

Abstract
Numerous studies have documented the mechanisms that regulate intracellular pH (pHi) in hippocampal neurons in response to an acid load. Here, we studied the response of pHi to depolarization in cultured hippocampal neurons. Elevation of external K+ (6–30 mm) elicited an acid transient followed by a large net alkaline shift. Similar responses were observed in acutely dissociated hippocampal neurons. In Ca2+-free media, the acid response was curtailed and the alkaline shift enhanced. DIDS blocked the alkaline response and revealed a prolonged underlying acidification that was highly dependent on Ca2+ entry. Similar alkaline responses could be elicited by AMPA, indicating that this rise in pHi was a depolarization-induced alkalinization (DIA). The DIA was found to consist of Cl−-dependent and Cl−-independent components, each accounting for approximately one-half of the peak amplitude. The Cl−-independent component was postulated to arise from operation of the electrogenic Na+–HCO3− cotransporter NBCe1. Quantitative PCR and single-cell multiplex reverse transcription-PCR demonstrated message for NBCe1 in our hippocampal neurons. In neurons cultured from Slc4a4 knock-out (KO) mice, the DIA was reduced by approximately one-half compared with wild type, suggesting that NBCe1 was responsible for the Cl−-independent DIA. In Slc4a4 KO neurons, the remaining DIA was virtually abolished in Cl−-free media. These data demonstrate that DIA of hippocampal neurons occurs via NBCe1, and a parallel DIDS-sensitive, Cl−-dependent mechanism. Our results indicate that, by activating net acid extrusion in response to depolarization, hippocampal neurons can preempt a large, prolonged, Ca2+-dependent acidosis.
Introduction
In hippocampal neurons, and other nerve cells, depolarizing stimuli cause a fall in intracellular pH (pHi), linked to the entry of Ca2+. This acid is generated because of Ca2+–H+ exchange by Ca2+-ATPases, located in the plasma membrane and endoplasmic reticulum, and may also arise from the generation of metabolic acids (for review, see Chesler, 2003). Most cells respond to a fall in pHi by actively extruding net H+ from the cytosol. In hippocampal neurons, this is accomplished primarily by electro-neutral mechanisms that include a Na+–H+ exchanger (NHE), a Na+-driven Cl−–HCO3− exchanger (NDCBE), and one or more additional Na+–HCO3− cotransporters. After an imposed acidification, these mechanisms allow pHi to return toward baseline over the course of several minutes (Raley-Susman et al., 1991; Schwiening and Boron, 1994; Baxter and Church, 1996; Bevensee et al., 1996; Cooper et al., 2005).
In addition to these electro-neutral processes, hippocampal neurons also express an electrogenic Na+–HCO3− cotransporter (Bevensee et al., 2000; Schmitt et al., 2000; Rickmann et al., 2007). The role of this carrier (NBCe1; gene name Slc4a4) in neuronal function is unknown. In astrocytes, and some invertebrate glia, this mechanism imports two HCO3− ions with one Na+ and thereby gives rise to a depolarization-induced alkalinization (DIA) (Deitmer and Szatkowski, 1990; Brookes and Turner, 1994; Pappas and Ransom, 1994) that is pronounced and rapid in vivo (Chesler and Kraig, 1987, 1989). The role of NBCe1 in neurons, however, has remained obscure.
In the present report, we identify a DIA of hippocampal neurons and directly address the involvement of NBCe1, using cells cultured from wild-type (WT) and Slc4a4 knock-out (KO) mice. We show that an initial depolarization-induced acid transient, dependent on Ca2+ entry, is markedly limited by the subsequent onset of a DIA. Using Slc4a4 knock-out mice, we demonstrate that a substantial component of this neuronal DIA is attributable to NBCe1. In addition, we describe the presence of a parallel Cl−-dependent mechanism that also contributes prominently to the DIA. These results indicate that hippocampal neurons respond to sustained membrane depolarization by activating net acid extrusion in advance of an ensuing Ca2+-dependent acid load, and thereby preempt much of the acidosis.
Materials and Methods
Hippocampal neuronal cultures.
All procedures were performed with approval of the Institutional Animal Care and Use Committee of the New York University School of Medicine. Primary neuronal cultures were prepared from the hippocampi of neonatal [postnatal day 1 (P1)] Swiss Webster (Taconic) mice of either gender, by dissociation with trypsin, followed by plating on poly-l-lysine-coated coverslips (Svichar et al., 2009). For Slc4a4 knock-out mouse primary culture, each hippocampus was individually processed to provide a single neuronal culture, which was subsequently identified by PCR analysis of tail DNA. Genotyping used the following primers (Gawenis et al., 2007): a forward primer from the deleted region of intron 9 (5′-TCACAAACCTTTCAGCAAAAGAGTGC-3′) that identified only the wild-type allele; a reverse primer from intron 9 (5′-CAAAGAGCAACAGTCAGACAGC-3′) that identified both wild-type and mutant alleles; and a primer from the neomycin resistance gene (5′-GACAATAGCAGGCATGCTGG-3′) that identified only the mutant allele. Breeding pairs of Slc4a4 heterozygous mice were kindly provided by Dr. Gary Schull (University of Cincinnati College of Medicine, Cincinnati, OH). For all neuronal cultures, physiological experiments were performed after 14–21 d in vitro.
Acutely dissociated hippocampal neurons.
Hippocampal neurons from area CA1 were dissociated from neonatal animals of either gender (P12–P14) as described previously (Svichar et al., 2009). Cells were plated onto concanavalin A-coated coverslips and left to settle for 1–2 h before use. Pyramidal neurons were identified morphologically by their large size, pyramidal shape, and distinct basal and apical dendritic arbors (Tse et al., 1992).
Experimental solutions and drugs.
Standard bicarbonate-buffered saline contained the following (in mm): 124 NaCl, 26 NaHCO3, 3.0 KCl, 1.0 NaH2PO4, 2.0 CaCl2, 1.5 MgCl2, and 10 glucose. Solutions were gassed with 95% O2 and 5% CO2 and had a nominal pH of 7.4. Saline with elevated K+ was made by equimolar replacement of NaCl with KCl. In nominally zero Ca2+ saline, CaCl2 was replaced by MgCl2. EGTA was omitted from zero Ca2+ saline, as neurons did not survive for the duration of the experiments when it was present. In zero Cl− saline, the NaCl, KCl, CaCl2, and MgCl2 were substituted with 124 Na+-methanesulfonate, K+-methanesulfonate, CaSO4, and MgSO4, respectively. After exposure of neurons to zero Cl− saline, it was necessary to lower the pHi (see below) using a 3–5 min prepulse of 10 mm NH4+ (Boron and De Weer, 1976). For this purpose, 10 mm Na+-methanesulfonate was replaced by 10 mm NH4+-methanesulfonate. Experimental solutions were warmed to 32°C and superfused at 2 ml per minute, exchanging the chamber volume in 20–30 s. AMPA, cyclothiazide, and bafilomycin were respectively obtained from Tocris Biosciences, Research Biochemicals International, and LC Laboratories. 3-Methylsulfonyl-4-piperidinobenzoyl guanidine hydrochloride (HOE-694) and cariporide were generous gifts from Sanofi-Aventis. All other reagents were purchased from Sigma-Aldrich.
