, 17]. Moreover, somatic mutations in PTPN11 occur in about 35% of the patients with juvenile myelomonocytic leukemia (JMML) [18, 19], a childhood myeloproliferative disorder (MPD). In addition, PTPN11 mutations have been identified in pediatric acute leukemias, such as myelodysplastic syndrome (MDS) (10%) [18, 19], B cell acute lymphoblastic leukemia (B-ALL) (7%) [20, 63], and acute myeloid leukemia (AML) (4%) [64]. In contrast, PTPN11 mutations occur rarely in adult patients with MDS, AML, or chronic myelomonocytic leukemia (CMML) [65–67]. The reason for this is unclear. PTPN11 mutations found in NS and LS are clustered in the PTP domain, whereas most of the leukemia-associated PTPN11 mutations are located in the N-SH2 domain. NS and leukemia mutations cause changes in amino acids located at the interface formed by the N-SH2 and PTP domains in the self-inhibited SHP-2 conformation, disrupting the inhibitory intramolecular interaction, leading to hyperactivation of the catalytic activity [18]. Biochemical analyses have shown that SHP-2 mutants found in leukemias are more enzymatically active than those in NS [18, 68], suggesting that low levels of SHP-2 activation result in NS, whereas higher levels of SHP-2 activity may be required for leukemogenesis. PTPN11 mutations found in LS are located in the PTP domain, also causing changes in amino acids located at the interface formed by the N-SH2 and PTP domains and disrupting the intramolecular interaction. However, these mutations result in inactivation of the enzymatic activity due to the changes in key catalytic amino acid residues in the phosphatase active site [69].
Recent studies have begun to elucidate the pathogenesis of PTPN11 mutation-associated diseases. Heterozygous PTPN11 D61G and PTPN11 Y279C knock-in mice develop NS, and LS phenotypes, respectively, strongly suggesting a causal role of PTPN11 D61G and PTPN11 Y279C mutations in the pathogenesis of these two diseases [70, 71]. Moreover, PTPN11D61G/+ mice develop moderate MPD, characterized by excessive myeloid cell expansion and hepatosplenomegaly [70]. Myeloid progenitors from these mutant mice are hypersensitive to cytokines (GM-CSF and IL-3), reminiscent of JMML, the hallmark of which is the hypersensitive pattern of myeloid progenitor colony growth in response to GM-CSF [72]. Our recent studies showed that GM-CSF and IL-3-induced ERK and AKT activation was enhanced in PTPN11D61G/+ macrophages and mast cells [73]. Clearly, aberrantly enhanced cytokine signaling contributes to the excess expansion of myeloid cells in these mutant animals. More importantly, PTPN11D61G/+ mutation also aberrantly activates HSCs. This mutation accelerates HSC cycling, thereby increasing the stem cell pool and elevating short-term and long-term repopulating capabilities [73]. Inducible knock-in mice expressing the leukemogenic allele PTPN11D61Y in hematopoietic cells develop fatal MPD [74]. PTPN11D61Y/+ mutation decreases the HSC pool and the percentage of HSCs in G0 phase in the bone marrow, whereas the number of HSCs was markedly increased and larger proportion of HSCs are quiescent in the spleen [74]. These data suggests PTPN11D61Y/+ mutation drives HSCs in the bone marrow out of the quiescence, and they migrate from bone marrow to the spleen. Although global PTPN11D61G/+ mice similarly show the increased HSCs in the spleen, and quiescent HSCs are also decreased in the bone marrow, the overall HSC pool in PTPN11D61G/+ bone marrow is increased [73]. Since the D61Y mutation is more potent than the D61G mutation in enhancing the catalytic activity of SHP-2, the difference in HSC phenotypes caused by these two mutations suggests that the pathogenic effects of PTPN11 mutations on HSC homeostasis is dependent on the level of SHP-2 catalytic activity. In addition, stem cell microenvironments of these two mouse models may also contribute to the different HSC phenotypes. Unlike PTPN11D61Y/+ inducible knock-in mice where the mutation is primarily within the hematopoietic compartment, PTPN11D61G/+global knock-in mice carry the same mutation in all tissues and cells, including endothelial, osteoblasts, and other stromal cells that compromise the microenvironment for HSCs. PTPN11D61G/+ mutation in the microenvironment may exert certain detrimental effects on HSC homeostasis by altering cytokine/growth factor secretion. Thus, the HSC phenotypes caused by PTPN11D61G/+ germline mutation may represent a combined effect of PTPN11 mutation in HSCs and in the microenvironment.
The molecular mechanisms by which PTPN11 mutations induce hematopoietic malignancies are not fully understood. Hyperactive RAS signaling is the central event in the abnormal growth of malignant myeloid cells. Somatic RAS point mutations are found in ~20% of JMML [75] and ~40% of CMML cases [76], and NRAS or KRAS mutations occur in ~20% of AML specimens [77]. Given the positive role of SHP-2 in the RAS pathway, it is possible that PTPN11 mutations contribute to hematopoietic malignancies also by deregulating RAS-ERK signaling. However, other signaling pathways may also contribute to PTPN11-associated leukemogenesis. Studies from our laboratory and others have shown that leukemia-associated PTPN11 mutations also enhance multiple other hematopoietic signaling cascades, such as PI3K/AKT and JAK/STAT pathways [73, 74, 78, 79]. Mutant SHP-2 also has increased interactions with Gab2, Grb2, and p85, contributing to enhanced activation of these pathways [78]. Intriguingly, catalytically inactive SHP-2 E76K with an additional C459S mutation retained the capability to increase interaction with Gab2 and to enhance activation of the PI3K pathway, suggesting that in addition to the elevated catalytic activity, fundamental changes in physical and functional interactions between gain-of-function (GOF) mutant SHP-2 and signaling partners also play an important role in cytokine hypersensitivity. This notion is further supported by the fact that both NS and LS are associated with increased risk of hematologic malignancies although PTPN11 mutations found in NS and LS activate and inactivate SHP-2 enzymatic activity, respectively. It, thus, appears that catalytic activity of mutant SHP-2 is not fully responsible for the pathogenic effects. Rather, increased adaptor function of mutant SHP-2 also contributes to the enhanced downstream signaling. Consistent with this idea, LS mutations, like NS mutations, also enhance SHP-2 binding to signaling partners due to its protein conformational changes resulting from the mutations [69]. It would be interesting to see how LS mutation PTPN11 Y279C affects hematopoietic cell development in the mutation knock-in mice [71].
Other recent studies provide additional new insights into the mechanisms by which PTPN11 mutations induce hematologic malignancies. Konieczna et al. have found that the effect of leukemia-associated SHP-2 mutants on myeloid cell transformation involves inactivation of interferon consensus sequence-binding protein (ICSBP), which is an interferon-regulatory transcription factor that functions as a leukemia tumor suppressor [80]. ICSBP tyrosine phosphorylation during myelopoiesis is required for transcription of NF1, which subsequently inactivates cytokine-activated RAS, thereby creating a negative feedback mechanism for cytokine-induced proliferation [81]. Consequently, dephosphorylation of ICSBP by constitutively active mutants of SHP-2 inhibits ICSBP-dependent NF1 transcription, thereby impairing this negative feedback mechanism on cytokine-activated RAS and contributing to the proliferative phenotype in myeloid malignancies. In addition, the same group reported that HoxA10, a homeodomain transcription factor that represses myeloid differentiation genes, is also a substrate for SHP-2. As tyrosine phosphorylation of HoxA10 decreases its DNA-binding activity, enhanced dephosphorylation of HoxA10 by constitutively active SHP-2 synergizes with HoxA10 overexpression to accelerate disease progression to AML [82].
6. Perspectives
Since its discovery, tremendous progress has been made in understanding the physiological functions of SHP-2 and its clinical relevance to human diseases. However, there are still numerous questions that remain to be addressed. All of the available data support that GOF mutations in PTPN11 play a causal role in NS, LS, and JMML; however, whether the contribution of PTPN11 mutations to acute leukemias represents a primary event or a second hit acquired during disease progression is not conclusive. Moreover, upregulating the RAS pathway may be necessary but not sufficient for PTPN11 mutations to cause leukemias. Further studies are needed to find new signaling pathways and molecules disturbed by leukemia-associated mutant SHP-2. Finally, our current understanding is very limited regarding the role of SHP-2 in maintaining HSC functions. Fully characterizing how PTPN11 mutations deregulate HSC activity may provide new insights into the pathogenesis of PTPN11 mutation-associated acute leukemias and improve stem cell-based therapies.
Acknowledgments
This work was supported by the National Institutes of Health Grants nos. HL068212 and HL095657 (to C.-K. Qu) and Case Comprehensive Cancer Center Cancer Stem Cell Pilot Grant (to C.-K. Qu).
References
- 1.Freeman RM, Jr., Plutzky J, Neel BG. Identification of a human src homology 2-containing protein-tyrosine-phosphatase: a putative homolog of Drosophila corkscrew. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(23):11239–11243. doi: 10.1073/pnas.89.23.11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adachi M, Sekiya M, Miyachi T, et al. Molecular cloning of a novel protein-tyrosine phosphatase SH-PTP3 with sequence similarity to the src-homology region 2. FEBS Letters. 1992;314(3):335–339. doi: 10.1016/0014-5793(92)81500-l. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad S, Banville D, Zhao Z, Fischer EH, Shen SH. A widely expressed human protein-tyrosine phosphatase containing src homology 2 domains. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(6):2197–2201. doi: 10.1073/pnas.90.6.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng GS, Hui CC, Pawson T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science. 1993;259(5101):1607–1611. doi: 10.1126/science.8096088. [DOI] [PubMed] [Google Scholar]
- 5.Vogel W, Lammers R, Huang J, Ullrich A. Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science. 1993;259(5101):1611–1614. doi: 10.1126/science.7681217. [DOI] [PubMed] [Google Scholar]
- 6.Yi T, Cleveland JL, Ihle JN. Identification of novel protein tyrosine phosphatases of hematopoietic cells by polymerase chain reaction amplification. Blood. 1991;78(9):2222–2228. [PubMed] [Google Scholar]
- 7.Yi TL, Cleveland JL, Ihle JN. Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13. Molecular and Cellular Biology. 1992;12(2):836–846. doi: 10.1128/mcb.12.2.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neel BG, Gu H, Pao L. The ’Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends in Biochemical Sciences. 2003;28(6):284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 9.Qu CK. Role of the SHP-2 tyrosine phosphatase in cytokine-induced signaling and cellular response. Biochimica et Biophysica Acta. 2002;1592(3):297–301. doi: 10.1016/s0167-4889(02)00322-1. [DOI] [PubMed] [Google Scholar]
- 10.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nature Reviews Molecular Cell Biology. 2006;7(11):833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 11.Tang TL, Freeman RM, Jr., O’Reilly AM, Neel BG, Sokol SY. The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell. 1995;80(3):473–483. doi: 10.1016/0092-8674(95)90498-0. [DOI] [PubMed] [Google Scholar]
- 12.O’Reilly AM, Neel BG. Structural determinants of SHP-2 function and specificity in Xenopus mesoderm induction. Molecular and Cellular Biology. 1998;18(1):161–177. doi: 10.1128/mcb.18.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998;6(3):249–254. doi: 10.1016/s0969-2126(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 14.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92(4):441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 15.Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nature Genetics. 2001;29(4):465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 16.Digilio MC, Conti E, Sarkozy A, et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. American Journal of Human Genetics. 2002;71(2):389–394. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, Fryns JP. PTPN11 mutations in LEOPARD syndrome. Journal of Medical Genetics. 2002;39(8):571–574. doi: 10.1136/jmg.39.8.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tartaglia M, Niemeyer CM, Fragale A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nature Genetics. 2003;34(2):148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 19.Loh ML, Vattikuti S, Schubbert S, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103(6):2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 20.Tartaglia M, Martinelli S, Cazzaniga G, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004;104(2):307–313. doi: 10.1182/blood-2003-11-3876. [DOI] [PubMed] [Google Scholar]
- 21.Bentires-Alj M, Paez JG, David FS, et al. Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Research. 2004;64(24):8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 22.Chughtai N, Schimchowitsch S, Lebrun JJ, Ali S. Prolactin induces SHP-2 association with Stat5, nuclear translocation, and binding to the β-casein gene promoter in mammary cells. Journal of Biological Chemistry. 2002;277(34):31107–31114. doi: 10.1074/jbc.M200156200. [DOI] [PubMed] [Google Scholar]
- 23.Yuan L, Yu WM, Yuan Z, Haudenschild CC, Qu CK. Role of SHP-2 tyrosine phosphatase in the DNA damage-induced cell death response. Journal of Biological Chemistry. 2003;278(17):15208–15216. doi: 10.1074/jbc.M211327200. [DOI] [PubMed] [Google Scholar]
- 24.Salvi M, Stringaro A, Brunati AM, et al. Tyrosine phosphatase activity in mitochondria: presence of Shp-2 phosphatase in mitochondria. Cellular and Molecular Life Sciences. 2004;61(18):2393–2404. doi: 10.1007/s00018-004-4211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Frontiers in Bioscience. 2008;13(13):4925–4932. doi: 10.2741/3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. Journal of Biological Chemistry. 1998;273(33):21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]
- 27.Yo K, Iwata S, Hashizume Y, et al. SHP-2 inhibits tyrosine phosphorylation of Cas-L and regulates cell migration. Biochemical and Biophysical Research Communications. 2009;382(1):210–214. doi: 10.1016/j.bbrc.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 28.Wang S, Yu WM, Zhang W, McCrae KR, Neel BG, Qu CK. Noonan syndrome/leukemia-associated gain-of-function mutations in SHP-2 phosphatase (PTPN11) enhance cell migration and angiogenesis. Journal of Biological Chemistry. 2009;284(2):913–920. doi: 10.1074/jbc.M804129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu JX, Cao G, Williams JT, Delisser HM. SHP-2 phosphatase activity is required for PECAM-1-dependent cell motility. American Journal of Physiology. 2010;299(4):C854–C865. doi: 10.1152/ajpcell.00436.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang FM, Liu HQ, Liu SR, Tang SP, Yang L, Feng GS. SHP-2 promoting migration and metastasis of MCF-7 with loss of E-cadherin, dephosphorylation of FAK and secretion of MMP-9 induced by IL-1β in vivo and in vitro. Breast Cancer Research and Treatment. 2005;89(1):5–14. doi: 10.1007/s10549-004-1002-z. [DOI] [PubMed] [Google Scholar]
- 31.Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor β to Ras. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(15):7335–7339. doi: 10.1073/pnas.91.15.7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Nishimura R, Kashishian A, et al. A new function for a phosphotyrosine phosphatase: linking GRB2-Sos to a receptor tyrosine kinase. Molecular and Cellular Biology. 1994;14(1):509–517. doi: 10.1128/mcb.14.1.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu WM, Hawley TS, Hawley RG, Qu CK. Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling. Oncogene. 2003;22(38):5995–6004. doi: 10.1038/sj.onc.1206846. [DOI] [PubMed] [Google Scholar]
- 34.Zhang SQ, Yang W, Kontaridis MI, et al. Shp2 regulates Src family kinase activity and Ras/Erk activation by controlling Csk recruitment. Molecular Cell. 2004;13(3):341–355. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- 35.Matozaki T, Murata Y, Saito Y, Okazawa H, Ohnishi H. Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes ras activation. Cancer Science. 2009;100(10):1786–1793. doi: 10.1111/j.1349-7006.2009.01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tartaglia M, Gelb BD. Germ-line and somatic PTPN11 mutations in human disease. European Journal of Medical Genetics. 2005;48(2):81–96. doi: 10.1016/j.ejmg.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 37.You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Molecular and Cellular Biology. 1999;19(3):2416–2424. doi: 10.1128/mcb.19.3.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohtani T, Ishihara K, Atsumi T, et al. Dissection of signaling cascades through gp130 in vivo: reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity. 2000;12(1):95–105. doi: 10.1016/s1074-7613(00)80162-4. [DOI] [PubMed] [Google Scholar]
- 39.Qu CK, Feng GS. Shp-2 has a positive regulatory role in ES cell differentiation and proliferation. Oncogene. 1998;17(4):433–439. doi: 10.1038/sj.onc.1201920. [DOI] [PubMed] [Google Scholar]
- 40.Wu TR, Hong YK, Wang XD, et al. SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. Journal of Biological Chemistry. 2002;277(49):47572–47580. doi: 10.1074/jbc.M207536200. [DOI] [PubMed] [Google Scholar]
- 41.Zhang W, Chan RJ, Chen H, et al. Negative regulation of Stat3 by activating PTPN11 mutants contributes to the pathogenesis of Noonan syndrome and juvenile myelomonocytic leukemia. Journal of Biological Chemistry. 2009;284(33):22353–22363. doi: 10.1074/jbc.M109.020495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakob S, Schroeder P, Lukosz M, et al. Nuclear protein tyrosine phosphatase Shp-2 is one important negative regulator of nuclear export of telomerase reverse transcriptase. Journal of Biological Chemistry. 2008;283(48):33155–33161. doi: 10.1074/jbc.M805138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan L, Yu WM, Qu CK. DNA damage-induced G2/M Checkpoint in SV40 Large T antigen-immortalized embryonic fibroblast cells requires SHP-2 tyrosine phosphatase. Journal of Biological Chemistry. 2003;278(44):42812–42820. doi: 10.1074/jbc.M305075200. [DOI] [PubMed] [Google Scholar]
- 44.Yuan L, Yu WM, Xu M, Qu CK. SHP-2 phosphatase regulates DNA damage-induced apoptosis and G2/M arrest in catalytically dependent and independent manners, respectively. Journal of Biological Chemistry. 2005;280(52):42701–42706. doi: 10.1074/jbc.M506768200. [DOI] [PubMed] [Google Scholar]
- 45.Arachiche A, Augereau O, Decossas M, et al. Localization of PTP-1B, SHP-2, and Src exclusively in rat brain mitochondria and functional consequences. Journal of Biological Chemistry. 2008;283(36):24406–24411. doi: 10.1074/jbc.M709217200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee I, Pecinova A, Pecina P, et al. A suggested role for mitochondria in Noonan syndrome. Biochimica et Biophysica Acta. 2010;1802(2):275–283. doi: 10.1016/j.bbadis.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyazaki T, Neff L, Tanaka S, Horne WC, Baron R. Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. Journal of Cell Biology. 2003;160(5):709–718. doi: 10.1083/jcb.200209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salvi M, Brunati AM, Bordin L, la Rocca N, Clari G, Toninello A. Characterization and location of Src-dependent tyrosine phosphorylation in rat brain mitochondria. Biochimica et Biophysica Acta. 2002;1589(2):181–195. doi: 10.1016/s0167-4889(02)00174-x. [DOI] [PubMed] [Google Scholar]
- 49.Shultz LD, Schweitzer PA, Rajan TV, et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73(7):1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 50.Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nature Genetics. 1993;4(2):124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 51.Qu CK, Shi ZQ, Shen R, Tsai FY, Orkin SH, Feng GS. A deletion mutation in the SH2-N domain of Shp-2 severely suppresses hematopoietic cell development. Molecular and Cellular Biology. 1997;17(9):5499–5507. doi: 10.1128/mcb.17.9.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qu CK, Yu WM, Azzarelli B, Cooper S, Broxmeyer HE, Feng GS. Biased suppression of hematopoiesis and multiple developmental defects in chimeric mice containing Shp-2 mutant cells. Molecular and Cellular Biology. 1998;18(10):6075–6082. doi: 10.1128/mcb.18.10.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qu CK, Nguyen S, Chen J, Feng GS. Requirement of Shp-2 tyrosine phosphatase in lymphoid and hematopoietic cell development. Blood. 2001;97(4):911–914. doi: 10.1182/blood.v97.4.911. [DOI] [PubMed] [Google Scholar]
- 54.Chan RJ, Li Y, Hass MN, et al. Shp-2 heterozygous hematopoietic stem cells have deficient repopulating ability due to diminished self-renewal. Experimental Hematology. 2006;34(9):1230–1239. doi: 10.1016/j.exphem.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 55.Chan G, Cheung LS, Yang W, et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood. 2011;117(16):4253–4261. doi: 10.1182/blood-2010-11-319517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, Yu WM, Bunting KD, Qu CK. A negative role of SHP-2 tyrosine phosphatase in growth factor-dependent hematopoietic cell survival. Oncogene. 2004;23(20):3659–3669. doi: 10.1038/sj.onc.1207471. [DOI] [PubMed] [Google Scholar]
- 57.Salmond RJ, Alexander DR. SHP2 forecast for the immune system: fog gradually clearing. Trends in Immunology. 2006;27(3):154–160. doi: 10.1016/j.it.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 58.Frearson JA, Alexander DR. The phosphotyrosine phosphatase SHP-2 participates in a multimeric signaling complex and regulates T cell receptor (TCR) coupling to the Ras/mitogen-activated protein kinase (MAPK) pathway in jurkat T cells. Journal of Experimental Medicine. 1998;187(9):1417–1426. doi: 10.1084/jem.187.9.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamasaki S, Nishida K, Hibi M, et al. Docking protein Gab2 is phosphorylated by ZAP-70 and negatively regulates T cell receptor signaling by recruitment of inhibitory molecules. Journal of Biological Chemistry. 2001;276(48):45175–45183. doi: 10.1074/jbc.M105384200. [DOI] [PubMed] [Google Scholar]
- 60.Kwon J, Qu CK, Maeng JS, Falahati R, Lee C, Williams MS. Receptor-stimulated oxidation of SHP-2 promotes T-cell adhesion through SLP-76-ADAP. The EMBO Journal. 2005;24(13):2331–2341. doi: 10.1038/sj.emboj.7600706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salmond RJ, Huyer G, Kotsoni A, Clements L, Alexander DR. The src homology 2 domain-containing tyrosine phosphatase 2 regulates primary T-dependent immune responses and Th cell differentiation. Journal of Immunology. 2005;175(10):6498–6508. doi: 10.4049/jimmunol.175.10.6498. [DOI] [PubMed] [Google Scholar]
- 62.Nguyen TV, Ke Y, Zhang EE, Feng GS. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. Journal of Immunology. 2006;177(9):5990–5996. doi: 10.4049/jimmunol.177.9.5990. [DOI] [PubMed] [Google Scholar]
- 63.Yamamoto T, Isomura M, Xu Y, et al. PTPN11, RAS and FLT3 mutations in childhood acute lymphoblastic leukemia. Leukemia Research. 2006;30(9):1085–1089. doi: 10.1016/j.leukres.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 64.Tartaglia M, Martinelli S, Iavarone I, et al. Somatic PTPN11 mutations in childhood acute myeloid leukaemia. British Journal of Haematology. 2005;129(3):333–339. doi: 10.1111/j.1365-2141.2005.05457.x. [DOI] [PubMed] [Google Scholar]
- 65.Johan MF, Bowen DT, Frew ME, et al. Mutations in PTPN11 are uncommon in adult myelodysplastic syndromes and acute myeloid leukaemia. British Journal of Haematology. 2004;124(6):843–844. doi: 10.1111/j.1365-2141.2004.04862.x. [DOI] [PubMed] [Google Scholar]
- 66.Loh ML, Martinelli S, Cordeddu V, et al. Acquired PTPN11 mutations occur rarely in adult patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leukemia Research. 2005;29(4):459–462. doi: 10.1016/j.leukres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 67.Watkins F, Fidler C, Boultwood J, Wainscoat JS. Mutations in PTPN11 are rare in adult myelodysplastic syndromes and acute myeloid leukemia. American Journal of Hematology. 2004;76(4):p. 417. doi: 10.1002/ajh.20134. [DOI] [PubMed] [Google Scholar]
- 68.Marin MC, Jost CA, Irwin MS, DeCaprio JA, Caput D, Kaelin WG. Viral oncoproteins discriminate between p53 and the p53 homolog p73. Molecular and Cellular Biology. 1998;18(11):6316–6324. doi: 10.1128/mcb.18.11.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. Journal of Biological Chemistry. 2006;281(10):6785–6792. doi: 10.1074/jbc.M513068200. [DOI] [PubMed] [Google Scholar]
- 70.Araki T, Mohi MG, Ismat FA, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nature Medicine. 2004;10(8):849–857. doi: 10.1038/nm1084. [DOI] [PubMed] [Google Scholar]
- 71.Marin TM, Keith K, Davies B, et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. The Journal of Clinical Investigation. 2011;121(3):1026–1043. doi: 10.1172/JCI44972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Emanuel PD, Shannon KM, Castleberry RP. Juvenile myelomonocytic leukemia: molecular understanding and prospects for therapy. Molecular Medicine Today. 1996;2(11):468–475. doi: 10.1016/1357-4310(96)10044-7. [DOI] [PubMed] [Google Scholar]
- 73.Xu D, Wang S, Yu WM, et al. A germline gain-of-function mutation in Ptpn11 (Shp-2) phosphatase induces myeloproliferative disease by aberrant activation of hematopoietic stem cells. Blood. 2010;116(18):3611–3621. doi: 10.1182/blood-2010-01-265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chan G, Kalaitzidis D, Usenko T, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009;113(18):4414–4424. doi: 10.1182/blood-2008-10-182626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miyauchi J, Asada M, Sasaki M, Tsunematsu Y, Kojima S, Mizutani S. Mutations of the N-ras gene in juvenile chronic myelogenous leukemia. Blood. 1994;83(8):2248–2254. [PubMed] [Google Scholar]
- 76.Braun BS, Shannon K. Targeting ras in myeloid leukemias. Clinical Cancer Research. 2008;14(8):2249–2252. doi: 10.1158/1078-0432.CCR-07-1005. [DOI] [PubMed] [Google Scholar]
- 77.Bowen DT, Frew ME, Hills R, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood. 2005;106(6):2113–2119. doi: 10.1182/blood-2005-03-0867. [DOI] [PubMed] [Google Scholar]
- 78.Yu WM, Daino H, Chen J, Bunting KD, Qu CK. Effects of a leukemia-associated gain-of-function mutation of SHP-2 phosphatase on interleukin-3 signaling. Journal of Biological Chemistry. 2006;281(9):5426–5434. doi: 10.1074/jbc.M507622200. [DOI] [PubMed] [Google Scholar]
- 79.Mohi MG, Williams IR, Dearolf CR, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7(2):179–191. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 80.Konieczna I, Horvath E, Wang H, et al. Constitutive activation of SHP2 in mice cooperates with ICSBP deficiency to accelerate progression to acute myeloid leukemia. The Journal of Clinical Investigation. 2008;118(3):853–867. doi: 10.1172/JCI33742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang W, Saberwal G, Horvath E, Zhu C, Lindsey S, Eklund EA. Leukemia-associated, constitutively active mutants of SHP2 protein tyrosine phosphatase inhibit NF1 transcriptional activation by the interferon consensus sequence binding protein. Molecular and Cellular Biology. 2006;26(17):6311–6332. doi: 10.1128/MCB.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang H, Lindsey S, Konieczna I, et al. Constitutively active SHP2 cooperates with HoxA10 overexpression to induce acute myeloid leukemia. Journal of Biological Chemistry. 2009;284(4):2549–2567. doi: 10.1074/jbc.M804704200. [DOI] [PMC free article] [PubMed] [Google Scholar]