

Abstract
The nonreceptor protein tyrosine kinase Src plays a crucial role in the signal transduction pathways involved in cell division, motility, adhesion, and survival in both normal and cancer cells. Although the Src family kinases (SFKs) are activated in various types of cancers, the exact mechanisms through which they contribute to the progression of individual tumors remain to be defined. The activation of Src in human cancers may occur through a variety of mechanisms that include domain interaction and structural remodeling in response to various activators or upstream kinases and phosphatastes. Because of Src's prominent roles in invasion and tumor progression, epithelial-to-mesenchymal transition, angiogenesis, and the development of metastasis, Src is a promising target for cancer therapy. Several small molecule inhibitors of Src are currently being investigated in clinical trials. In this article, we will summarize the mechanisms regulating Src kinase activity in normal and cancer cells and discuss the status of Src inhibitor development against various types of cancers.
1. Introduction
Francis Peyton Rous was awarded the Nobel prize in 1966 for his groundbreaking discovery that a virus could cause cancer [1]. In 1911, he was able to purify a substance from chickens that was later shown to be a sarcoma-causing virus (Rous sarcoma virus). The responsible oncogene was called v-Src [2, 3]. In 1976, J. M. Bishop and H. E. Varmus discovered a related gene in chickens, which showed a striking resemblance to v-Src. This normal cellular counterpart, cellular Src (known as c-Src or Src), was the first proto-oncogene to be identified, and its discovery led to the Nobel prize for medicine in 1989 [2]. Src was also the first gene product discovered to have intrinsic protein tyrosine kinase activity [4–6].
Src belongs to a family of 11 nonreceptor tyrosine kinases known as the Src family kinases (SFKs); the other ten are Fyn, Yes, Blk, Yrk, Frk (also known as Rak), Fgr, Hck, Lck, Srm, and Lyn. The human genome contains a Yes pseudogene (YESps), and Src, Yes, YESps, and Fyn are ubiquitously expressed in a variety of tissues [7, 8]. Srm is found in keratinocytes, whereas Blk, Fgr, Hck, Lck, and Lyn are found primarily in hematopoietic cells. Frk occurs chiefly in bladder, breast, brain, colon, and lymphoid cells. Like all members of the Src kinase family, the Frk kinase possesses an SH domain as well as conserved autoregulatory tyrosine residues in its catalytic domain [9, 10]. However, Frk differs significantly from the other Src family members in many structural features, including the presence of a putative bipartite nuclear localization signal and the lack of a consensus myristoylation motif [10, 11]. In fact, Frk has been shown to be a nuclear protein with growth-inhibitory effects when ectopically expressed in breast cancer cells [12]. Blk occurs chiefly in colon, prostate, and small intestine cells; however, it was initially isolated from a breast cancer cell line [13].
In this review, we will discuss the structure of SFKs, the regulation of their kinase activity, the involvement of SFKs in the development of cancer, and recent therapeutic advancements in targeting SFKs.
2. Structure of the Src Family Kinases
The ability of the avian viral oncoproteins v-Src and v-Yes to induce fibroblast transformation suggests that their cellular counterparts, Src and c-Yes, have the potential to contribute to human carcinogenesis. v-Src and v-Yes are encoded by avian retroviruses and are capable of inducing sarcomas in chickens and of transforming chicken embryo fibroblast cells in culture [14, 15]. To understand how these proteins are able to induce cell transformation, it is important to understand the functional domain architecture shared by all SFKs and the role of these domains in both regulating tyrosine kinase activity and recruiting additional proteins into signaling complexes. These aspects of SFK behavior have also been reviewed extensively elsewhere [8, 16].
Src is a 60-KDa protein composed of several functional domains [13, 17, 18]. Src contains a 14-carbon myristic acid moiety attached to an SH4 domain, a unique domain, an SH3 domain followed by an SH2 domain, an SH2-kinase linker, a protein tyrosine kinase domain (also known as an SH1 domain), and a C-terminal regulatory segment [19] (Figure 1). During cotranslational modification, the N-terminal methionine is removed and the resulting N-terminal glycine is myristoylated by myristoyl-coA. Myristoylation facilitates attachment to the inner surface of the cell membrane [19]. N-myristoylation is required for Src membrane association and its ability to transform cells [13, 20]. The differential state of palmitoylation at the SH4 domain of SFKs regulates subcellular trafficking of different SFKs in intact cell. All SFKs are cotranslationally myristoylated at Cly2 with the exception of Src and Blk, which are post translationally palmitoylated at Cys3, Cys5 or Cys6 [21]. Fatty acylation of SFKs has been shown to regulate their interaction with the cell membrane and their subcellular distribution [22, 23]. The poorly conserved unique domain is believed to provide unique functions and specificity to each SFK member.
Figure 1.
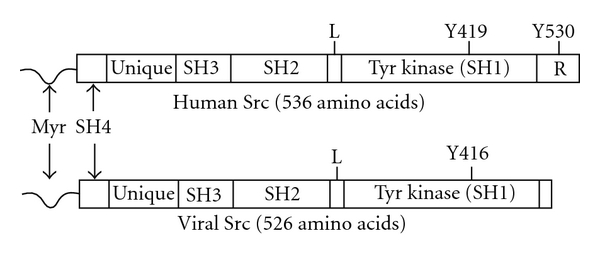
Schematic of the structural domain of human Src (Upper) and v-Src (Lower). The Src molecule is composed of an N-terminal myristoylation sequence (Myr) attached to the SH4 domain, a unique region followed by SH3 and SH2 domains, a linker region (L), a kinase domain (SH1 domain) that contains Tyr419, and a C-terminal regulatory domain (R) that contains Tyr530. v-Src protein differs from Src in a number of ways, with one major difference being the lack of a regulatory domain (R) at the C-terminal sequence.
The SH3 domain, composed of ~60 amino acid residues, is able to bind proline-rich sequences facilitating SFK-substrate or intramolecular interactions [19, 24]. The SH2 domain is composed of ~100 amino acids that can bind to phosphorylated tyrosine residues on either its own C-terminal regulatory domain or those of other proteins. Songyang and Cantley analyzed the binding of a library of phosphopeptides to the SH2 domain to define the preferred docking sequence [25]. The SH2 domain of each SFK member has distinct peptide preferences towards its binding partners [26]. The linker domain is involved in intramolecular binding with the SH3 domain. The catalytic domain is composed of two subdomains separated by a catalytic cleft, in which the adenosine-5′-triphosphate (ATP) and substrate-binding sites reside and phosphate transfer occurs. The cleft forms an activation loop that contains Tyrosine 419 (Tyr419; human Src) which is the positive regulatory site responsible for maximizing kinase activity [19]. Phosphorylation at the C-terminal end Tyr530 (human Src), which is a negative regulatory residue, leads to binding of this region to the SH2 domain; thus a “closed” or inactive conformation is attained, which is inaccessible to external ligands. In the closed conformation, the activation loop attains a compact structure, which fills the catalytic cleft and masks Tyr419 residues, thus preventing Tyr419 autophosphorylation and subsequent activation (Figure 2).
Figure 2.
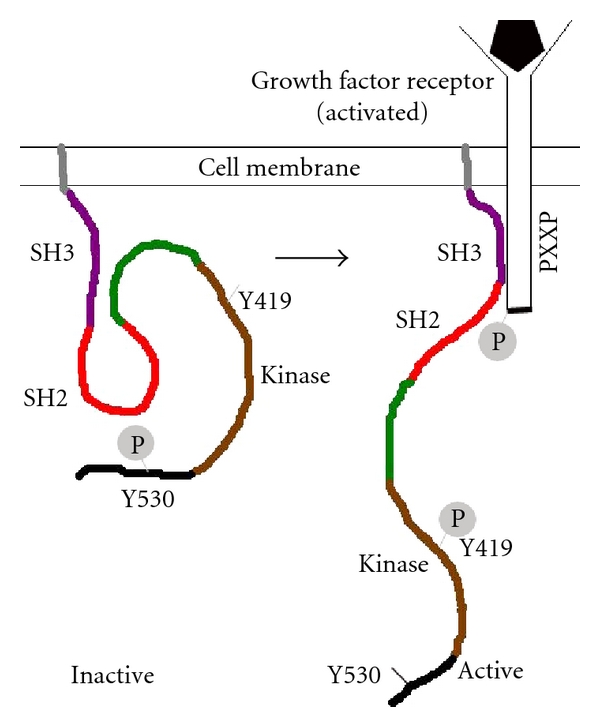
Cartoon representation of Src kinase regulation by differential phosphorylation at kinase domain as well as C-terminal regulatory domain.
3. Src Activation in Cancer
Src actions on mammalian cells are pleiotropic and include effects on cell morphology, adhesion, migration, invasion, proliferation, differentiation, and survival. Src kinase activation is common in various types of cancers although activating mutations and genomic amplifications are very rare. Thus, Src activation is usually accomplished by structural alteration mediated by upstream kinases or phosphatases. There are several ways SFKs activities are regulated, which include interactions that influence its intramolecular interactions and localizations. The net phosphorylation status of Src at its regulatory residues determines the activation status of Src, which is dependent upon a balance between phosphatase and kinase enzymes.
3.1. Regulation through the C-Terminal Negative Regulatory Domain
There are several ways Src kinase activity can be regulated, and any one of these might contribute to its activation in cancer cells. These include the phosphorylation of Tyr530, deletion or mutation of the C-terminal regulatory region, displacement of the SH3- and SH2 domain mediated by intramolecular interactions with higher affinity ligands, and phosphorylation of Tyr419. Independent biochemical and X-ray crystallographic analyses have revealed that Src maintains its inactive condition by various internal interactions. The interactions between the SH2 domain and the C-terminal Tyr530, as well as interactions between the SH3 domain and the SH2-kinase linker, modulate SFK activity [27].
Phosphorylation of the C-terminal negative regulatory tyrosine (Tyr530, human Src) is one of the mechanisms for the regulation of SFK activity. Due to the loss of the C-terminal residues, the viral proteins v-Src and v-Yes, are no longer able to be regulated by intramolecular interaction and become constitutively active and transformation competent [14, 15]. Regulation through the phosphorylation of Tyr530 in Src is accomplished by several kinases and phosphatases.
Two important protein tyrosine kinases in this process are Csk (c-Src kinase) and its homolog Csk-homologous kinase (Chk), which are both able to phosphorylate Tyr530 and to inactivate Src [28–30]. Reduced expression of Csk might play a role in the activation of Src in some cancers. In hepatocellular carcinoma, Csk levels are reduced compared to those in normal liver tissue and this reduced expression correlates with enhanced Src activity [31]. Evidence suggests that overexpression of Csk also appears to reduce tumor metastasis in colon cancer [32]. In addition to the reduced expression of Csk seen in cancer cells, other modes of regulating Csk are now being identified. Csk is structurally similar to Src, but its mode of regulation is distinct in that it lacks the regulatory tyrosine residue at the C-terminal end to control its activity [33].
Another mechanism of the regulation of Csk is through the transmembrane adaptor protein Cbp (Csk-binding protein or protein associated with glycosphingolipid-enriched microdomains [PAG]), a lipid raft-associated binding partner of Csk. Following phosphorylation by Src, Cbp can bind to the SH2 domain of Csk, thus allowing its recruitment to the plasma membrane where active Src resides. This creates a negative regulatory loop in which Cbp mediates the cross-linking of active Src with its suppressor, Csk [34]. An independent study by Oneyama et al. showed that membrane-bound adaptor protein Cbp suppress the Src-mediated cell transformation and tumorigenesis by binding and sequestering Src within lipid rafts [35]. Interestingly, this Cbp-mediated Src suppression was Csk independent. They have shown that Csk−/− mouse embryonic fibroblast cells underwent malignant transformation in the presence of Src [36]. The authors first noted that the levels of endogenous Cbp messenger RNA and protein were reduced when activated Src was expressed. They then made the seminal observation that overexpression of exogenous Cbp reversed the oncogenic effect of Src. They found that Cbp did not have any effect on Src tyrosine kinase activity; instead, it altered Src localization. The SH2 domain of Src binds to tyrosine phosphorylated Cbp and moves to the raft region and becomes inaccessible to kinase action. The cytoplasmic domain of Cbp has two proline-rich SH3 binding motifs and ten tyrosine residues, nine of which are Src targets. Oneyama found that phosphorylated Cbp could recruit SH2 domain-containing proteins such as Csk, SFKs, and suppressor of cytokine signaling 1 (SOCS1) to lipid rafts [34]. This finding further complicated our understanding of lipid rafts. Previous evidence had suggested that lipid rafts acted as positive hubs for activated signaling molecules and their associated SFKs. In order to mediate signals, SFKs need to be localized to the raft region [37]. Moreover, two independent studies have shown that SFKs remain active and can drive cancer cell growth even when bound to lipid-raft associated Cbps [38, 39]. This conflict can be addressed by studying the differences in fatty acylation status, cell types, and extent of Cbp interaction with SFKs.
4. Regulation of Src Activity by Phosphatases
Several protein tyrosine phosphatases (PTPs) are able to dephosphorylate Src Tyr530 and are responsible for the regulation of its kinase activity, such as PTPα, PTPγ, SHP-1 and -2, and PTP1B. PTPα is ubiquitously expressed and enriched in brain tissue [40–42] and is also able to dephosphorylate Tyr419, as evidenced by the lack of pSrcTyr419 in PTPα-overexpressing cells [40, 43]. Overexpression of PTPα also can dephosphorylate Src in A431 cell lines and cause enhancements in cell adhesion [44, 45]. A general question arises from these studies as to whether PTPα acts as an activator or repressor of Src molecules. Antisense studies of PTPα in 3T3-L1 adipocytes [46] and PTPα−/− murine studies [47, 48] show that Src kinase activity is linearly correlated with levels of PTPα protein in cells.
PTPγ was first identified from chicken brain tissue as a homolog of CD45 capable of dephosphorylating the SFK Lck [49]. It is expressed in the spleen and intestine and is able to dephosphorylate both Tyr530 and Tyr419 residues in Src. Chappel et al. have shown that PTPγ can modulate Src activity in osteoclast precursor cells treated with 1,25-dihydroxyvitamine D3; there was a dramatic increase in Src kinase activity without an increase in total protein levels. This change was accompanied by a decrease in phosphorylation at Tyr530 Interestingly both PTPγ mRNA and PTPγ protein levels were upregulated upon 1,25-dihydroxyvitamine D3 treatment suggesting the possibility that PTPg might be responsible for elevated Src kinase activity [50].
SHP1 is another member of the protein tyrosine phosphatase protein family that is also known as PTP-1c. It is a cytosolic two-SH2 domain containing PTP expressed in epithelial and hematopoetic cells [51]. Somani et al. have shown that SHP1 is responsible for the dephosphorylation and subsequent activation of Src, and it is much more specific for Src Tyr530 than Tyr419. This observation has been validated in transgenic mice that expressed the mutated loss of function form of SHP1, which has an increased level of Tyr530-phosphorylated Src [52].
SHP2 is a cytoplasmic SH2 domain containing PTP, which is also able to dephosphorylate Tyr530 [53]. SHP2 is very specific for the C-terminal regulatory tyrosine residue of Src. An independent study by Walter et al. demonstrated that SHP2 overexpression led to the activation of Src without significant changes in tyrosine phosphorylation at either residue (Tyr419 or Tyr530). In addition, the phosphatase-inactive mutant of SHP2 was also capable of Src activation. Further studies on the mechanism of Src activation by SHP2 revealed that the SH2 domain of SHP2 associates with Src by binding to the Src-SH3 domain and results in the allosteric activation of Src without involving Src dephosphorylation [54].
Another tyrosine phosphatase known as PTP-1B (also known as PTPN1) was first identified by Charbonneau et al. and first cloned and purified from human placenta [55–59]. Later Bjorge et al. demonstrated that PTP-1B was associated with Src activation in breast cancer cell lines [60]. PTP-1B is capable of both in vitro and in vivo activation of Src kinase activity as a result of its specificity towards tyrosine residues at the C-terminal tail. Human melanocyte [61] and several breast cancer cell lines [62] have elevated Src activity with concomitant hypophosphorylation of Tyr530. Biochemical analyses showed that these cells have elevated levels of PTP activity, which correlates with reduced phosphorylation on the C-terminal residue of Src and may have an important role in controlling Src kinase activity. The ability of PTP-1B to modulate Src activity has been demonstrated in mouse L-cell fibroblasts [63].
Rare activating mutations in Src that are truncated at codon 531 have been reported in some cases of advanced colon cancer patients [64]. The Src 531 mutation resulted in the production of a stop at codon 531, one residue beyond the regulatory Tyr530. Due to the lack of a C-terminal regulatory region, phosphorylation of Tyr530 did not result in a closed conformation and the mutated Src remained constitutively active.
5. Regulation of Src Activity by Receptor Tyrosine Kinases (RTKs)
Src can acts as an upstream or downstream modulator of several receptor molecules, as well as nonreceptor tyrosine kinases, which are responsible for the robustness and persistence of RTK signaling [65]. Src acts as a signal transducer from the cell surface receptors by sequential phosphorylation of tyrosine residues on substrates [66]. Src participates in the activation of various downstream signaling pathways through molecular interactions with growth factor receptors such as the epidermal growth factor receptor (EGFR) family, hepatocyte growth factor receptor (Met), integrin cell adhesion receptors, steroid hormone receptors, G protein-coupled receptors, focal adhesion kinase (FAK) and cytoskeleton components [65, 67]. Src can activate the phosphatidylinositol 3-kinase (PI3K)-Akt, growth factor receptor-bound protein 2 (Grb2)-Ras-Raf-mitogen-activated protein kinase (MAPK), Jak-signal transducers and activators of transcription (STAT) as well as FAK-paxillin-p130-Crk-associated substrate (Cas) cascades that are most crucial for cell cycle progression, survival, and proliferation [68–72]. Aberrant expression and activation of Src occurs in several tumor types and has been correlated with poor clinical outcome, which has stimulated interest in using Src kinase inhibitors as therapeutic cancer agents, some of which have entered the clinical trial stage [73, 74].
A variety of Src-binding proteins have been detected that compete for binding to the protein's SH domains and disturb the intramolecular interactions that allow the activation of Src kinase. v-Src cellular counterpart (c-Src) forms activated dimerized receptors via its SH2 domain binding to specific phosphotyrosine residues in the platelet-derived growth factor receptor (PDGFR) juxtamembrane region [75]. Other reports have suggested that activated PDGFR can phosphorylate tyrosine residues in the SH2/SH3 domain of Src and subsequently activate Src [76–79]. FAK is another kinase molecule able to bind to the Src-SH2 domain and activate the kinase activity [80–82]. Additional examples of regulators are FAK binding partners p130Cas [83, 84] and PTPα [85]. Recently, p130Cas, a protein that is thought to function as a docking protein because of its large number of binding motifs, has been demonstrated to bind to Src-SH2 and SH3 domains, resulting in Src activation [84]. Nef [86] and Sin [87] are examples of proteins that can bind to SH3 domains and activate the Src-family members Hck and Src, respectively.
There is also evidence to suggest that Src cooperates with EGFR in growth signaling [88, 89]. Src promotes EGF-induced anchorage-independent growth and tumorigenesis in nude mice. Cooperation between these two proteins depends on Src catalytic activity [90, 91]. EGFR leads to transient activation of Src kinase activity in glioma cells. Activation of Src leads to phosphorylation of Tyr845 on EGFR which is not an autophosphorylation site [92]. In an independent study on glioblastoma patients, Lu have shown that Src and Fyn act as effectors of oncogenic EGFR signaling and enhance invasion and tumor cell survival in vivo. Selective inhibition of Src and Fyn limited EGFR-dependent tumor cell motility. Src inhibition combined with an anti-EGFR monoclonal antibody further inhibited tumor growth and increased survival in an orthotopic glioblastoma mouser model [93]. Src is responsible for activation of STAT transcription factors after activation of ErbB1 by EGF, suggesting that EGF-induced mitogenesis might be mediated by the Src-STAT pathway which is independent of Jak [94]. Recently, we have shown that Src and c-Met interact differently in head and neck cancer cells that are sensitive or resistant to Src inhibition. Interestingly, however, in both cases c-Met acts as a direct Src substrate in an in vitro immunocomplex kinase assay system, which suggests that Src-dependent cell survival is also regulated by c-Met receptor activation, at least in head and neck cancer cells [95].
Another tier of Src regulation by RTKs was demonstrated by Jiang et al. who showed that EGFR, PDGFR, and fibroblast growth factor receptor (FGFR) phosphorylate Cbp upon ligand stimulation [96]. The EGFR mediated Cbp phosphorylation occurs via Src. Overexpression of Cbp blocks EGFR-mediated Src activation, signaling, and cell transformation, whereas loss of Cbp function has the opposite effect. Thus, Cbp may regulate the synergistic interactions between Src and EGFR in breast cancer.
6. Focal Adhesion
In a manner similar to many other signaling molecules, Src exerts its biologic action not only through its enzymatic activity and multidomain structure but also through its ability to interact with other signaling molecules in different cellular compartments [97]. Due to its N-terminal fatty acid moiety, Src associates with the plasma membrane as well as the perinuclear and endosomal membranes. The inactive form of Src has juxtanuclear localization. Upon activation by phosphorylation, Src SH3 domain associates with actin filaments, which then drive the translocation of Src to cell-cell and cell-matrix adhesion sites, where Src can interact with plasma membrane-bound molecular partners to take part in two major transduction events. These are (i) signaling from receptor tyrosine kinases, which mainly affects cell growth, proliferation, and migration and (ii) signaling from adhesion receptors, including integrins and E-cadherin, which mainly regulate cytoskeletal functions [98]. Constraints on the SH2 and SH3 domains that are released when the molecule is activated and are also likely to influence intracellular signaling by allowing the recruitment of high-affinity binding partners to specific intracellular sites. By this means, conformational activation of Src induces formation of SH2- and SH3-dependent multiprotein complexes at the cell periphery.
The primary role of tyrosine phosphorylation is to generate docking sites for proteins containing SH2 or phosphotyrosine binding (PTB) domains, thereby promoting protein-protein interaction and the formation of the macromolecular complexes responsible for signal transduction [99]. Many prominent Src substrates are found in focal adhesion junctions and include FAK, Cas, and tensins. Focal adhesion junctions are the sites of integrin-dependent substrate adhesion.
Tensins are the members of focal adhesion proteins that can serve as Src substrates. There are four members of the tensin family in mammals [100]. Tensins 1–3 contain three distinct regions: the N-terminal domain, which binds to F-actin and targets molecules for focal adhesion, a nonconserved central region, and the C-terminal SH2- PTB domain. The SH2 domains of Tensin-1 are required for promigratory functions [101], and the SH2 domains of Tensin 2 and 3 are responsible for binding with proangiogenic tyrosine (Tyr)-phosphorylated Cas and FAK. Qian et al. showed for the first time that the knockdown of Tensin-3 inhibited Src mediated cell transformation as well as cell migration and the growth of cancer cell lines [102].
Previously, Davis et al. showed that Tensin-1 is Tyr phosphorylated in Src-transformed chicken embryo fibroblasts [103]. Qian et al. observed that in a panel of human cancer cell lines, the level of phospho-Tensin-3 correlated roughly with both malignancy and with the levels of Src kinase activity [104]. Furthermore, the level of phospho-Tensin-3 was strongly reduced by specific inhibition of Src. Tensin-3 was also phosphorylated at Tyr in a mouse mammary tumor virus-(MMTV-)polyoma middle T (PyMT) murine model, in which endogenous Src was activated. This phosphorylation was reversed by Src inhibitor PP2. In addition, recombinant Src was also able to phosphorylate Tensin-3 in vitro [102]. They also have shown that the Tyr residue of SH2 domain of Tensin-3 at positions 1173/1206 and 1256 was phosphorylated by Src in a range of different types of cancers. Interestingly, Src inhibitors not only decreased the phosphorylation of Cas and the RNA-binding protein Sam68, but also decreased its interactions with Tensin-3.
7. Src Localization
Studies on the subcellular localization of Src reveal that it has been associated with the plasma, perinuclear, and endosomal membranes [105–109]. Although much evidence has been acquired regarding the role of Src at the plasma membrane and its interaction with growth factor receptors and integrin-nucleated focal adhesion complexes for regulating cell growth and proliferation [8, 66, 97], the functional significance of Src at other subcellular locations, such as cytoplasmic granules and perinuclear membranes, has not been as well characterized. The punctate staining pattern of Src in fibroblasts may represent the protein's association with membrane vesicles. Furthermore, analysis of Src function in Src-overexpressing fibroblasts indicates a possible association between Src with endosomal membranes [110]. Analysis of indirect immunofluorescence by three-dimensional optical sectioning microscopy revealed Src to be associated primarily with membranes at the microtubule organizing center, which represent a late stage in the endocytic pathway [109]. Moreover, Src is also associated with a number of microtubule-related structures including microtubule bundles at point of cell-cell contact and a region associated with the spindle pole during mitosis that regulates the transport or function of specialized secretory vesicles [109].
These data contrast with and extend previous reports of Src localization at the plasma membrane. One explanation for this discrepancy was that the biochemical fractionation techniques used in some prior studies did not differentiate between the plasma and endosomal membranes, which have similar densities and are thus likely to cofractionate [109]. The presence of Src in secretory organelles of chromaffin cells and platelets [111, 112], its association with endosomally derived synaptic vesicles in differentiated PC-12 cells [113], and the development of osteopetrosis in mice that are null for Src [114] further suggest a possible role for Src in protein-trafficking events.
7.1. Perinuclear and Nuclear Signaling
Src exhibits a predominantly perinuclear pattern of expression in malignant cells in contrast to a more evenly cytoplasmic distribution in normal breast epithelial cells [115]. The localization of Src to perinuclear membranes, endosomes and possibly even the nucleus suggests that Src is involved in nuclear-signal transduction events. The tyrosine kinase activity of Src is increased in mitotic cells arrested with nocodazole [116]. There is growing evidence that Src may play a role in cell cycle regulation especially at the G1/S transition [117, 118]. A 68 kDa phosphorylated protein (Src associated in mitosis, SAM68) is associated with Src in Src-activated mouse fibroblasts. An identical 70 kDa protein was identified as a tyrosine phosphorylated protein that was capable of binding to Lck and regulating T-cell activation. It has been postulated that Src regulates general splicing and mRNA transport via its effects on the expression at the posttranscriptional level of Sam68 [119]. Comparison of several modes of Src activation demonstrates that Src could either slow down the splicing rate or allow the export of partially spliced transcript [119]. Overexpression of Fyn in HEK293 cells interferes with the association of Sam68 with the splicing factor YT521-B and demonstrates Fyn's role in mRNA splicing [120]. Gondran and Dautry further strengthen the importance of Src in mRNA splicing and transport by inducing mutations at the SH2 and SH3 domains in Src [121]. There is evidence that Src can interact with different SH2 and SH3 domains containing signaling molecules such as PLCg-1, Grb2, NCK, Jak3, SHP1, Cbl, Grap (Grb2 like protein), p21 GTPase, p85 subunit of PI3K, p47 and Tec kinase family [122–130]. ASAP1, an ADP-ribosylation factor, is associated with Src [131]. ASAP1 is found primarily in the cytoplasm in a perinuclear, reticulate network. The association of Src with ASAP1, Arfs and PIP2 is thought to be important in coordinating membrane trafficking with actin cytoskeletal remodeling [131, 132]. Src associates with and phosphorylates various proteins responsible for vesicle transport at the perinuclear region; such as synapsin, dynamin, and so forth, [133–135]. Golgin67 has also been identified as a potential Src target, involved in vesicle docking and tethering [136]. Collectively this evidence suggests that Src might have a role in membrane trafficking events through transgolgi network [137].
8. Involvement of Src in Human Cancers
Src contribution to cell regulation and cancer development has been widely discussed in several review articles [74, 97, 138], so the discussion will be limited to a very short summary of a few relevant concepts and experimental findings.
There is a large body of evidence that has demonstrated that Src kinase activity and protein levels are elevated in several cancers, including those of the colon and breast. A correlation has often been observed between increases in Src kinase activity and the progression of malignancy [62, 64, 97, 115, 139–141]. Previously, we showed that Src promotes cancer cell survival in conjunction with STAT3 in head and neck squamous cell carcinoma (HNSCC) and non-small cell lung carcinoma (NSCLC) cells [142, 143].
Recently, Zhang et al. provided both clinical and experimental evidence that Src plays a critical role in the establishment of latent bone metastasis in breast cancer [144]. Using a bioinformatic approach that investigated the association between various signaling pathway-specific gene expression patterns and breast cancer, they identified a “Src activity gene expression signature” (Src responsive signature, SRS) that was highly associated with late onset of bone metastasis in breast cancer. To address the role of Src in the process of bone metastasis, they used two SRS-expressing breast cancer cell lines that possessed either aggressive or indolent metastatic bone tropism in a xenograft mouse model. In the cell line possessing aggressive metastatic bone tropism, stable knockdown of Src resulted in a significantly decreased rate of tumor outgrowth of bone lesions. In an indolent model of bone metastasis, knockdown of Src led to complete loss of bone metastatic activity, whereas the silencing of Src did not alter lung or lymph node metastatic activity, thus supporting a specific role for Src in bone metastasis. These prominent findings set the stage for the development of novel therapeutic strategies for eradicating breast cancer metastasis to bone.
In 2009, Yim et al. showed that the ectopic expression of Rak (also known as Frk) effectively suppressed breast cancer cell proliferation, invasion, and colony formation in vitro and tumor growth in vivo via its regulation of PTEN protein stability and function. Thus Rak may function as a tumor suppressor gene. Further understanding of its function may contribute to effective therapeutic approaches for both Rak- and PTEN-defective cancers [145].
Using integrated genomic and phosphoproteomic analysis of mouse lung primary and metastatic tumors, Carretero et al. demonstrated that loss of tumor suppressor LKB1 led to the activation of Src and FAK in a KrasG12D/Lkb1 murine model of lung tumor [146]. Src and FAK activation result in focal adhesion disassembly and turnover through the downregulation of Ras homolog gene family, member A (RhoA), which results in an increase in cellular motility and migration in the process of metastasis. They also confirmed the involvement of Src in the regulation of metastasis in KrasG12D/Lkb1 lung tumors by inhibiting Src, with concomitant increase in the sensitivity of tumor towards PI3K-MEK inhibition.
9. Clinical Trials of Src Inhibitors
A large body of evidence, including that discussed above, has identified Src as a key molecule in tumor progression that can provide oncogenic signals for cell survival, EMT, mitogenesis, and invasion and angiogenesis and metastasis [74, 147]. Due to the positive correlation between the development of cancer and the upregulation of Src activity, