

Abstract
Oxidative stress is involved in the pathogenesis of neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and Huntington's disease. Low levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) are important for maintenance of neuronal function, though elevated levels lead to neuronal cell death. A complex series of events including excitotoxicity, Ca2+ overload, and mitochondrial dysfunction contributes to oxidative stress-mediated neurodegeneration. As expected, many antioxidants like phytochemicals and vitamins are known to reduce oxidative toxicity. Additionally, growing evidence indicates that neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and estrogens significantly prevent neuronal damage caused by oxidative stress. Here, we review and discuss recent studies addressing the protective mechanisms of neurotrophic factors and estrogen within this system.
1. Introduction
It is well established that the brain consumes a large quantity of oxygen and glucose [1–5]. Brain neurons utilize such nutrients, requiring a consistent and steady supply in order to function appropriately. Not surprisingly, brain neurons are vulnerable to oxidative stress [6], which threatens the overall functionality of the brain. Though various systems protecting against oxidative toxicity exist in the brain at cellular and molecular levels, a disruption of the defensive system may be involved in neurological deficits observed in neurodegenerative diseases. Indeed, many studies suggest that oxidative toxicity is related to Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD) [7]. In addition, a correlation between an accumulation of oxidative stress and aging has also been established [8]. Thus, it is important to clarify the detailed relationship between oxidative stress and cellular damage in neurodegenerative diseases and the aging process. In the cellular and molecular mechanisms underlying oxidative stress-induced cell death, it is well known that excitotoxicity, Ca2+ overload, mitochondrial dysfunction, and the stimulation of intracellular signaling cascades play a role [9]. As expected, antioxidants including many phytochemicals and vitamins have been found to support the survival of neurons under oxidative stress.
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is known to be a strong survival-promoting factor against various neuronal insults. As a result, the molecular mechanisms underlying neurotrophin-dependent survival promotion when exposed to oxidative stress have been extensively studied. BDNF plays a critical role in cell proliferation, cell differentiation, neuronal protection, and the regulation of synaptic function in the central nervous system (CNS) via stimulating key intracellular signaling cascades [10, 11]. In addition to BDNF, glial cell line-derived neurotrophic factor (GDNF) and hepatocyte growth factor (HGF) are also effective for neuronal survival [12, 13]. Furthermore, estrogens, which regulate synaptic plasticity in addition to sex differentiation of the brain [14–16], are found to exert protective actions against toxic conditions such as oxidative stress [17]. Here, we review the current issues concerning protective functions of neurotrophic factors and estrogen on neurons under oxidative stress.
2. The Role of Oxidative Stress in Neurodegenerative Diseases
Low levels of ROS and RNS have a physiological effect on cellular functions including neuronal plasticity [18]. However, in excess, ROS/RNS cause oxidation/nitrosylation of lipids, proteins, and nucleic acids, resulting in neuronal cell death (Figure 1). Such damage occurs as a result of either overproduction of ROS/RNS or reduced activity of enzymatic and nonenzymatic antioxidants. Thus, the delicate balance between pro- and antioxidant reactions is critical for maintaining normal neuronal function.
Figure 1.
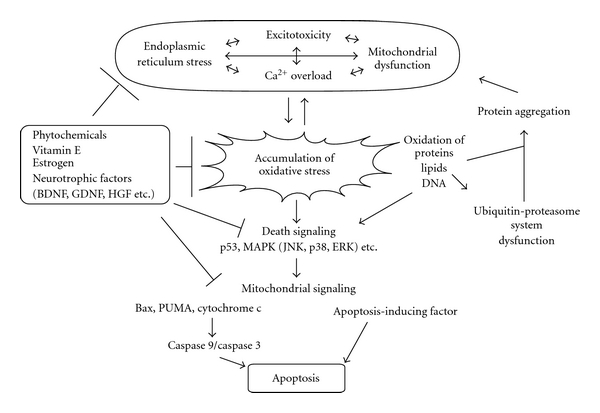
Mechanisms underlying oxidative stress-mediated neuronal apoptosis. Accumulation of oxidative stress is involved in the development/progression of neurodegenerative diseases. A number of events including excitotoxicity, mitochondrial dysfunction, Ca2+ overload, and endoplasmic reticulum stress are associated with excess reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation. High levels of ROS/RNS lead to oxidation of proteins, lipids, and DNA. Oxidized lipids induce damage of the ubiquitin-proteasome system (UPS). The UPS dysfunction and oxidation of proteins result in aggregation of proteins, recognized as a hallmark of several neurodegenerative diseases. Under oxidative stress, death signaling pathways (p53, mitogen-activated protein kinase (MAPK), etc.) are activated. Activation of p53 leads to induction of proapoptotic proteins such as Bax and p53-upregulated modulator of apoptosis (PUMA), followed by translocation of these proteins into mitochondria. Finally, mitochondrial cytochrome c is released, which then stimulates the activation of caspase 9/caspase 3. Alternatively, mitochondria secrete apoptosis-inducing factor (AIF), leading to caspase-independent apoptosis. As shown, recent studies suggest antioxidant effects of phytochemicals, vitamin E, estrogen, and neurotrophic factors including brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), and hepatocyte growth factor (HGF), leading to increased preservation of neuronal function.
Oxidative stress-mediated toxicity may be closely related to the pathogenesis of neurodegenerative diseases such as AD, PD, and HD [7]. For example, in AD brains, markers for protein oxidation (protein carbonyls and 3-nitro-tyrosine (3-NT)), lipid oxidation (4-hydroxy-2′-nonenal (4-HNE)), and DNA oxidation (8-hydroxy-2-deoxyoguanine (8-OHdG)) are elevated [19]. Indeed, the accumulation of amyloid beta (Aβ), a hallmark of AD, produces ROS including hydrogen peroxide (H2O2) in the presence of Fe3+ or Cu2+ [20–22], but see [23]. In PD brains, in which a selective and progressive loss of dopamine (DA) neurons in the substantia nigra pars compacta occurs, 4-HNE, protein carbonyls, 3-NT, and 8-OHdG are all increased while glutathione (GSH, a major intracellular antioxidant) is decreased [24]. Interestingly, 4-HNE covalently binds to alpha-synuclein (α-Syn), a central protein in PD pathogenesis, resulting in neurotoxic effects on DAergic and GABAergic neuronal cultures [25]. Similarly, HD brains (where significant neuronal loss in the striatum and cortex is observed) demonstrate elevated 3-NT, lipofuscin (a product of unsaturated fatty acid peroxidation), malondialdehyde (a marker for lipid oxidation), and 8-OHdG [26]. Reduced levels of GSH were also confirmed in cultured neurons from mice expressing mutant Huntingtin protein (Htt140Q/140Q) [27].
Oxidative toxicity is also involved in cerebral ischemia/reperfusion injury. Brain regions and types of neurons that are vulnerable to ischemia are limited. It may be because cerebral blood flow is highly spatiotemporally modulated [2], and this view could also be important to understand why specific types of neurons in different brain regions are affected in each neurodegenerative disease. In addition, a large body of evidence suggests that accumulation of oxidative stress-dependent damage occurs during normal aging, which may cause a noticeable decline in cognitive function [8, 28]. Considering that cognitive deficits are observed in neurodegenerative diseases such as AD as well, a common mechanism underlying oxidative stress-mediated neuronal cell death may exist. In the following section, we summarize the current knowledge concerning oxidative stress-mediated neuronal cell death.
3. Oxidative Stress-Mediated Neuronal Cell Death
3.1. Mitochondrial Dysfunction, Ca2+ Overload and Excitotoxicity
Apoptosis, a prototypic form of programmed cell death, is a major mode of cell death in neurodegenerative diseases. Various mechanisms including excitotoxicity, Ca2+ overload, mitochondrial dysfunction, endoplasmic reticulum stress, and oxidative stress have been found to contribute to apoptosis [9] (Figure 1). Mitochondria produce low levels of ROS in a process known as cellular respiration through the electron transport chain (ETC). The ETC consists of five protein complexes (I–V), and a disruption of this electron transport system leads to excess generation of ROS [29]. Importantly, a number of studies reported possible involvement of mitochondrial dysfunction, including altered activity of the ETC, in patients and animal models for AD [30], PD [31], HD [32], and stroke [33]. Some reports suggest that patients with psychiatric disorders, such as schizophrenia [34], depression [35], and bipolar disorder [36], also display mitochondrial dysfunction.
In addition, mitochondria regulate/impact/affect Ca2+ homeostasis by sequestering excess cytosolic Ca2+ into their matrix (named Ca2+ loading). However, an uncontrolled Ca2+ loading may be involved in neurodegeneration. In a study investigating striatal mitochondria of Hdh150 knock-in HD mice, a disrupted Ca2+ homeostasis was found [37]. Another study discovered that a deficiency of phosphatase and tensin homolog deleted on chromosome 10 (PTEN)-induced putative kinase 1 (PINK1, a mitochondrial kinase linked to familial PD) results in mitochondrial Ca2+ accumulation in cultured neurons [38]. Endoplasmic reticulum also regulates intracellular Ca2+ concentration through inositol-1,4,5-triphosphate receptors (InsP3Rs) and ryanodine receptors (RyRs). Interestingly, presenilin (PS) 1 and 2, genes involved in the pathogenesis of AD, acted as a passive endoplasmic reticulum Ca2+ channel to maintain steady-state Ca2+ levels, which was disrupted by mutant PS1-M146V and PS2-N141I [39, 40]. These PS mutants enhanced the gating activity of InsP3Rs, leading to Aβ generation [41]. Furthermore, it was shown that Aβ-containing senile plaques cause Ca2+ overload [42]. Taken together, it seems likely that mutant PSs and Aβ contribute to the disruption of Ca2+ homeostasis, which may cause mitochondrial dysfunction leading to neuronal degeneration [30].
Remarkably, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) may generate ROS in a mitochondria-independent manner. In cultured cortical neurons lacking p47(phox), a cytosolic subunit of Nox, extensive N-methyl-D-aspartic acid (NMDA) receptor activation failed to produce ROS, while H2O2 or the mitochondrial complex III inhibitor (antimycin) increased ROS [43]. Furthermore, ROS production and oxidative damage in the hippocampal CA1 neurons after ischemia were dramatically attenuated in mice either treated with Nox inhibitor or lacking gp91(phox), another Nox subunit [44]. Considering the fact that overactivation of NMDA receptors occurs in ischemia [45], it is possible that NMDA-mediated excitotoxicity may cause mitochondria-independent, but Nox-dependent, ROS production in cerebral ischemia/reperfusion injury.
3.2. Signaling Pathways in Apoptosis
p53, a transcription factor, is activated by ROS, and induces the upregulation of mitochondrial proapoptotic proteins including B-cell lymphoma-2-associated X protein (Bax) and members of the B-cell lymphoma-2-homology 3 (BH3) family consisting of BH3 interacting death agonist (Bid), Nox activator 1 (Noxa), and p53-upregulated modulator of apoptosis (PUMA) [33]. Indeed, oxidative stressors including H2O2 increased Noxa, Bim, and PUMA (but not Bid) in cultured cortical neurons [46]. Importantly, PUMA, but not Noxa or Bim, was involved in Bax-dependent apoptosis [46]. The contribution of p53/PUMA to delayed cell death of hippocampal neurons after stroke was also reported [47]. These studies suggest that p53-mediated PUMA expression may be a key event in neuronal apoptosis (Figure 1).
As the final step of apoptosis, cytochrome c is released from mitochondria via the permeability transition pore (PTP), which consists of the mitochondrial inner and outer membrane proteins including B-cell lymphoma-2 (Bcl-2) and Bax (Figure 1). Cytosolic cytochrome c participates in the formation of the apoptosome, a multiprotein complex including apoptosis protease-activating factor 1 (Apaf-1) and caspase-9, which activates caspase-3, an executioner in cell death [48]. On the other hand, apoptosis-inducing factor (AIF) is involved in mitochondria-mediated, but caspase-independent, apoptosis [49] (Figure 1).
3.3. Antioxidative Factors
Considering that oxidative stress may be associated with the pathogenesis of neurodegenerative diseases, a key therapeutic intervention would be to block or delay accumulating oxidative stress levels via increasing the function of endogenous antioxidants and/or suppressing ROS production (Figure 1). Well-known antioxidants include glutathione precursor [50, 51], polyphenols [52–54], catechins [55], flavonoids [56], and sulfated polysaccharides [57]. As the toxicity of phytochemicals is low, these substances offer a new therapeutic approach against neurodegenerative diseases [58]. On the other hand, whether oxidative stress is a cause or consequence of neurodegenerative disease remains to be elucidated [7]. A growing body of evidence suggests that oxidative stress directly initiates and progresses to neuronal cell death. However, it is possible that accumulation of oxidative stress is easily induced in neurons weakened by other insults. Indeed, in the apoptotic process, many cellular events including mitochondrial dysfunction, Ca2+ overload, and excitotoxicity activate death signaling cascades (Figure 1). Such negative feedback loops may influence cell viability. These events probably occur in parallel and have an additive or synergic effect in the induction of cell death. Therefore, in addition to blocking accumulation of oxidative stress, inhibiting death-signaling cascades and activating survival signaling would also be effective. In the following section, we focus specifically on neurotrophic factors and steroid hormones that may exert a beneficial influence.
4. Neurotrophins and Oxidative Stress in Neurodegenerative Diseases
As mentioned above, oxidative stress may be involved in the onset of HD, AD, PD, and amyotrophic lateral sclerosis (ALS) [7, 9]. Interestingly, neurotrophic factors, including neurotrophins, may also be associated with the pathology of these neurodegenerative diseases. For example, both mRNA and protein levels of BDNF are decreased in patients and animal models of HD [59–61]. In addition, the level of TrkB (tropomyosin-related kinase B), a high affinity receptor for BDNF, is also reduced in knockin HD striatal cells, in which mutant huntingtin with 111 glutamines (7 glutamines in normal) is expressed [62]. Following TrkB activation stimulated by BDNF, the mitogen-activated protein kinase/extracellular signal-regulated protein kinase (MAPK/ERK), phospholipase Cγ (PLCγ), and phosphatidylinositol 3-kinase (PI3K) pathways are primarily triggered [10]. In the knock-in HD striatal cells, a down-regulation of ERK signaling occurred, while PI3K/Akt and PLCγ pathways were intact. Such a decrease in ERK signaling in these striatal cells resulted in an increase in the cell death caused by H2O2 [63]. As expected, it was revealed that BDNF, neurotrophin-3 (NT-3), and NT-4/5 prevent neuronal cell death in an animal model of HD [64].
Recent reports suggest that the upregulation of BDNF expression/function plays a role in neuroprotection within AD models. Counts and Mufson showed that noradrenaline (NA) is neuroprotective against Aβ-dependent toxicity in human NTera-2N (hNT) neurons and rat hippocampal neurons [65]. NA prevented an increase in ROS caused by Aβ. Notably, coapplication with functional blocking antibodies for BDNF or nerve growth factor (NGF) significantly inhibited the NA-dependent protective effect against Aβ toxicity [65]. As AD is well known as an age-related neurodegenerative illness, the senescence-accelerated mouse prone 8 (SAMP8) mice, which show age-related impairment of cognitive function, is a useful model of AD [66]. Using the SAMP8 mice, Zhao et al. investigated the effect of ginsenoside, a component of ginseng, on memory [67]. They reported that chronic treatment with ginsenoside prevented loss of memory in aged SAMP8 mice. Such a treatment with ginsenoside decreased the Aβ and, in turn, increased antioxidation and synaptic plasticity-related proteins such as BDNF [67].
Oxidative stress may damage nigral DA neurons, resulting in the onset of PD. Under oxidative stress, heme oxygenase-1 (HO-1) increases and exerts a positive effect on nigral DA neurons. Overexpression of HO-1 in rat substantia nigra rescued DA neurons from cell death caused by 1-methyl-4-phenylpyridinium (MPP(+)), which is an inhibitor for mitochondrial complex I and is well known to produce PD symptoms. After HO-1 overexpression, GDNF, in addition to BDNF, was upregulated [68]. Additionally, it was reported that bilirubin, a downstream product of HO-1, increased GDNF and BDNF expression through ERK and PI3K/Akt pathways [69]. These results suggest that HO-1 protects neurons through increasing these neurotrophic factors. A role of the novel DA D3 receptor agonist D-264 in neuroprotection was reported [70]. In the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP, an inhibitor of mitochondrial complex I)-induced neurodegeneration mouse model for PD, D-264 treatment improved behavioral performance and reduced neuronal loss. Remarkably, the D-264 treatment induced an upregulation of BDNF and GDNF in MPTP-treated animals [70]. Finally, using an in vitro system, L-theanine (a glutamate analog) was shown to promote SH-SY5Y cell survival and inhibited downregulation of both BDNF and GDNF under neurotoxicant (rotenone and dieldrin) application [71]. Generally, GDNF and BDNF are important for survival/morphological change of DA neurons, and both have a recovery effect on PD-like behavior [12, 72, 73]. Taken together, it is possible that upregulation of growth factors including BDNF and GDNF is necessary for the prevention of DA neuronal damage.
5. BDNF and Oxidative Stress-Induced Cell Death
BDNF exerts protective effects against neuronal cell death by activating intracellular signaling cascades via TrkB [10, 11, 74]. Interestingly, trypanosome trans-sialidase (TS, sialic acid-transferring enzyme) mimics neurotrophins. Woronowicz et al. showed that TS induced phosphorylation of TrkB in rat pheochromocytoma (PC12) cells expressing TrkB and promoted cell survival under H2O2 stress [75]. The PI3K pathway was important for TS-mediated survival promotion. On the other hand, BDNF protects cultured cortical neurons from NMDA- or H2O2-induced cell death via suppressing the MAPK pathway [76]. Once exposed to NMDA or H2O2, retinoblastoma protein and E2F1 transcription factor, which are cell cycle regulators, were stimulated. BDNF inhibited such activation of cell cycle regulators, suggesting that the prevention of cell cycle reentry is involved in BDNF function during oxidative stress [76]. Moreover, the activation of cyclic adenosine 3′,5′-monophosphate (cAMP)-responsive element-binding protein (CREB) is involved in BDNF neuroprotection. Transgenic mice expressing A-CREB, a dominant negative form of CREB, showed a significant increase in vulnerability to seizure activity. The A-CREB mice demonstrated increased ROS levels and decreased neuroprotection by BDNF application, suggesting that CREB is an essential upstream effector of neuroprotection against oxidative toxicity [77]. Importantly, CREB also regulates the transcriptional production of BDNF [78]. The BDNF gene consists of nine exons, and exon IX corresponds to the common open reading frame of the protein. The remaining exons have distinct promoters, respectively. Thus, the transcript of BDNF consists of one of eight 5′ untranslated exons (exon I~VIII) and 3′ exon IX [79]. Interestingly, the action of CREB via promoter IV is critical for experience-dependent production of BDNF [80]. Therefore, positive-feedback mechanisms may be involved in BDNF-mediated neuroprotection.
As mentioned, BDNF seems to be beneficial in the therapeutic approach to neurodegenerative diseases. However, previous clinical trials have revealed numerous side effects of neurotrophins as well as their poor penetration through the blood brain barrier, making it very difficult to use these proteins as a drug [81]. Therefore, many studies have been performed in an effort to find a drug that upregulates BDNF. In SH-SY5Y cells after H2O2 application, tripterygium regelii extract (TRE), a traditional herbal medicine, increased tyrosine hydroxylase, a dopaminergic marker, and BDNF [82]. TRE was shown to repress the upregulation of proapoptotic proteins Bax and caspase-3, while inhibiting downregulation of antiapoptotic Bcl-2 under H2O2 application [82]. Sonic hedgehog (SHH) protein, a member of the Hedgehog family of signaling molecules [83], is putatively involved as a neuroprotective agent in oxidative stress-related neurodegenerative disease and ischemia. After H2O2 exposure, the SHH pathway was stimulated in cultured cortical neurons, and the increase in SHH pathway activation was noticeably protective against cell death caused by H2O2 [84]. In that in vitro system, exogenous SHH increased levels of vascular endothelial growth factor (VEGF) and BDNF, as well as activity of superoxide dismutase (SOD) and Bcl-2 expression [84].
Positive effects of the antioxidant vitamin E on oxidative stress-mediated toxicity in vitro [85–87] and in vivo [88, 89] have been reported. Vitamin E has also been shown to exert beneficial effects against neurodegenerative diseases [90, 91]. Our research demonstrates that pretreatment with vitamin E analogs including α- and γ-tocopherol (αT and γT, respectively) and α- and γ-tocotrienol (αT3 and γT3) protected cultured cortical neurons against H2O2-mediated neuronal cell death [92]. In our cultures, αT stimulated the activation of both the ERK and PI3K pathways and caused the upregulation of Bcl-2. Importantly, αT-mediated survival and Bcl-2 upregulation disappeared in the presence of inhibitors for ERK and PI3K signaling, suggesting the involvement of both pathways in neuroprotection by vitamin E analogs. However, the neuroprotection was not via BDNF signaling, as αT unchanged TrkB activation and BDNF expression [92]. It would be interesting to examine possible contributions from other neurotrophic factors.
It is now critical to further investigate the mechanisms underlying the upregulation of BDNF and/or other effective growth factors in order to discover more efficacious medications. In general, BDNF levels are regulated by neuronal activity. In addition to the influx of Ca2+, neuronal activity, including glutamatergic regulation, contributes to the production and secretion of BDNF [93–98]. Change in the production and secretion of BDNF is thought to be involved in the activity-dependent synaptic plasticity in the CNS [99, 100]. Interestingly, two recent studies have demonstrated the role of synaptic activity in neuroprotection. In cultured hippocampal neurons, action potential bursting reduced the levels of p53, PUMA and Apaf1 [101]. Furthermore, NMDA receptor stimulation inhibited PUMA-mediated apoptosis via reducing levels of Apaf1 and procaspase-9 [102]. In support of these current studies, a previous study demonstrated that transcranial magnetic stimulation, which is well known to potentiate neuronal activity, inhibited toxic effects of 3-nitropropionic acid (3-NPA) (protein/lipid oxidations, reduction in activities of catalase, GSH peroxidase and succinate dehydrogenase, and GSH deficiency) and rescued the striatal neuronal loss in rats [103]. It is necessary to investigate whether or not such neuronal activity-mediated protection occurs via the upregulation of BDNF. Additionally, future studies investigating the role of neuronal activity in the expression of neurotrophic factors that are influenced by molecules that cross the blood brain barrier are needed.
Transplantation of growth factor-secreting cells may serve as an alternative method to treat neurodegenerative diseases. Indeed, the grafting of neurotrophin-secreting cell lines has been shown to protect neurons against quinolinate-induced cell death in an animal model of HD [64]. In addition, it was shown that erythropoietin-transduced human mesenchymal stromal cells (EPO-MSCs) played a neuroprotective role in the rat model for ischemic stroke [104]. In the EPO-MSCs, neurotrophic factors including BDNF and HGF were upregulated. The implantation of EPO-MSCs into ischemic rats reversed impairment in neurological function and infarct volumes [104]. Finally, a gene transfer approach may be a potentially effective strategy as well. In an in vivo cognitive dysfunction model induced by Aβ injection, HGF gene transfer improved impairment of cognitive behavior. It was suggested that BDNF upregulation was involved in the positive action of HGF gene transfer [105]. Further investigation on the possible mechanisms underlying the BDNF upregulation is interesting.
6. Estrogen Signaling and Oxidative Stress
Estrogen, one of the sex steroids, has various roles in sex differentiation, neuroprotection, and synaptic plasticity [14–16, 106]. Furthermore, estrogenic protection from toxicity including excitotoxicity and oxidative stress is well studied [107–109]. Importantly, the maintenance of mitochondrial function is linked to estrogenic protection under toxic stress. Protein phosphatases influence activation levels of kinase signaling and of mitochondrial apoptosis-related proteins, and such intracellular mechanisms are closely associated with estrogenic protection [110].
Generally, estrogens are believed to regulate transcription of target genes via estrogen receptor α (ERα) and ERβ. Estrogens bind to ERα and ERβ, exerting various effects via initiating diverse intracellular signaling cascades. Specifically, the discovery of ERβ prompted major developments leading towards the understanding of estrogenic function [111, 112]. In addition, it has been recently suggested that estrogens also exert their effects via ER-mediated nongenomic or non-ER-mediated functions.
Estrogens protect neurons from severe conditions including oxidative stress. 17β-estradiol (E2), one of the estrogens, reduces CA1 hippocampal cell death following global cerebral ischemia [113]. In that in vivo system, Nox activity and superoxide production in the hippocampal region were repressed by E2 application. Interestingly, extranuclear ERα-dependent nongenomic function, including the activation of Akt, is involved in the E2 effect [113]. Xia et al. examined the effect of selective ER ligands on glutamate toxicity. In cultured cortical neurons, R,R-tetrahydrochrysene (R,R-THC, ERβ antagonist and ERα agonist) displays a neuroprotective effect against glutamate-induced cell death [114], suggesting an important role of ERα in estrogen-mediated neuroprotection. On the other hand, a knockdown of ERβ induced a lower resting mitochondrial membrane potential in immortal hippocampal and primary hippocampal neurons [115]. The ERβ knockdown resulted in maintenance of adenosine 5′-triphosphate (ATP) concentration, and decreased mitochondrial superoxide levels under H2O2 stress. As expected, the neuronal loss of ERβ knockdown cells diminished in the presence of oxidative stress caused by glutamate or H2O2 [115]. Recently, the novel function of GPR30, a G protein-coupled ER, has been reported. Gingerich et al. found that pretreatment with E2 decreased cell death caused by glutamate, which may be partially mediated by GPR30 [116].
It is possible that ERβ regulates neuronal activity. As a result of neurotransmission, spontaneous Ca2+ oscillations occured and our group previously showed potentiation in glutamate-mediated Ca2+ oscillation after BDNF addition [117]. In our cortical cultures, voltage-dependent Ca2+ channels and ionotropic glutamate receptors contributed to the spontaneous Ca2+ oscillations, and BDNF-induced glutamate release was critical for the potentiation in the oscillations. Recently, Zhang et al. found that selective ERβ agonists (not ERα agonists) rapidly potentiated Ca2+ oscillations in neurons derived from embryonic stem cells and activated protein kinase C (PKC), Akt, and ERK pathways. Interestingly, nifedipine, a blocker of L-type voltage-dependent Ca2+ channels, abolished these ERβ actions [118], suggesting that estrogen regulates neuronal function via ERβ. Remarkably, membrane-localized ERα activates mGluR5 signaling (one of the metabotropic glutamate receptors) to stimulate CREB in striatal neurons. Furthermore, both ERα and ERβ activate mGluR3 to attenuate L-type voltage-dependent Ca2+ channel-mediated CREB activation [119]. Considering that CREB is involved in the transcriptional production of BDNF [78], the action of these ERs may affect BDNF levels in neuronal cells.
7. Estrogen and Ca2+ Homeostasis under Oxidative Stress
Using cultured cortical neurons, we demonstrated the protective effect of E2 against cell death under oxidative stress caused by H2O2 [120] (Figure 2). Members of the MAPK family including c-jun N-terminal kinase (JNK) [121], p38 [122], and ERK [123, 124] play pivotal roles in neuronal apoptosis [125] (Figure 1). In our system, the exposure to H2O2 triggered the overactivation of the ERK pathway, leading to an abnormal increase in intracellular Ca2+ concentration (Figure 3). In general, perturbations of Ca2+ homeostasis are related to apoptosis in various cell populations [126–131]. In our neurons, the abnormal Ca2+ accumulation caused by H2O2 was significantly decreased by E2 pretreatment, or in the presence of U0126, an inhibitor for ERK signaling [120]. Recently, we reported that ERK signaling plays a role in maintaining adequate expression levels of glutamate receptors [132–134]. Importantly, chronic E2 treatment induced the downregulation of ionotropic glutamate receptor subunits including NR2A and GluR2/3. Such a decrease in glutamate receptor expression levels was also confirmed after U0126 addition. Indeed, such E2 treatment suppressed the overactivation of ERK pathway stimulated by H2O2. Furthermore, inhibitors of ionotropic glutamate receptors blocked cell death caused by H2O2. Taken together, it is possible that E2 exerts survival-promoting effects through repressing glutamate receptor-mediated Ca2+ influx [120] (Figure 3). As described, ERK signaling is essential for maintenance of glutamate receptor levels, making it interesting to investigate how estrogens influence ERK signaling.
Figure 2.
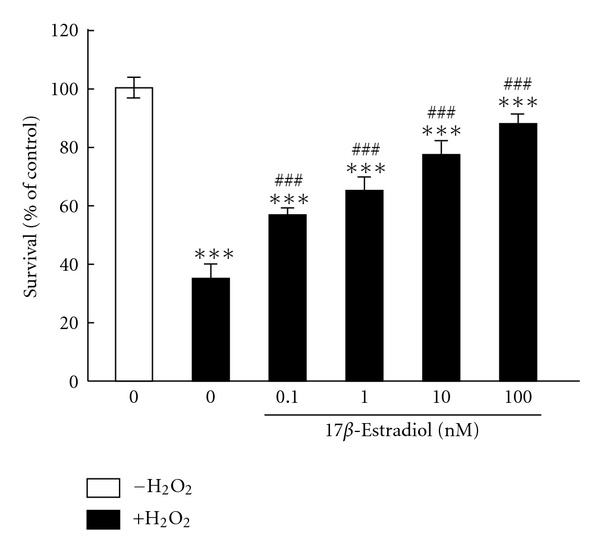
17β-estradiol prevents cortical neurons from cell death caused by H2O2 exposure. Dissociated cortical neurons were prepared from cerebral cortex of postnatal 2-day-old rats. At 6 days in vitro, 17β-estradiol was applied at indicated concentrations. Twenty-four hours later, H2O2 (final 50 μM) was added to induce cell death. Following an additional twelve-hour culture, cell survival was determined using an MTT (tetrazolium salt) assay. Data represent mean ± S.D. (n = 6). ***P < .001 versus control (no H2O2). ### P < .001 versus no estradiol + H2O2.
Figure 3.
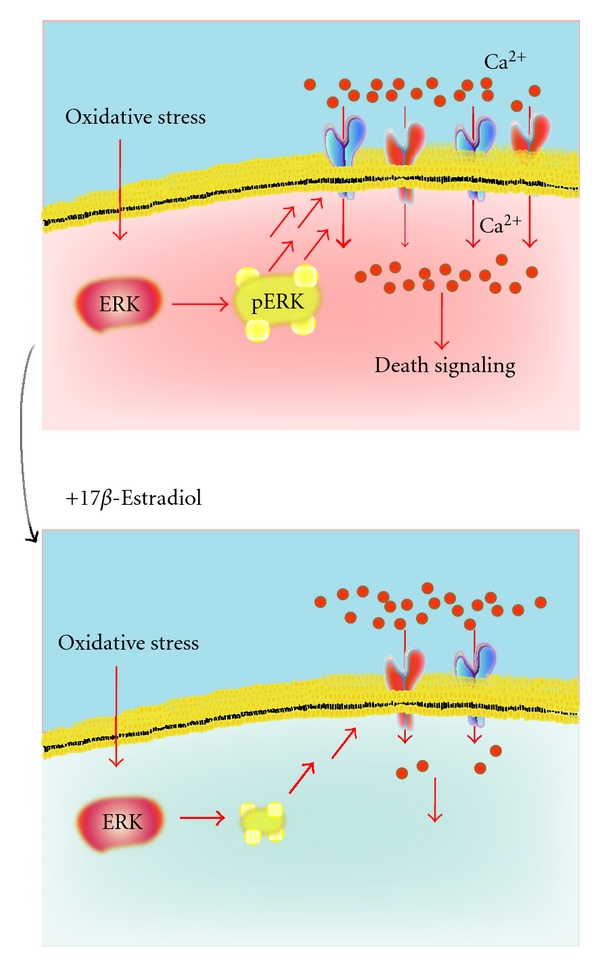
17β-estradiol inhibits neuronal cell death under oxidative stress via reducing the series of events evoked by exposure to H2O2, including overactivation of the ERK signaling and overload of Ca2+. Upper: After H2O2 addition, marked phosphorylated (activated) ERK (pERK) and resultant increase in intracellular Ca2+ concentration were observed, resulting in cell death. Lower: Pretreatment with 17β-estradiol induced downregulation of ionotropic glutamate receptors via decreasing ERK activation, while also serving to decrease levels of Ca2+ influx triggered by H2O2. Such a decrease in glutamate receptor expression and intracellular Ca2+ was also confirmed in the presence of U0126, an inhibitor of ERK signaling. As expected, chronic 17β-estradiol reduced levels of pERK stimulated by H2O2. A blockade of glutamate receptors rescued cortical cells from H2O2-dependent death. Therefore, it is possible that 17β-estradiol promotes survival via suppressing glutamate receptor-mediated Ca2+ influx, due to downregulation of ionotropic glutamate receptors [120].
p66Shc also generates mitochondrial ROS (H2O2), causes impairment in Ca2+ homeostasis, and is associated with apoptosis [135, 136]. Almeida et al. found that H2O2 stimulates PKCβ/p66Shc/NF-κB signaling to apoptosis in osteoblastic cells, and that E2 prevents H2O2-dependent activation of p66Shc and NF-κB via repressing phosphorylation of PKCβ, resulting in protection from cell death [137].
8. Estrogen In Vivo Approach
In 6-hydroxydopamine (6-OHDA, a PD mimetic)-lesioned rats, a neuroprotective effect of silymarin (SM, one of flavonolignans) was shown [138]. SM administration protected neurons of the substantia nigra pars compacta from 6-OHDA toxicity, while fulvestrant, an ER antagonist, partially blocked the effects of SM. Additionally, the effect of oral estrogen on ROS generation was reported. Wing et al. demonstrated a beneficial effect of chronic oral estrogen treatment on oxidative stress and atherosclerosis in apoE-deficient mice [139]. Using ovariectomized apoE-deficient mice, it was revealed that atherosclerosis was reduced when treated with E2 (6 μg/day) for 12 weeks. Importantly, after E2 treatment, superoxide anion and expression of Nox decreased, while Cu/ZnSOD and MnSOD increased [139]. Last, Schwann cells (SC) play a critical role in spinal cord injury repair, though SC survival after transplantation is very difficult. Current research is focused on discovering if E2 pretreatment protects SC, in an effort to generate more successful spinal cord transplantation procedures [140]. In primary SC cultures, strong expression of ERα and ERβ, and overall E2-dependent survival mechanisms against H2O2 exposure were confirmed, though ICI182780 (an ER antagonist) had no influence on E2 effects. These findings suggest that genomic signaling via ERs is not involved. Importantly, in spinal cord injury, sustained E2 administration was found to be an effective treatment improving SC transplantation [140].
9. Conclusion
An increase in neuronal damage at the cellular and molecular level may be involved in the pathogenesis of brain illness, including neurodegenerative disease. It is possible that oxidative stress leads to neuronal cell death via increasing glutamate-mediated excitotoxicity, intracellular Ca2+ concentration, mitochondrial dysfunction, activation of death-signaling cascades, and decreasing overall survival signaling. Several drug candidates, which were found to attenuate deleterious symptoms in various models of neurodegenerative disease, are reported to upregulate the expression of neurotrophic factors including BDNF. Considering this, it seems pertinent to further investigate the possible mechanisms underlying such neurotrophic factor upregulation. On the other hand, estrogenic survival promotion is also well studied, though further investigation addressing how each ER contributes to neuronal protection against oxidative toxicity is needed. Finally, as a close relationship between steroid hormones and BDNF in various neuronal functions including cell survival is known [141], detailed studies concerning how estrogen and BDNF interact with each other in CNS neurons under oxidative stress are important.
Acknowledgments
This work was supported by a grant from the Core Research for Evolutional Science and Technology Program (CREST) Japan Science and Technology Agency (JST) (T. N., T. M., S. Y. and H. K.), the Takeda Science Foundation (T. N.), the Japan Health Sciences Foundation (Research on Health Sciences focusing on Drug Innovation) (H. K.), Health and Labor Sciences Research Grants (Comprehensive Research on Disability, Health, and Welfare) (H. K.), Intramural Research Grants (20-3, 21-9) for Neurological and Psychiatric Disorders of NCNP (H. K.), and Grants-in-Aid for Scientific Research (B) (grant number 20390318) (H. K.) and Young Scientists (A) (21680034) (T. N.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1.Sakurada O, Kennedy C, Jehle J, Brown JD, Carbin GL, Sokoloff L. Measurement of local cerebral blood flow with iodo [14C] antipyrine. The American Journal of Physiology. 1978;234(1):H59–H66. doi: 10.1152/ajpheart.1978.234.1.H59. [DOI] [PubMed] [Google Scholar]
- 2.Cremer JE, Seville MP. Regional brain blood flow, blood volume, and haematocrit values in the adult rat. Journal of Cerebral Blood Flow and Metabolism. 1983;3(2):254–256. doi: 10.1038/jcbfm.1983.35. [DOI] [PubMed] [Google Scholar]
- 3.Scremin OU, Sonnenschein RR, Rubinstein EH. Cholinergic cerebral vasodilatation: lack of involvement of cranial parasympathetic nerves. Journal of Cerebral Blood Flow and Metabolism. 1983;3(3):362–368. doi: 10.1038/jcbfm.1983.52. [DOI] [PubMed] [Google Scholar]
- 4.Sokoloff L, Reivich M, Kennedy C, et al. The [C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. Journal of Neurochemistry. 1977;28(5):897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- 5.Sokoloff L. Localization of functional activity in the central nervous system by measurement of glucose utilization with radioactive deoxyglucose. Journal of Cerebral Blood Flow and Metabolism. 1981;1(1):7–36. doi: 10.1038/jcbfm.1981.4. [DOI] [PubMed] [Google Scholar]
- 6.Satoh T, Enokido Y, Kubo T, Yamada M, Hatanaka H. Oxygen toxicity induces apoptosis in neuronal cells. Cellular and Molecular Neurobiology. 1998;18(6):649–666. doi: 10.1023/A:1020633919115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nature Medicine. 2004;10:S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 8.Harman D. Free radical theory of aging: an update—increasing the functional life span. Annals of the New York Academy of Sciences. 2006;1067(1):10–21. doi: 10.1196/annals.1354.003. [DOI] [PubMed] [Google Scholar]
- 9.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nature Medicine. 2004;10(7):S2–S9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 10.Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annual Review of Biochemistry. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 11.Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H. BDNF function and intracellular signaling in neurons. Histology and Histopathology. 2010;25(2):237–258. doi: 10.14670/HH-25.237. [DOI] [PubMed] [Google Scholar]
- 12.Gill SS, Patel NK, Hotton GR, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nature Medicine. 2003;9(5):589–595. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- 13.Maina F, Klein R. Hepatocyte growth factor, a versatile signal for developing neurons. Nature Neuroscience. 1999;2(3):213–217. doi: 10.1038/6310. [DOI] [PubMed] [Google Scholar]
- 14.Brinton RD. Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends in Pharmacological Sciences. 2009;30(4):212–222. doi: 10.1016/j.tips.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee AW, Pfaff DW. Hormone effects on specific and global brain functions. Journal of Physiological Sciences. 2008;58(4):213–220. doi: 10.2170/physiolsci.RV007008. [DOI] [PubMed] [Google Scholar]
- 16.Tobet S, Knoll JG, Hartshorn C, et al. Brain sex differences and hormone influences: a moving experience? Journal of Neuroendocrinology. 2009;21(4):387–392. doi: 10.1111/j.1365-2826.2009.01834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simpkins JW, Yi KD, Yang SH, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Biochimica et Biophysica Acta. 2010;1800(10):1113–1120. doi: 10.1016/j.bbagen.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxidants and Redox Signaling. 2007;9(2):233–244. doi: 10.1089/ars.2007.9.ft-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radical Biology and Medicine. 2007;43(5):658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang X, Atwood CS, Hartshorn MA, et al. The Aβ peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38(24):7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 21.Huang X, Cuajungco MP, Atwood CS, et al. Cu(II) potentiation of Alzheimer Aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. Journal of Biological Chemistry. 1999;274(52):37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 22.Jiang D, Men L, Wang J, et al. Redox reactions of copper complexes formed with different β-amyloid peptides and their neuropathalogical relevance. Biochemistry. 2007;46(32):9270–9282. doi: 10.1021/bi700508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nadal RC, Rigby SEJ, Viles JH. Amyloid β-Cu2+ complexes in both monomeric and fibrillar forms do not generate H2O2 catalytically but quench hydroxyl radicals. Biochemistry. 2008;47(44):11653–11664. doi: 10.1021/bi8011093. [DOI] [PubMed] [Google Scholar]
- 24.Zhou C, Huang Y, Przedborski S. Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance. Annals of the New York Academy of Sciences. 2008;1147:93–104. doi: 10.1196/annals.1427.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin Z, Hu D, Han S, Reaney SH, Di Monte DA, Fink AL. Effect of 4-hydroxy-2-nonenal modification on α-synuclein aggregation. Journal of Biological Chemistry. 2007;282(8):5862–5870. doi: 10.1074/jbc.M608126200. [DOI] [PubMed] [Google Scholar]
- 26.Stack EC, Matson WR, Ferrante RJ. Evidence of oxidant damage in Huntington’s disease: translational strategies using antioxidants. Annals of the New York Academy of Sciences. 2008;1147:79–92. doi: 10.1196/annals.1427.008. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Valencia A, Sapp E, et al. Aberrant rab 11-dependent trafficking of the neuronal glutamate transporter EAACl causes oxidative stress and cell death in huntington’s disease. Journal of Neuroscience. 2010;30(13):4552–4561. doi: 10.1523/JNEUROSCI.5865-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464(7288):529–535. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Annals of the New York Academy of Sciences. 2008;1147:53–60. doi: 10.1196/annals.1427.002. [DOI] [PubMed] [Google Scholar]
- 30.Adam-Vizi V, Starkov AA. Calcium and mitochondrial reactive oxygen species generation: how to read the facts. Journal of Alzheimer’s Disease. 2010;20(supplment 2):S413–S426. doi: 10.3233/JAD-2010-100465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henchcliffe C, Beal FM. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nature Clinical Practice Neurology. 2008;4(11):600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 32.Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington’s disease. European Journal of Neuroscience. 2008;27(11):2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- 33.Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. Journal of Neurochemistry. 2009;109(1):133–138. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Shachar D, Laifenfeld D. Mitochondria, synaptic plasticity, and schizophrenia. International Review of Neurobiology. 2004;59:273–296. doi: 10.1016/S0074-7742(04)59011-6. [DOI] [PubMed] [Google Scholar]
- 35.Gardner A, Boles RG. Beyond the serotonin hypothesis: mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2011;35(3):730–743. doi: 10.1016/j.pnpbp.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 36.Kato T. Role of mitochondrial DNA in calcium signaling abnormality in bipolar disorder. Cell Calcium. 2008;44(1):92–102. doi: 10.1016/j.ceca.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Oliveira JMA, Jekabsons MB, Chen S, et al. Mitochondrial dysfunction in Huntington’s disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. Journal of Neurochemistry. 2007;101(1):241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- 38.Gandhi S, Wood-Kaczmar A, Yao Z, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Molecular Cell. 2009;33(5):627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tu H, Nelson O, Bezprozvanny A, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126(5):981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. Journal of Neuroscience. 2010;30(25):8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheung KH, Shineman D, Müller M, et al. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58(6):871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59(2):214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brennan AM, Suh SW, Won SJ, et al. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nature Neuroscience. 2009;12(7):857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. Journal of Cerebral Blood Flow and Metabolism. 2009;29(7):1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi DW. Neurodegeneration: cellular defences destroyed. Nature. 2005;433(7027):696–698. doi: 10.1038/433696a. [DOI] [PubMed] [Google Scholar]
- 46.Steckley D, Karajgikar M, Dale LB, et al. Puma is a dominant regulator of oxidative stress induced bax activation and neuronal apoptosis. Journal of Neuroscience. 2007;27(47):12989–12999. doi: 10.1523/JNEUROSCI.3400-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2009;40(2):618–625. doi: 10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nature Reviews Molecular Cell Biology. 2007;8(5):405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 49.Candé C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? Journal of Cell Science. 2002;115(24):4727–4734. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 50.Aoyama K, Sang WS, Hamby AM, et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nature Neuroscience. 2006;9(1):119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- 51.Berman AE, Chan WY, Brennan AM, et al. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1(-/-) mouse. Annals of Neurology. 2011;69(3):509–520. doi: 10.1002/ana.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kairisalo M, Bonomo A, Hyrskyluoto A, et al. Resveratrol reduces oxidative stress and cell death and increases mitochondrial antioxidants and XIAP in PC6.3-cells. Neuroscience Letters. 2011;488(3):263–266. doi: 10.1016/j.neulet.2010.11.042. [DOI] [PubMed] [Google Scholar]
- 53.Fallarini S, Miglio G, Paoletti T, et al. Clovamide and rosmarinic acid induce neuroprotective effects in in vitro models of neuronal death. British Journal of Pharmacology. 2009;157(6):1072–1084. doi: 10.1111/j.1476-5381.2009.00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimojo Y, Kosaka K, Noda Y, Shimizu T, Shirasawa T. Effect of rosmarinic acid in motor dysfunction and life span in a mouse model of familial amyotrophic lateral sclerosis. Journal of Neuroscience Research. 2010;88(4):896–904. doi: 10.1002/jnr.22242. [DOI] [PubMed] [Google Scholar]
- 55.Chao J, Lau WKW, Huie MJ, et al. A pro-drug of the green tea polyphenol (-)-epigallocatechin-3-gallate (EGCG) prevents differentiated SH-SY5Y cells from toxicity induced by 6-hydroxydopamine. Neuroscience Letters. 2010;469(3):360–364. doi: 10.1016/j.neulet.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 56.Lagoa R, Lopez-Sanchez C, Samhan-Arias AK, Gañan CM, Garcia-Martinez V, Gutierrez-Merino C. Kaempferol protects against rat striatal degeneration induced by 3-nitropropionic acid. Journal of Neurochemistry. 2009;111(2):473–487. doi: 10.1111/j.1471-4159.2009.06331.x. [DOI] [PubMed] [Google Scholar]
- 57.Luo D, Zhang Q, Wang H, et al. Fucoidan protects against dopaminergic neuron death in vivo and in vitro. European Journal of Pharmacology. 2009;617(1–3):33–40. doi: 10.1016/j.ejphar.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 58.Kim J, Lee HJ, Lee KW. Naturally occurring phytochemicals for the prevention of Alzheimer’s disease. Journal of Neurochemistry. 2010;112(6):1415–1430. doi: 10.1111/j.1471-4159.2009.06562.x. [DOI] [PubMed] [Google Scholar]
- 59.Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrer I, Goutan E, Marín C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Research. 2000;866(1-2):257–261. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- 61.Gines S, Seong IS, Fossale E, et al. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Human Molecular Genetics. 2003;12(5):497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- 62.Ginés S, Bosch M, Marco S, et al. Reduced expression of the TrkB receptor in Huntington’s disease mouse models and in human brain. European Journal of Neuroscience. 2006;23(3):649–658. doi: 10.1111/j.1460-9568.2006.04590.x. [DOI] [PubMed] [Google Scholar]
- 63.Ginés S, Paoletti P, Alberch J. Impaired TrkB-mediated ERK1/2 activation in huntington disease knock-in striatal cells involves reduced p52/p46 Shc expression. Journal of Biological Chemistry. 2010;285(28):21537–21548. doi: 10.1074/jbc.M109.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perez-Navarro E, Canudas AM, Akerud P, Alberch J, Arenas E. Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 prevent the death of striatal projection neurons in a rodent model of Huntington’s disease. Journal of Neurochemistry. 2000;75(5):2190–2199. doi: 10.1046/j.1471-4159.2000.0752190.x. [DOI] [PubMed] [Google Scholar]
- 65.Counts SE, Mufson EJ. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. Journal of Neurochemistry. 2010;113(3):649–660. doi: 10.1111/j.1471-4159.2010.06622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng H, Yu J, Jiang Z, et al. Acupuncture improves cognitive deficits and regulates the brain cell proliferation of SAMP8 mice. Neuroscience Letters. 2008;432(2):111–116. doi: 10.1016/j.neulet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 67.Zhao H, Li Q, Zhang Z, Pei X, Wang J, Li Y. Long-term ginsenoside consumption prevents memory loss in aged SAMP8 mice by decreasing oxidative stress and up-regulating the plasticity-related proteins in hippocampus. Brain Research. 2009;1256(C):111–122. doi: 10.1016/j.brainres.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 68.Hung SY, Liou HC, Kang KH, Wu RM, Wen CC, Fu WM. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Molecular Pharmacology. 2008;74(6):1564–1575. doi: 10.1124/mol.108.048611. [DOI] [PubMed] [Google Scholar]
- 69.Hung SY, Liou HC, Fu WM. The mechanism of heme oxygenase-1 action involved in the enhancement of neurotrophic factor expression. Neuropharmacology. 2010;58(2):321–329. doi: 10.1016/j.neuropharm.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 70.Li C, Biswas S, Li X, Dutta AK, Le W. Novel D3 dopamine receptor-preferring agonist D-264: evidence of neuroprotective property in Parkinson’s disease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin. Journal of Neuroscience Research. 2010;88(11):2513–2523. doi: 10.1002/jnr.22405. [DOI] [PubMed] [Google Scholar]
- 71.Cho HS, Kim S, Lee SY, Park JA, Kim SJ, Chun HS. Protective effect of the green tea component, l-theanine on environmental toxins-induced neuronal cell death. NeuroToxicology. 2008;29(4):656–662. doi: 10.1016/j.neuro.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 72.Takai S, Yamada M, Araki T, Koshimizu H, Nawa H, Hatanaka H. Shp-2 positively regulates brain-derived neurotrophic factor-promoted survival of cultured ventral mesencephalic dopaminergic neurons through a brain immunoglobulin-like molecule with tyrosine-based activation motifs/Shp substrate-1. Journal of Neurochemistry. 2002;82(2):353–364. doi: 10.1046/j.1471-4159.2002.00960.x. [DOI] [PubMed] [Google Scholar]
- 73.Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, Gill SS. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nature Medicine. 2005;11(7):703–704. doi: 10.1038/nm0705-703. [DOI] [PubMed] [Google Scholar]
- 74.Numakawa T, Kumamaru E, Adachi N, Yagasaki Y, Izumi A, Kunugi H. Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-γ signaling for glutamate release via a glutamate transporter. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(2):647–652. doi: 10.1073/pnas.0800888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woronowicz A, Amith SR, Davis VW, et al. Trypanosome trans-sialidase mediates neuroprotection against oxidative stress, serum/glucose deprivation, and hypoxia-induced neurite retraction in Trk-expressing PC12 cells. Glycobiology. 2007;17(7):725–734. doi: 10.1093/glycob/cwm034. [DOI] [PubMed] [Google Scholar]
- 76.Boutahar N, Reynaud E, Lassabliere F, Borg J. Brain-derived neurotrophic factor inhibits cell cycle reentry but not endoplasmic reticulum stress in cultured neurons following oxidative or excitotoxic stress. Journal of Neuroscience Research. 2010;88(10):2263–2271. doi: 10.1002/jnr.22384. [DOI] [PubMed] [Google Scholar]
- 77.Lee B, Cao R, Choi YS, et al. The CREB/CRE transcriptional pathway: protection against oxidative stress-mediated neuronal cell death. Journal of Neurochemistry. 2009;108(5):1251–1265. doi: 10.1111/j.1471-4159.2008.05864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35(4):605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 79.Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. Journal of Neuroscience Research. 2007;85(3):525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60(4):610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allen SJ, Dawbarn D. Clinical relevance of the neurotrophins and their receptors. Clinical Science. 2006;110(2):175–191. doi: 10.1042/CS20050161. [DOI] [PubMed] [Google Scholar]
- 82.Choi BS, Sapkota K, Kim S, Lee HJ, Choi HS, Kim SJ. Antioxidant activity and protective effects of tripterygium regelii extract on hydrogen peroxide-induced injury in human dopaminergic cells, SH-SY5Y. Neurochemical Research. 2010;35(8):1269–1280. doi: 10.1007/s11064-010-0185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hooper JE, Scott MP. Communicating with hedgehogs. Nature Reviews Molecular Cell Biology. 2005;6(4):306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 84.Dai RL, Zhu SY, Xia YP, et al. Sonic hedgehog protects cortical neurons against oxidative stress. Neurochemical Research. 2011;36(1):67–75. doi: 10.1007/s11064-010-0264-6. [DOI] [PubMed] [Google Scholar]
- 85.Khanna S, Roy S, Ryu H, et al. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. Journal of Biological Chemistry. 2003;278(44):43508–43515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sherer TB, Betarbet R, Testa CM, et al. Mechanism of toxicity in rotenone models of Parkinson’s disease. Journal of Neuroscience. 2003;23(34):10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Osakada F, Hashino A, Kume T, Katsuki H, Kaneko S, Akaike A. α-tocotrienol provides the most potent neuroprotection among vitamin E analogs on cultured striatal neurons. Neuropharmacology. 2004;47(6):904–915. doi: 10.1016/j.neuropharm.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 88.Conte V, Uryu K, Fujimoto S, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. Journal of Neurochemistry. 2004;90(3):758–764. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- 89.Nakashima H, Ishihara T, Yokota O, et al. Effects of α-tocopherol on an animal model of tauopathies. Free Radical Biology and Medicine. 2004;37(2):176–186. doi: 10.1016/j.freeradbiomed.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 90.Kontush A, Schekatolina S. Vitamin E in neurodegenerative disorders: Alzheimer’s disease. Annals of the New York Academy of Sciences. 2004;1031:249–262. doi: 10.1196/annals.1331.025. [DOI] [PubMed] [Google Scholar]
- 91.Praticò D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: lights and shadows. Annals of the New York Academy of Sciences. 2008;1147:70–78. doi: 10.1196/annals.1427.010. [DOI] [PubMed] [Google Scholar]
- 92.Numakawa Y, Numakawa T, Matsumoto T, et al. Vitamin E protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Journal of Neurochemistry. 2006;97(4):1191–1202. doi: 10.1111/j.1471-4159.2006.03827.x. [DOI] [PubMed] [Google Scholar]
- 93.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20(4):727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 94.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20(4):709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 95.Gwag BJ, Springer JE. Activation of NMDA receptors increases brain-derived neurotrophic factor (BDNF) mRNA expression in the hippocampal formation. NeuroReport. 1993;5(2):125–128. doi: 10.1097/00001756-199311180-00007. [DOI] [PubMed] [Google Scholar]
- 96.Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO Journal. 2001;20(21):5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. Journal of Neuroscience. 2000;20(19):7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. Journal of Neuroscience. 2002;22(23):10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Greenberg ME, Xu B, Lu B, Hempstead BL. New insights in the biology of BDNF synthesis and release: implications in CNS function. Journal of Neuroscience. 2009;29(41):12764–12767. doi: 10.1523/JNEUROSCI.3566-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annual Review of Neuroscience. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lau D, Bading H. Synaptic activity-mediated suppression of p53 and induction of nuclear calcium-regulated neuroprotective genes promote survival through inhibition of mitochondrial permeability transition. Journal of Neuroscience. 2009;29(14):4420–4429. doi: 10.1523/JNEUROSCI.0802-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Léveillé F, Papadia S, Fricker M, et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. Journal of Neuroscience. 2010;30(7):2623–2635. doi: 10.1523/JNEUROSCI.5115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Túnez I, Drucker-Colín R, Jimena I, et al. Transcranial magnetic stimulation attenuates cell loss and oxidative damage in the striatum induced in the 3-nitropropionic model of Huntington’s disease. Journal of Neurochemistry. 2006;97(3):619–630. doi: 10.1111/j.1471-4159.2006.03724.x. [DOI] [PubMed] [Google Scholar]
- 104.Cho GW, Koh SH, Kim MH, et al. The neuroprotective effect of erythropoietin-transduced human mesenchymal stromal cells in an animal model of ischemic stroke. Brain Research. 2010;1353:1–13. doi: 10.1016/j.brainres.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 105.Takeuchi D, Sato N, Shimamura M, et al. Alleviation of Aβ-induced cognitive impairment by ultrasound-mediated gene transfer of HGF in a mouse model. Gene Therapy. 2008;15(8):561–571. doi: 10.1038/sj.gt.3303094. [DOI] [PubMed] [Google Scholar]
- 106.Brinton RD. Cellular and molecular mechanisms of estrogen regulation of memory function and neuroprotection against Alzheimer’s disease: recent insights and remaining challenges. Learning and Memory. 2001;8(3):121–133. doi: 10.1101/lm.39601. [DOI] [PubMed] [Google Scholar]
- 107.Kurata K, Takebayashi M, Morinobu S, Yamawaki S. β-estradiol, dehydroepiandrosterone, and dehydroepiandrosterone sulfate protect against N-methyl-D-aspartate-induced neurotoxicity in rat hippocampal neurons by different mechanisms. Journal of Pharmacology and Experimental Therapeutics. 2004;311(1):237–245. doi: 10.1124/jpet.104.067629. [DOI] [PubMed] [Google Scholar]
- 108.Simpkins JW, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Brain Research Reviews. 2008;57(2):421–430. doi: 10.1016/j.brainresrev.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 109.Liu S, Mauvais-Jarvis F. Minireview: estrogenic protection of β-cell failure in metabolic diseases. Endocrinology. 2010;151(3):859–864. doi: 10.1210/en.2009-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Simpkins JW, Yi KD, Yang SH. Role of protein phosphatases and mitochondria in the neuroprotective effects of estrogens. Frontiers in Neuroendocrinology. 2009;30(2):93–105. doi: 10.1016/j.yfrne.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kuiper GGJM, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(12):5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen signaling via estrogen receptor β . The Journal of Biological Chemistry. 2010;285(51):39575–39579. doi: 10.1074/jbc.R110.180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang QG, Raz L, Wang R, et al. Estrogen attenuates ischemic oxidative damage via an estrogen receptor α-mediated inhibition of NADPH oxidase activation. Journal of Neuroscience. 2009;29(44):13823–13836. doi: 10.1523/JNEUROSCI.3574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xia Y, Xing JZ, Krukoff TL. Neuroprotective effects of R,R-tetrahydrochrysene against glutamate-induced cell death through anti-excitotoxic and antioxidant actions involving estrogen receptor-dependent and -independent pathways. Neuroscience. 2009;162(2):292–306. doi: 10.1016/j.neuroscience.2009.04.068. [DOI] [PubMed] [Google Scholar]
- 115.Yang SH, Sarkar SN, Liu R, et al. Estrogen receptor β as a mitochondrial vulnerability factor. Journal of Biological Chemistry. 2009;284(14):9540–9548. doi: 10.1074/jbc.M808246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gingerich S, Kim GL, Chalmers JA, et al. Estrogen receptor alpha and G-protein coupled receptor 30 mediate the neuroprotective effects of 17β-estradiol in novel murine hippocampal cell models. Neuroscience. 2010;170(1):54–66. doi: 10.1016/j.neuroscience.2010.06.076. [DOI] [PubMed] [Google Scholar]
- 117.Numakawa T, Yamagishi S, Adachi N, et al. Brain-derived neurotrophic factor-induced potentiation of Ca2+ oscillations in developing cortical neurons. Journal of Biological Chemistry. 2002;277(8):6520–6529. doi: 10.1074/jbc.M109139200. [DOI] [PubMed] [Google Scholar]
- 118.Zhang L, Blackman BE, Schonemann MD, et al. Estrogen receptor β-selective agonists stimulate calcium oscillations in human and mouse embryonic stem cell-derived neurons. PLoS ONE. 2010;5(7, article e11791) doi: 10.1371/journal.pone.0011791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Grove-Strawser D, Boulware MI, Mermelstein PG. Membrane estrogen receptors activate the metabotropic glutamate receptors mGluR5 and mGluR3 to bidirectionally regulate CREB phosphorylation in female rat striatal neurons. Neuroscience. 2010;170(4):1045–1055. doi: 10.1016/j.neuroscience.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Numakawa Y, Matsumoto T, Yokomaku D, et al. 17β-estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of mitogen-activated protein kinase and in the accumulation of intracellular calcium. Endocrinology. 2007;148(2):627–637. doi: 10.1210/en.2006-1210. [DOI] [PubMed] [Google Scholar]
- 121.Inamura N, Enokido Y, Hatanaka H. Involvement of c-Jun N-terminal kinase and caspase 3-like protease in DNA damage-induced, p53-mediated apoptosis of cultured mouse cerebellar granule neurons. Brain Research. 2001;904(2):270–278. doi: 10.1016/s0006-8993(01)02472-6. [DOI] [PubMed] [Google Scholar]
- 122.Vallés SL, Borrás C, Gambini J, et al. Oestradiol or genistein rescues neurons from amyloid beta-induced cell death by inhibiting activation of p38. Aging Cell. 2008;7(1):112–118. doi: 10.1111/j.1474-9726.2007.00356.x. [DOI] [PubMed] [Google Scholar]
- 123.Ishikawa Y, Kusaka E, Enokido Y, Ikeuchi T, Hatanaka H. Regulation of Bax translocation through phosphorylation at Ser-70 of Bcl-2 by MAP kinase in NO-induced neuronal apoptosis. Molecular and Cellular Neuroscience. 2003;24(2):451–459. doi: 10.1016/s1044-7431(03)00203-3. [DOI] [PubMed] [Google Scholar]
- 124.Yamagishi S, Matsumoto T, Numakawa T, et al. ERK1/2 are involved in low potassium-induced apoptotic signaling downstream of ASK1-p38 MAPK pathway in cultured cerebellar granule neurons. Brain Research. 2005;1038(2):223–230. doi: 10.1016/j.brainres.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 125.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochimica et Biophysica Acta. 2010;1802(4):396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 126.Mattson MP. Apoptosis in neurodegenerative disorders. Nature Reviews Molecular Cell Biology. 2000;1(2):120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 127.Stanciu M, Wang Y, Kentor R, et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. Journal of Biological Chemistry. 2000;275(16):12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- 128.Crossthwaite AJ, Hasan S, Williams RJ. Hydrogen peroxide-mediated phosphorylation of ERK1/2, AKt/PKB and JNK in cortical neurones: dependence on Ca2+ and PI3-kinase. Journal of Neurochemistry. 2002;80(1):24–35. doi: 10.1046/j.0022-3042.2001.00637.x. [DOI] [PubMed] [Google Scholar]
- 129.Matsumoto Y, Yamamoto S, Suzuki Y, et al. Na+/H+ exchanger inhibitor, SM-20220, is protective against excitotoxicity in cultured cortical neurons. Stroke. 2004;35(1):185–190. doi: 10.1161/01.STR.0000106910.42815.C2. [DOI] [PubMed] [Google Scholar]
- 130.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. Journal of Neuroscience. 1993;13(5):1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Weiss JH, Hartley DM, Koh J, Choi DW. The calcium channel blocker nifedipine attenuates slow excitatory amino acid neurotoxicity. Science. 1990;247(4949 I):1474–1477. doi: 10.1126/science.247.4949.1474. [DOI] [PubMed] [Google Scholar]
- 132.Matsumoto T, Numakawa T, Yokomaku D, et al. Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: different dependency on signaling pathways and neuronal activity. Molecular and Cellular Neuroscience. 2006;31(1):70–84. doi: 10.1016/j.mcn.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 133.Tuerxun T, Numakawa T, Adachi N, et al. SA4503, a sigma-1 receptor agonist, prevents cultured cortical neurons from oxidative stress-induced cell death via suppression of MAPK pathway activation and glutamate receptor expression. Neuroscience Letters. 2010;469(3):303–308. doi: 10.1016/j.neulet.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 134.Kawashima H, Numakawa T, Kumamaru E, et al. Glucocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the suppression of microRNA-132 expression. Neuroscience. 2010;165(4):1301–1311. doi: 10.1016/j.neuroscience.2009.11.057. [DOI] [PubMed] [Google Scholar]
- 135.Giorgio M, Migliaccio E, Orsini F, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122(2):221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 136.Pellegrini M, Finetti F, Petronilli V, et al. p66Shc promotes T cell apoptosis by inducing mitochondrial dysfunction and impaired Ca2+ homeostasis. Cell Death and Differentiation. 2007;14(2):338–347. doi: 10.1038/sj.cdd.4401997. [DOI] [PubMed] [Google Scholar]
- 137.Almeida M, Han L, Ambrogini E, Bartell SM, Manolagas SC. Oxidative stress stimulates apoptosis and activates NF-κB in osteoblastic cells via a PKCβ/p66Shc signaling cascade: counter regulation by estrogens or androgens. Molecular Endocrinology. 2010;24(10):2030–2037. doi: 10.1210/me.2010-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Baluchnejadmojarad T, Roghani M, Mafakheri M. Neuroprotective effect of silymarin in 6-hydroxydopamine hemi-parkinsonian rat: involvement of estrogen receptors and oxidative stress. Neuroscience Letters. 2010;480(3):206–210. doi: 10.1016/j.neulet.2010.06.038. [DOI] [PubMed] [Google Scholar]
- 139.Wing LYC, Chen YC, Shih YY, Cheng JC, Lin YJ, Jiang MJ. Effects of oral estrogen on aortic ROS-generating and -scavenging enzymes and atherosclerosis in apoE-deficient mice. Experimental Biology and Medicine. 2009;234(9):1037–1046. doi: 10.3181/0811-RM-332. [DOI] [PubMed] [Google Scholar]
- 140.Siriphorn A, Chompoopong S, Floyd CL. 17β-estradiol protects Schwann cells against H2O2-induced cytotoxicity and increases transplanted Schwann cell survival in a cervical hemicontusion spinal cord injury model. Journal of Neurochemistry. 2010;115(4):864–872. doi: 10.1111/j.1471-4159.2010.06770.x. [DOI] [PubMed] [Google Scholar]
- 141.Numakawa T, Yokomaku D, Richards M, Hori H, Adachi N, Kunugi H. Functional interactions between steroid hormones and neurotrophin BDNF. World Journal of Biological Chemistry. 2010;1(5):133–143. doi: 10.4331/wjbc.v1.i5.133. [DOI] [PMC free article] [PubMed] [Google Scholar]