Recording of intracellular pH.
Coverslips with attached neurons served as the floor of a submersion chamber mounted on the stage of a Zeiss Axiovert inverted microscope equipped for epifluorescence. Neurons were loaded with pH-sensitive fluorophore BCECF [2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein] by incubation with 2 μm of the acetoxy-methylester (Invitrogen) for 10 min at room temperature. A 75 W xenon lamp and a monochromater provided alternate 490 and 440 nm fluorescence excitation. For each excitation wavelength, the respective emissions above 535 nm (F490 and F440, respectively) were collected via a 40× oil-immersion objective and an intensified CCD camera. Averaged fluorescence from regions of interest around single neurons was imaged at 15 s intervals using ImageMaster software (Photon Technology International). In each experiment, the F490:F440 ratios were displayed as simultaneous line traces from multiple neurons. Experimental ratios were converted to pHi using the nigericin single point technique (Boyarsky et al., 1988a), using a HEPES-buffered calibration solution (150 mm K+ and 3 μm nigericin, pH 7.0) applied at the end of each experiment. Data were referenced to a calibration curve previously constructed using nigericin–150 mm K+ solutions buffered with PIPES or HEPES over the pH range of 6.0–8.0.
Determination of intracellular buffering capacity.
The total intracellular buffering capacity of neurons, βT, was calculated as the sum of the intrinsic buffering capacity (βi) and the bicarbonate-derived buffering capacity (βb) (Roos and Boron, 1981). The βi was determined by a variation of the method of Bevensee et al. (1996): in bicarbonate-free saline (using 26 mm HEPES, pH 7.4), the pHi was lowered by transition to Na+-free saline and 20, 10, 5, and 2.5 mm NH4+ were then added sequentially to produce stepwise changes in pHi. Data were pooled for pHi values over increments of 0.2 pH units and the results fit to straight line that served as a standard calibration for βi values. The βb was calculated as 2.3 × [HCO3−]i based on the Henderson–Hasselbalch equation, with CO2 concentration assumed to be equal across the cell membrane.
Determination of net molar fluxes.
The response to elevation of external K+ consisted of an initial acid transient, followed by a net alkaline shift (see Results) (see Fig. 1). The net acid flux was defined as the product of the initial acid-directed dpHi/dt and βT, with βT determined at the initial baseline pHi. The initial net alkaline flux was defined as the product of the initial alkaline-directed dpHi/dt and βT, with βT determined at the onset of the alkaline response where the rate of rise became constant.
Figure 1.

Responses induced by 30 mm K+ and intracellular buffering capacity of hippocampal neurons. A, Typical response of a cultured mouse hippocampal neuron consisted of early acid transient, which was followed by a net alkaline shift. B, Peak amplitudes and net molar fluxes for acid and alkaline shifts in cultured hippocampal neurons. C, Typical response of an acutely dissociated CA1 pyramidal neuron from a juvenile mouse. D, Peak amplitudes and net molar fluxes for acid and alkaline shifts in CA1 pyramidal neurons acutely dissociated from juvenile mice. E, Example of repetitive acid-alkaline transients elicited in cultured neurons. F, Relationship of intrinsic (non-bicarbonate) buffering capacity (βi in millimolar concentration) and pHi. Parentheses contain number of cells for each mean value. For the pHi traces, the acid and alkaline responses during application of high K+ are emphasized in black, with baseline pHi and pHi recoveries in gray. All data are in standard 2 mm Ca2+ saline. Error bars indicate SEM.
Comparisons in zero Cl− saline.
In hippocampal neurons, replacement of extracellular Cl− produces a large increase in pHi, because of the reversal of Cl−–HCO3− exchange (Boyarsky et al., 1988b; Raley-Susman et al., 1993; Baxter and Church, 1996). To assess pHi responses at comparable baselines, we lowered the pHi after exposure to Cl−-free solution. This was accomplished by applying a short pulse of 10 mm NH4+ followed by 100 μm HOE-694 after the resulting peak acidification, thereby blocking the contribution of NHE to recovery of pHi from the imposed acid shift. In some cases, pHi could be lowered with application of 100 μm HOE alone. In later experiments, HOE-694 became unavailable, and the congener cariporide was used instead (Weichert et al., 1997; Xue et al., 2008).
Whole-cell recording.
The somata of neurons were visualized under infrared differential interference contrast microscopy using a Zeiss Axioskop 2 Plus, fixed-stage microscope, fitted with a 40×, water-immersion objective (0.75 numerical aperture), and an Olympus Optical 150 video camera. Patch pipettes were pulled from 1.5 mm outer diameter × 1.12 mm inner diameter borosilicate tubing (World Precision Instruments) using a Narishige PP-830 two-stage puller (Japan). The intracellular filling solution contained the following (in mm): 120 K-gluconate, 20 KCl, 2.0 MgCl2, 25 Na-HEPES, and 2.0 Mg2+-ATP. After adjusting the pH to 7.3 with KOH, the final osmolarity was 280–290 mOsm. Pipettes had resistances of 3–5 mΩ. Data were acquired using an Axopatch 1D amplifier and Digidata board 1320A, controlled by Clampex 8.2, and analyzed using ClampFit (Molecular Devices).
RNA extraction, reverse transcription, and PCR.
Total RNA from neuronal cultures was isolated using RNeasy mini kit (QIAGEN) and reverse transcribed into a single-stranded cDNA with a SuperScript II Reverse Transcriptase kit (Invitrogen) as per the manufacturer's instructions. The resulting first-strand cDNA was then used as a template for SYBR Green real-time PCR. Intron-spanning primers [Table 1, Quantitative PCRs (QPCRs)] were designed with Primer 3.0 (Whitehead Institute for Biomedical Research) using sequences obtained from the National Center for Biotechnology Information GenBank database. The amplification efficiencies for each PCR were calculated from the respective kinetic curves. Initial gene transcript levels (per 1000 transcripts of β-actin; gene Actb) were derived as per Liu and Saint (2002).
Table 1.
Primer sequences
| Gene | Accession no. | Fragment | Forward primer | Reverse primer |
|---|---|---|---|---|
| QPCRs | ||||
| Slc4a4 | NM_018760 | 222–325 | TCCGACAAATCTGATGTGGA | TCCTCTCCCAAGATGAATCG |
| Slc4a8 | NM_021530 | 763–744 | CTGGACATGCGTGCAAGTAG | CTTTAACCCTCACGCTGTCG |
| Slc9a1 | NM_016981 | 1189–1170 | ATCCTTGTCTTCGGGGAGTC | TGCCCACAGAGTCATAGCTG |
| ActB | NM_007393 | 858–911 | GCTCTTTTCCAGCCTTCCTT | AGTTTCATGGATGCCACAGG |
| Multiplex PCRs | ||||
| Slc4a4 | NM_018760 | 76–697 | ATGTGTGTGATGAAGAAGAAGTAGAAG | GACCGAAGGTTGGATTTCTTG |
| ActB | NM_007393 | 305–877 | TGTTACCAACTGGGACGACA | AAGGAAGGCTGGAAAAGAGC |
| Nested PCRs | ||||
| Slc4a4 | NM_018760 | 359–577 | GGAACTCGATGAGCTTCTGG | ACCAGCTGTGGGAGAGAAGA |
| ActB | NM_007393 | 470–696 | AGCCATGTACGTAGCCATCC | TCTCAGCTGTGGTGGTGAAG |
For single-cell multiplex reverse transcription (RT)-PCR, the cytoplasm of a neuron was aspirated into a patch pipette filled with a 5× buffer (Promega) containing RNase inhibitor (Eppendorf; 20 U/ml) and then expelled into a microcentrifuge tube containing a reverse transcription reaction mixture. First-strand cDNA was synthesized with ImProm II Reverse Transcriptase (Promega) following manufacturer's instructions. After reverse transcription, the cDNAs for Slc4a4 and β-actin were amplified in a multiplex PCR with primers listed in Table 1 (Multiplex PCRs). The final product was diluted tenfold and nested PCR performed using the primers in Table 1 (Nested PCRs), performed for each gene separately. The products of nested PCR were separated and visualized on an ethidium bromide-stained agarose gel.
Data analysis.
Data were presented as means with standard error of the mean (SEM). Values of n were given as number of cells/number of coverslips. Statistical comparisons were made with a paired or unpaired two-tailed Student's t test, or repeated-measures ANOVA with a Tukey post hoc test, as appropriate.
Results
pHi shifts of hippocampal neurons elicited by high K+
Changes in pHi of cultured hippocampal neurons were elicited by elevation of bath K+ from the baseline of 3 mm to 30 mm. In separate current-clamp recordings, this caused a peak depolarization of 39 ± 3.1 mV from a resting potential of −52 ± 3.4 mV (n = 5). From an initial baseline pHi of 7.07 ± 0.02, the typical response (Fig. 1A) consisted of an initial acid transient, followed by a net alkaline shift that persisted for the duration of the K+ elevation. On return to 3 mm K+, the pHi recovered toward baseline and was sometimes followed by an undershoot. The size and speed of the responses varied considerably among cultures. In one representative culture, the early acid transient averaged −0.13 ± 0.01 with an initial molar acid flux of −4.04 mm · min−1 (n = 47/4). This included nine neurons that did not display an acid transient. In the same data set, the peak alkaline shift averaged 0.14 ± 0.03 unit pH above initial baseline, with an initial molar flux of 10.4 mm · min−1. This included eight cells, in which only an acid transient occurred (Fig. 1B). Qualitatively similar responses to 30 mm K+ were elicited from CA1 pyramidal neurons acutely dissociated from juvenile (Fig. 1C,D) and adult mice (n = 17/6). These acid-alkaline responses could be evoked sequentially (Fig. 1E) with no significant difference (p > 0.05) in either the first and second acid shifts (−0.03 ± 0.02 vs −0.03 ± 0.02, respectively) or the first and second alkaline shifts (0.22 ± 0.02 vs 0.29 ± 0.03, respectively). Among cultured, acutely dissociated juvenile, and acutely dissociated adult neurons, the baseline pHi did not differ significantly (p > 0.05) and was normal in distribution for each group, with average values of 7.07 ± 0.02 (n = 47/4), 7.09 ± 0.03 (n = 15/4), and 7.17 ± 0.07 (n = 17/6), respectively.
The intrinsic buffering capacity of the cultured neurons as a function of pHi is shown in Figure 1F. The values and relationship to pHi were comparable with those of acutely dissociated hippocampal CA1 neurons obtained by Yao et al. (1999) but were somewhat higher than the values reported by Bevensee et al. (1996). Subsequent experiments were performed exclusively on cultured hippocampal neurons.
Ca2+ dependence of K+-evoked pHi shifts
Numerous studies have shown that the hippocampal neurons acidify because of entry of Ca2+ (Irwin et al., 1994; Wang et al., 1994; Trapp et al., 1996; Cheng et al., 2008). Accordingly, when 30 mm K+ was applied in zero Ca2+ saline, the acid transient was diminished (Fig. 2A), and in 19 of 47 neurons, no acid transient was observed. Absence of external Ca2+ reduced the acid transient and the molar acid flux by 53 ± 6.3 (n = 23/3) and 54 ± 11%, respectively (n = 47/4) (Fig. 2B,C).
Figure 2.

The K+-evoked acid shifts were Ca2+ dependent. A, Transition to zero Ca2+ saline reduced the early acid transient and increased the alkaline shift. The inset shows overlay of responses to emphasize late effect of zero Ca2+ saline on the alkaline shift. B, Mean effect of zero Ca2+ saline on the peak amplitude of the acid and alkaline shifts. C, Mean effect of zero Ca2+ saline on the net acid and alkaline molar fluxes. D, Sequential responses to 30 mm K+ in a hippocampal neuron. The repetitive alkaline responses were highly consistent. For statistical comparisons, p values here and elsewhere are represented as follows: *p < 0.05; **p < 0.01; ***p < 0.001. Error bars indicate SEM.
The reduction in the acid response in the absence of bath Ca2+ was accompanied by an 84 ± 11% increase in the alkaline shift (n = 27/3) (Fig. 2B,C), which was highly consistent over sequential trials (Fig. 2D). Thus, in paired responses (at an interval of 26.8 ± 4.3 min), the respective amplitude of the first and second DIA was 0.42 ± 0.04 versus 0.42 ± 0.05 unit pH (p = 0.89), with initial rates of 0.16 ± 0.01 versus 0.17 ± 0.01 pH · min−1 (p = 0.83) and net alkaline fluxes of 8.36 ± 0.52 versus 8.31 ± 0.82 mm · min−1 (p = 0.95; n = 19/3). The alkalosis was enhanced mainly in its late phase (superimposed records) (Fig. 2A), suggesting that the mechanism of Ca2+-dependent acidification was far more prolonged than the observed acid transients. The long duration of this acid source was revealed in subsequent experiments using 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS).
The effect of acid transport inhibitors on the evoked alkaline shifts
In the presence of the HCO3− transport blocker DIDS (100 μm), the alkaline component of the response was abolished. Instead, a large, prolonged acidification occurred, which persisted for the duration of the high K+ pulse. Figure 3A shows a pronounced example of this effect. On average, a peak acid transient of −0.09 ± 0.02 unit pH in control saline was transformed into a sustained fall of −0.16 ± 0.04 unit pH in DIDS (n = 15/3) (Fig. 3B). The initial molar acid flux was not significantly changed by DIDS (−5.4 ± 1.4 vs −3.9 ± 0.7 mm · min−1; p = 0.73), suggesting that it had little or no effect on the acid transient. Thus, by blocking the alkaline shift, DIDS revealed the prolonged nature of the acid source.
Figure 3.

Effect of DIDS on the K+-evoked pHi shifts. A, DIDS abolished the alkaline shift and revealed a prolonged acid response. B, Mean effect of DIDS on the peak amplitude of the K+-evoked acid transient and alkaline shift. C, The prolonged acidosis revealed by DIDS was mostly decreased in zero Ca2+ saline. D, Mean effect of DIDS on the response amplitudes in zero Ca2+ saline. Error bars indicate SEM.
The prolonged acid source was primarily dependent on the entry of Ca2+, since it was greatly reduced (to −0.04 ± 0.01 unit pH; p < 0.01 compared with standard saline) when the DIDS protocol was performed in zero Ca2+ media. Thus, the generation of the alkaline shift acted to curtail and terminate the Ca2+-dependent fall in pHi. Since the alkalosis was variably diminished by the ongoing acid source, most of the subsequent experiments that addressed the nature of the alkalosis were performed in zero Ca2+ saline, so as to maximize the amplitude, and consistency of the responses.
The complete block of the alkaline shift by DIDS is consistent with the properties of an HCO3− transporter. Moreover, two principal HCO3−-independent acid extrusion mechanisms could be ruled out. NHE was not involved, as the DIA was undiminished by 100 μm HOE-694 (0.29 ± 0.02 control vs 0.33 ± 0.03 in HOE-694; n = 9/1; p > 0.05), which blocks the amiloride-insensitive Na+–H+ exchanger of these neurons (Yao et al., 1999), nor was it reduced by 100 μm cariporide (0.32 ± 0.03 control vs 0.36 ± 0.02 cariporide; n = 13/2; p > 0.05), a related, but more selective NHE inhibitor (Weichert et al., 1997; Xue et al., 2008). A plasma membrane H+-ATPase could also be ruled out as a cause of the DIA, since 100 nm bafilomycin did not block the response (0.47 ± 0.02 control vs 0.53 ± 0.02 bafilomycin; n = 25/3; p < 0.05). The slight increase in the DIA in the presence of bafilomycin is unexplained but suggests that proton pumps can modestly limit its amplitude.
The response to elevated K+ is a graded DIA
If the K+-evoked rise in pHi were a DIA, rather than a response to elevation of K+ per se, then another depolarizing agent would be expected to elicit a similar alkalosis. Exposure to 50 μm AMPA caused pHi to increase by 0.30 ± 0.01 (Fig. 4A) at a rate of 0.16 ± 0.01 unit pH · min−1 (n = 39/5), strongly suggesting that the alkaline shift was a DIA (Fig. 4A). When 30 mm K+ was added with 50 μm AMPA (Fig. 4B), the resulting DIA (0.31 ± 0.03) was larger than the AMPA response alone (0.23 ± 0.03; p < 0.05; n = 6/2), consistent with a larger net depolarization; however, the pHi recovered only slightly after the paired exposure (Fig. 4B). The cause of the curtailed recovery was not investigated.
Figure 4.
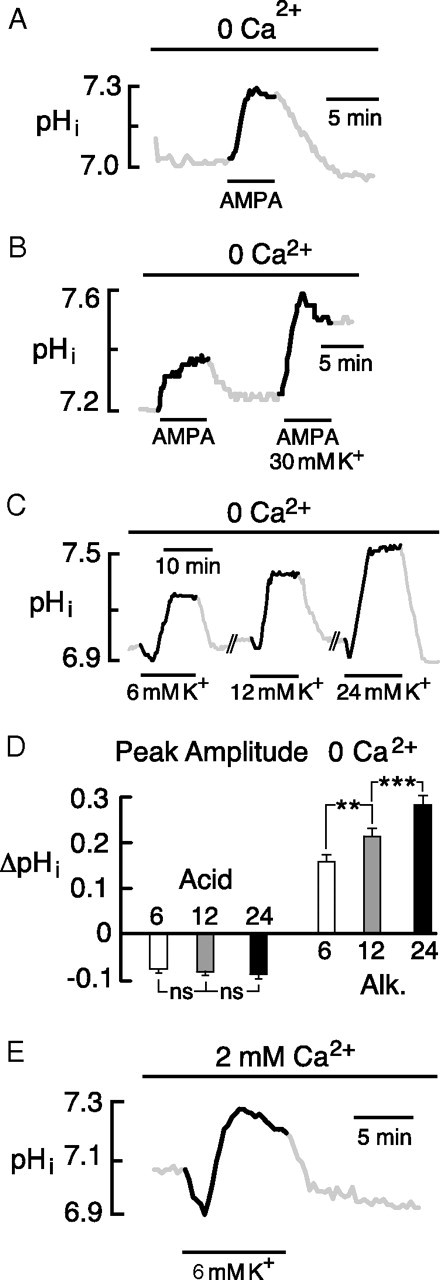
The alkaline shift is a DIA. A, Depolarization by 50 μm AMPA produced an alkaline shift similar to the K+-evoked responses. B, AMPA with 30 mm K+ caused a larger DIA than AMPA alone but failed to recover to baseline. C, Graded elevation of K+ elicited DIAs with stepwise increases in peak amplitude. Responses were separated by intervals of 20–25 min. D, Mean responses elicited by 6, 12, and 24 mm K+. Acid shifts did not significantly increase beyond the response seen in 6 mm K+. Corresponding alkaline shifts were significantly elevated compared with the response at the preceding lower K+ concentration. E, Acid and alkaline transients elicited by transition from 3 to 6 mm K+ in standard saline containing 2 mm Ca2+. AMPA experiments used 100 μm cyclothiazide to prevent receptor desensitization. Error bars indicate SEM.
Since depolarization could arise in part from increased synaptic activity and firing during the application of elevated K+, we examined responses before and after exposure to saline containing 1 μm tetrodotoxin plus APV (dl-2-amino-5-phosphonovalerate), CNQX (6-cyano-7-nitro-nitroquinoxaline-2,3-dione), and picrotoxin (all 50 μm). This mixture of inhibitors caused just a small decrease in the DIA to 96 ± 8% of the control (n = 34/4; p < 0.01), indicating that the response was triggered almost entirely via direct, K+-mediated depolarization.
We tested whether more physiological (i.e., smaller) elevations of K+ could cause a DIA. In the absence of external Ca2+, sequential elevation of K+ to 6, 12, and 24 mm elicited graded DIAs (Fig. 4C,D). In separate measurements on four neurons, these transitions were associated with depolarizations of 12 ± 1.7, 16 ± 2.6, and 25 ± 3.6 mV, respectively. The more physiological elevation to 6 mm K+ could also elicit a significant alkaline shift in saline containing 2 mm Ca2+, where it caused a DIA of 0.21 ± 0.04 pH (n = 25/2) (Fig. 4E).
Role of chloride in the DIA
An intracellular chloride load associated with depolarization might indirectly raise pHi by reducing efflux of HCO3− by a Cl−–HCO3− exchanger. In one report (Slemmer et al., 2004), depolarization-associated Cl− influx in hippocampal neurons was blocked by 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB). In our cells, however, 100 μm NPPB had no effect on the DIA elicited by 30 mm K+ (data not shown). The DIA was similarly unaffected by the Cl− transport blockers bumetinide (100 μm), furosemide (100 μm), niflumic acid (300 μm), and anthracene-9-carboxylic acid (100 μm).
To fully eliminate the possibility of Cl− entry, we tested the effect of zero Cl− saline. Withdrawal of Cl− from the bath caused a rise in pHi, commonly attributed to reversal of Cl−–HCO3− exchange (Boyarsky et al., 1988b; Raley-Susman et al., 1993; Baxter and Church, 1996). To assess DIA amplitudes at a comparable pHi and buffering capacity, the pHi was subsequently reduced, using a 3–5 min, 10 mm NH4+ prepulse in the presence of HOE-694 (or cariporide) to block NHE. In 27 neurons, the subsequent DIA was always significantly reduced compared with control (Fig. 5A), and in 13 of these cells, where the baseline pHi was exactly matched to the control value, the DIA and molar alkaline flux were 49 ± 5.7 and 45 ± 6.6% of controls, respectively (three coverslips; p < 0.001 for both) (Fig. 5B). This suggested that both a Cl−-dependent and a Cl−-independent mechanism contribute significantly to the DIA.
Figure 5.

The DIA has Cl−-dependent and Cl−-independent components. A, In the absence of external Cl−, the amplitude of the DIA was approximately one-half of the control value. B, Mean effect of zero Cl− saline on the DIA amplitude and initial alkaline molar flux. Since Cl−-free saline caused a rise in pHi, an NH4+ prepulse and HOE-694 were used to bring pHi down to control level before initiating the second DIA. The NH4+ prepulse and recovery were omitted for clarity. Error bars indicate SEM.
Role of NBCe1 in the neuronal DIA
We postulated that the component of the DIA that persisted in the absence of external Cl− was attributable to NBCe1. In support of this hypothesis, mRNA for Slc4a4, measured by QPCR (relative to β-actin), was abundant in our cultures (Fig. 6A), and on the same order as message for the acid extruders Slc4a8 (NDCBE) and Slc9a1 (NHE1). In addition, single-cell RT-PCR confirmed the presence of Slc4a4 message in individual neurons (Fig. 6B).
Figure 6.

Presence and function of NBCe1 in cultured cells. A, QPCR of neuronal cultures revealed NBCe1 message. Levels were same order of magnitude as messages for the Na+–HCO3− cotransporter NDCBE (gene Slc4a8) and the Na+-H+ exchanger NHE1 (Slc9a1). B, Single-neuron RT-PCR. Message for NBCe1 was detected in two individual neurons. C, Astrocytes in WT cultures displayed a rapid DIA. No DIA could be elicited in astrocytes cultured from Slc4a4 KO mice. D, Representative DIAs in hippocampal neurons cultured from WT and Slc4a4 KO mice. E, Peak amplitude of pHi shifts in neurons from WT versus Slc4a4 KO mice. Mean acid shifts were not significantly different. In the neurons from KO mice, the DIA was approximately one-half of the amplitude seen in WT cells. F, Net molar fluxes in WT versus Slc4a4 KO neurons. Net acid flux was no different in WT versus KO. In Slc4a4 KO neurons, the net alkaline flux was approximately one-fourth of the flux observed in WT cells. Error bars indicate SEM.
To test whether the DIA was reliant on NBCe1, we studied responses in cells cultured from WT mice versus animals with a global KO of Slc4a4. Some initial experiments were performed on scattered astrocytes in the neuronal cultures, identified as flat cells without processes that stained positive for glial fibrillary acidic protein. Since the pronounced DIA in astrocytes has been widely attributed to NBCe1 in physiological studies (Deitmer and Rose, 1996), these glial cells could be expected to provide a physiological correlate of the KO. Indeed, in astrocytes cultured from WT mice, 30 mm K+ evoked a DIA of 0.16 ± 0.02 unit pH (n = 8/2), but failed to elicit any pHi change in astrocytes from Slc4a4 KO animals (n = 29/3) (Fig. 6C). The astrocyte DIA was therefore fully attributable to NBCe1, as expected.
Analogous experiments performed on neurons from WT versus KO mice (in standard saline with 2 mm Ca2+) are shown in Figure 6D. Early acid transients were unchanged (Fig. 6E,F), and there was no significant difference in baseline pHi in the two groups (WT, 6.97 ± 0.03, n = 48/5; KO, 6.99 ± 0.02, n = 53/6; p = 0.42). In neurons from KO mice, however, the mean alkaline shift and the corresponding alkaline flux were approximately one-half and one-quarter of their respective magnitude in WT cells, suggesting a prominent, but not exclusive role of NBCe1 in generation of the neuronal DIA.
We postulated that the remaining DIA in the neurons from Slc4a4 KO mice would be mostly attributable to the previously noted Cl−-dependent mechanism, and accordingly tested whether this response required external Cl−. To maximize the alkaline responses, external Ca2+ was omitted in these experiments. As predicted, in zero Cl− saline, the DIA of the KO neurons was virtually abolished, with 96% reduction in mean peak amplitude compared with WT neurons in the same media (p < 0.001; WT, n = 24/5; KO, n = 35/5) (Fig. 7A,B). In the KO neurons, the absence of much of the DIA was associated with an increase in the initial acid transient (Fig. 7B). In a few instances (2 of 35 cells), a small DIA remained in the neurons from KO mice (Fig. 7C).
Figure 7.

DIA in Slc4a4 KO neurons was Cl− dependent. A, In zero Cl− media, a DIA persisted in WT neurons but was virtually abolished in KO neurons. B, Mean amplitude of acid transients and DIA in WT versus KO neurons. The mean acid shift was larger in the KO neurons, whereas the mean alkaline shift was nearly zero in the KO cells. C, Example of a DIA that persisted in 0 Cl− media in 2 of 35 KO neurons. Experiments were conducted in the presence of 100 μm HOE-694 to lower pHi after withdrawal of bath Cl−. Error bars indicate SEM.
Discussion
A principal finding of these studies is that hippocampal neurons respond to depolarization by the activation of a DIDS-sensitive, acid extrusion process that elicits a net increase in pHi during sustained depolarization. This behavior derives in part from the activation of neuronal NBCe1 and, in this respect, is similar to the long-recognized DIA of astrocytes. Whereas the glial response relies exclusively on NBCe1, as shown in Figure 6C, a component of the neuronal DIA is generated by a parallel DIDS-sensitive mechanism dependent on external Cl−. Accordingly, in the absence of extracellular Cl−, the DIA was virtually abolished in neurons lacking Slc4a4. Given the presence of multiple components to the response, which could have overlapping ionic dependencies, use of the NBCe1 KO was essential and provided specific, compelling evidence for the operation of this transporter.
Experiments using DIDS indicated that, without these mechanisms to generate a DIA, depolarization could cause a large, prolonged fall in pHi. As such, the neuronal DIA functions to preempt much of the acidosis. This behavior differs from classic pH regulatory responses, wherein acid extrusion is activated only subsequent to a fall in pHi (Boron, 2004). Mechanisms that activate net acid extrusion in response to depolarization would appear particularly adaptive for neurons that are subject to large acid loads generated by the influx of Ca2+, given the sensitivity of numerous channels to pHi, and the deleterious effects of prolonged acidosis (Ying et al., 1999; Ding et al., 2000).
In several previous reports, depolarizing agents were found to cause only a short-latency, prolonged acidosis of hippocampal neurons (Hartley and Dubinsky, 1993; Irwin et al., 1994; Wang et al., 1994). The absence of an early alkaline response may have been attributable to more than one factor. First, these studies either omitted or used little bicarbonate in the saline, which would have limited intracellular buffering power, thereby magnifying the acidosis. Second, activation of NBCe1 as well as the Cl−-dependent mechanism (see below) would have been precluded or curtailed in low bicarbonate media. Third, there may have been an especially large Ca2+-dependent acid source, particularly when NMDA receptors were activated. These acid responses were likely caused in part by the PMCA (plasma membrane Ca2+ ATPase) (Schwiening et al., 1993; Trapp et al., 1996; Cheng et al., 2008), as this transporter imports H+ in exchange for cytosolic Ca2+ (Di Leva et al., 2008). Metabolic sources may also have contributed to the acidosis in this and the previous studies, given the persistence of a small acid shift in the nominal absence of extracellular Ca2+.
Cheng et al. (2008) noted the occurrence of a DIA in hippocampal neurons in response to elevation of K+ in a range from 25 to 140 mm. It was postulated that these pH shifts arose by an H+ efflux through a proton conductance. The DIA described in the present report cannot be ascribed to such a mechanism, as a large component was clearly generated by NBCe1, and a second major component was attributable to a DIDS-sensitive process that required external Cl−, features inconsistent with properties of an H+ conductance (Decoursey, 2003). It also appears unlikely that DIA mediated by a conductive pathway could occur in response to depolarization of just 10–15 mV (Fig. 4), since the driving force for H+ would have remained inward. In the study by Cheng et al. (2008), the DIA was studied exclusively in HCO3−-free media buffered with HEPES and was therefore elicited under conditions of lower intracellular buffering capacity, which would have amplified the pH shifts. Their relationship to the DIA of the present study is therefore unclear.
A prominent component of the neuronal DIA was due to a DIDS-sensitive process that required external Cl−. A number of known mechanisms linked to Cl− may be considered (Romero et al., 2004). NDCBE (gene Slc4a8) is a key acid extruder of hippocampal neurons that uses the energy of the Na+ gradient to power the export of cytoplasmic Cl− in exchange for HCO3−. However, this mechanism is not electrogenic and therefore should be insensitive to depolarization. Moreover, the activity of NDCBE is not readily inhibited in the absence of external Cl−, as this anion is well retained in the cytosol of these cells. For example, after 3 h in the absence of external Cl−, acid extrusion attributed to NDCBE remained functional in hippocampal neurons (Schwiening and Boron, 1994). In the present experiments, the DIAs were elicited within 20 min after transition to Cl−-free media.
In principle, a Cl−-dependent DIA sensitive to DIDS could be generated by a downturn in constitutive acid loading by a Cl−–HCO3− exchanger that might lead to Cl− loading. NPPB had no effect on the DIA, however, nor did a number of other Cl− transport inhibitors. To address this possibility further, additional studies should be performed using neurons from Slc4a4 KO mice, so as to permit the study of the Cl−-dependent DIA in relative isolation.
The majority of experiments in this report were performed using 30 mm K+ to elicit a DIA. Elevation of K+ of this order occurs under conditions such as brain ischemia, traumatic brain injury, and spreading depression (Hansen, 1985; Martins-Ferreira et al., 2000). Depolarization-induced acid extrusion may therefore be relevant to neuronal survival, since an untoward acidosis can foster injury by the facilitation of apoptotic pathways and the generation of reactive oxygen species (Ying et al., 1999; Ding et al., 2000). During seizure activity, the elevation of extracellular K+ is less severe but can approach 12 mm (Somjen, 1979), a level associated with a considerable DIA in our studies. Whether a net DIA occurred under such conditions would depend on the degree of concurrent acid generation. However, even where acidosis was predominant, as in brain ischemia (Rehncrona et al., 1981), depolarization-induced acid extrusion could still serve to limit the fall in neuronal pHi. In this respect, it is notable that hippocampal pyramidal neurons are especially susceptible to hypoxic and ischemic insults (Pulsinelli et al., 1982; Schurr and Rigor, 1992). Therefore, it would be interesting to determine whether these DIA mechanisms exist in neurons from other brain regions, and whether the level of their expression is different.
In addition to limiting acidosis, the neuronal DIA may function to increase the neuronal metabolic rate under conditions of prolonged depolarization. Phosphofructokinase, the rate-limiting enzyme of the glycolytic pathway, is highly sensitive to pHi, and exhibits a steep increase in its activity as pHi rises (Trivedi and Danforth, 1966). Thus, the neuronal DIA might serve as a signal to raise neuronal glucose utilization under conditions requiring increased generation of ATP, such as seizure, spreading depression, and ischemia. Indeed, in astrocytes, the suggestion of coupling between a DIA and the glycolytic rate has received recent experimental support (Bittner et al., 2010).
A key question raised by these studies is whether depolarization-activated acid extrusion plays a role under physiological conditions. A rise of K+ from 3 to 6 mm can occur during normal synchronous activity (Somjen, 1979), and this increase was found to cause a significant activation of acid extrusion (Fig. 4). Prolonged positive shifts in membrane potential, which might cause a sustained net alkaline shift, are in fact normal occurrences. For example, a depolarization of 10 mV or more, lasting several minutes, occurs in cortical neurons in the transition from sleep to wakefulness (Steriade et al., 2001). It is conceivable that a significant rise in pHi might accompany these transitions, and thereby modulate metabolic rate and excitability. However, it remains to be determined whether neurons from other brain regions display a DIA similar to the response of hippocampal pyramidal cells.
In summary, we show that the regulation of intracellular pH in hippocampal neurons is strongly governed by membrane potential, because of two separate, DIDS-sensitive mechanisms that serve to limit the acidosis linked to Ca2+ entry. Our results demonstrate that the electrogenic Na+–HCO3− cotransporter NBCe1 contributes prominently to this response. In addition, these experiments have uncovered a parallel second mechanism of DIA, which requires external Cl−. The full ionic basis and molecular identity of the Cl−-dependent process remain to be established. The evolution of DIA in neurons may have been an adaptation to preempt untoward acidification from large intracellular Ca2+ loads, while maintaining or accelerating the rate of glucose utilization through the glycolytic pathway.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS032123 and The Attilio and Olympia Ricciardi Fund.
References
- Baxter KA, Church J. Characterization of acid extrusion mechanisms in cultured fetal rat hippocampal neurones. J Physiol. 1996;493:457–470. doi: 10.1113/jphysiol.1996.sp021396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevensee MO, Cummins TR, Haddad GG, Boron WF, Boyarsky G. pH regulation in single CA1 neurons acutely isolated from the hippocampi of immature and mature rats. J Physiol. 1996;494:315–328. doi: 10.1113/jphysiol.1996.sp021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevensee MO, Schmitt BM, Choi I, Romero MF, Boron WF. An electrogenic Na+-HCO3− cotransporter (NBC) with a novel COOH-terminus, cloned from rat brain. Am J Physiol Cell Physiol. 2000;278:C1200–C1211. doi: 10.1152/ajpcell.2000.278.6.C1200. [DOI] [PubMed] [Google Scholar]
- Bittner CX, Loaiza A, Ruminot I, Larenas V, Sotelo-Hitschfeld T, Gutiérrez R, Córdova A, Valdebenito R, Frommer WB, Barros LF. High resolution measurement of the glycolytic rate. Front Neuroenergetics. 2010;2 doi: 10.3389/fnene.2010.00026. pii:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron WF. Regulation of intracellular pH. Adv Physiol Educ. 2004;28:160–179. doi: 10.1152/advan.00045.2004. [DOI] [PubMed] [Google Scholar]
- Boron WF, De Weer P. Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J Gen Physiol. 1976;67:91–112. doi: 10.1085/jgp.67.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyarsky G, Ganz MB, Sterzel RB, Boron WF. pH regulation in single glomerular mesangial cells. I. Acid extrusion in absence and presence of HCO3. Am J Physiol. 1988a;255:C844–C856. doi: 10.1152/ajpcell.1988.255.6.C844. [DOI] [PubMed] [Google Scholar]
- Boyarsky G, Ganz MB, Sterzel RB, Boron WF. pH regulation in single glomerular mesangial cells. II. Na+-dependent and -independent Cl−-HCO3− exchangers. Am J Physiol. 1988b;255:C857–C869. doi: 10.1152/ajpcell.1988.255.6.C857. [DOI] [PubMed] [Google Scholar]
- Brookes N, Turner RJ. K+-induced alkalinization in mouse cerebral astrocytes mediated by reversal of electrogenic Na+-HCO3− cotransport. Am J Physiol. 1994;267:C1633–C1640. doi: 10.1152/ajpcell.1994.267.6.C1633. [DOI] [PubMed] [Google Scholar]
- Cheng YM, Kelly T, Church J. Potential contribution of a voltage-activated proton conductance to acid extrusion from rat hippocampal neurons. Neuroscience. 2008;151:1084–1098. doi: 10.1016/j.neuroscience.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kraig RP. Intracellular pH of astrocytes increases rapidly with cortical stimulation. Am J Physiol. 1987;253:R666–R670. doi: 10.1152/ajpregu.1987.253.4.R666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M, Kraig RP. Intracellular pH transients of mammalian astrocytes. J Neurosci. 1989;9:2011–2019. doi: 10.1523/JNEUROSCI.09-06-02011.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DS, Saxena NC, Yang HS, Lee HJ, Moring AG, Lee A, Choi I. Molecular and functional characterization of the electroneutral Na/HCO3 cotransporter NBCn1 in rat hippocampal neurons. J Biol Chem. 2005;280:17823–17830. doi: 10.1074/jbc.M408646200. [DOI] [PubMed] [Google Scholar]
- Decoursey TE. Voltage-gated proton channels and other proton transfer pathways. Physiol Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, Rose CR. pH regulation and proton signalling by glial cells. Prog Neurobiol. 1996;48:73–103. doi: 10.1016/0301-0082(95)00039-9. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, Szatkowski M. Membrane potential dependence of intracellular pH regulation by identified glial cells in the leech central nervous system. J Physiol. 1990;421:617–631. doi: 10.1113/jphysiol.1990.sp017965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leva F, Domi T, Fedrizzi L, Lim D, Carafoli E. The plasma membrane Ca2+ ATPase of animal cells: structure, function and regulation. Arch Biochem Biophys. 2008;476:65–74. doi: 10.1016/j.abb.2008.02.026. [DOI] [PubMed] [Google Scholar]
- Ding D, Moskowitz SI, Li R, Lee SB, Esteban M, Tomaselli K, Chan J, Bergold PJ. Acidosis induces necrosis and apoptosis of cultured hippocampal neurons. Exp Neurol. 2000;162:1–12. doi: 10.1006/exnr.2000.7226. [DOI] [PubMed] [Google Scholar]
- Gawenis LR, Bradford EM, Prasad V, Lorenz JN, Simpson JE, Clarke LL, Woo AL, Grisham C, Sanford LP, Doetschman T, Miller ML, Shull GE. Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/HCO3− cotransporter. J Biol Chem. 2007;282:9042–9052. doi: 10.1074/jbc.M607041200. [DOI] [PubMed] [Google Scholar]
- Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- Hartley Z, Dubinsky JM. Changes in intracellular pH associated with glutamate excitotoxicity. J Neurosci. 1993;13:4690–4699. doi: 10.1523/JNEUROSCI.13-11-04690.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin RP, Lin SZ, Long RT, Paul SM. N-Methyl-d-aspartate induces a rapid, reversible, and calcium-dependent intracellular acidosis in cultured fetal rat hippocampal neurons. J Neurosci. 1994;14:1352–1357. doi: 10.1523/JNEUROSCI.14-03-01352.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Saint DA. Validation of a quantitative method for real time PCR kinetics. Biochem Biophys Res Commun. 2002;294:347–353. doi: 10.1016/S0006-291X(02)00478-3. [DOI] [PubMed] [Google Scholar]
- Martins-Ferreira H, Nedergaard M, Nicholson C. Perspectives on spreading depression. Brain Res Brain Res Rev. 2000;32:215–234. doi: 10.1016/s0165-0173(99)00083-1. [DOI] [PubMed] [Google Scholar]
- Pappas CA, Ransom BR. Depolarization-induced alkalinization (DIA) in rat hippocampal astrocytes. J Neurophysiol. 1994;72:2816–2826. doi: 10.1152/jn.1994.72.6.2816. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Raley-Susman KM, Cragoe EJ, Jr, Sapolsky RM, Kopito RR. Regulation of intracellular pH in cultured hippocampal neurons by an amiloride-insensitive Na+/H+ exchanger. J Biol Chem. 1991;266:2739–2745. [PubMed] [Google Scholar]
- Raley-Susman KM, Sapolsky RM, Kopito RR. Cl−/HCO3− exchange functions differs in adult and fetal rat hippocampal neurons. Brain Res. 1993;614:308–314. doi: 10.1016/0006-8993(93)91049-x. [DOI] [PubMed] [Google Scholar]
- Rehncrona S, Rosén I, Siesjö BK. Brain lactic acidosis and ischemic cell damage: 1. Biochemistry and neurophysiology. J Cereb Blood Flow Metab. 1981;1:297–311. doi: 10.1038/jcbfm.1981.34. [DOI] [PubMed] [Google Scholar]
- Rickmann M, Orlowski B, Heupel K, Roussa E. Distinct expression and subcellular localization patterns of Na+/HCO3− cotransporter (SLC 4A4) variants NBCe1-A and NBCe1-B in mouse brain. Neuroscience. 2007;146:1220–1231. doi: 10.1016/j.neuroscience.2007.02.061. [DOI] [PubMed] [Google Scholar]
- Romero MF, Fulton CM, Boron WF. The SLC4 family of HCO3− transporters. Pflugers Arch. 2004;447:495–509. doi: 10.1007/s00424-003-1180-2. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Schmitt BM, Berger UV, Douglas RM, Bevensee MO, Hediger MA, Haddad GG, Boron WF. Na/HCO3 cotransporters in rat brain: expression in glia, neurons, and choroid plexus. J Neurosci. 2000;20:6839–6848. doi: 10.1523/JNEUROSCI.20-18-06839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, Rigor BM. The mechanism of cerebral hypoxic-ischemic damage. Hippocampus. 1992;2:221–228. doi: 10.1002/hipo.450020303. [DOI] [PubMed] [Google Scholar]
- Schwiening CJ, Boron WF. Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na+-dependent Cl−-HCO3− exchange. J Physiol. 1994;475:59–67. doi: 10.1113/jphysiol.1994.sp020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Kennedy HJ, Thomas RC. Calcium-hydrogen exchange by the plasma membrane Ca-ATPase of voltage-clamped snail neurons. Proc Biol Sci. 1993;253:285–289. doi: 10.1098/rspb.1993.0115. [DOI] [PubMed] [Google Scholar]
- Slemmer JE, Matsushita S, De Zeeuw CI, Weber JT, Knöpfel T. Glutamate-induced elevations in intracellular chloride concentration in hippocampal cell cultures derived from EYFP-expressing mice. Eur J Neurosci. 2004;19:2915–2922. doi: 10.1111/j.0953-816X.2004.03422.x. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Extracellular potassium in the mammalian central nervous system. Annu Rev Physiol. 1979;41:159–177. doi: 10.1146/annurev.ph.41.030179.001111. [DOI] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Grenier F. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 2001;85:1969–1985. doi: 10.1152/jn.2001.85.5.1969. [DOI] [PubMed] [Google Scholar]
- Svichar N, Waheed A, Sly WS, Hennings JC, Hübner CA, Chesler M. Carbonic anhydrases CA4 and CA14 both enhance AE3-mediated Cl−–HCO3− exchange in hippocampal neurons. J Neurosci. 2009;29:3252–3258. doi: 10.1523/JNEUROSCI.0036-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Lückermann M, Kaila K, Ballanyi K. Acidosis of hippocampal neurones mediated by a plasmalemmal Ca2+/H+ pump. Neuroreport. 1996;7:2000–2004. doi: 10.1097/00001756-199608120-00029. [DOI] [PubMed] [Google Scholar]
- Trivedi B, Danforth WH. Effect of pH on the kinetics of frog muscle phosphofructokinase. J Biol Chem. 1966;241:4110–4112. [PubMed] [Google Scholar]
- Tse FW, Fraser DD, Duffy S, MacVicar BA. Voltage-activated K+ currents in acutely isolated hippocampal astrocytes. J Neurosci. 1992;12:1781–1788. doi: 10.1523/JNEUROSCI.12-05-01781.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GJ, Randall RD, Thayer SA. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J Neurophysiol. 1994;72:2563–2569. doi: 10.1152/jn.1994.72.6.2563. [DOI] [PubMed] [Google Scholar]
- Weichert A, Faber S, Jansen HW, Scholz W, Lang HJ. Synthesis of the highly selective Na+/H+ exchange inhibitors cariporide mesilate and (3-methanesulfonyl-4-piperidino-benzoyl)guanidine methanesulfonate. Arzneimittelforschung. 1997;47:1204–1207. [PubMed] [Google Scholar]
- Xue J, Zhou D, Yao H, Haddad GG. Role of transporters and ion channels in neuronal injury under hypoxia. Am J Physiol Regul Integr Comp Physiol. 2008;294:R451–R457. doi: 10.1152/ajpregu.00528.2007. [DOI] [PubMed] [Google Scholar]
- Yao H, Ma E, Gu XQ, Haddad GG. Intracellular pH regulation of CA1 neurons in Na+/H+ isoform 1 mutant mice. J Clin Invest. 1999;104:637–645. doi: 10.1172/JCI6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Han SK, Miller JW, Swanson RA. Acidosis potentiates oxidative neuronal death by multiple mechanisms. J Neurochem. 1999;73:1549–1556. doi: 10.1046/j.1471-4159.1999.0731549.x. [DOI] [PubMed] [Google Scholar]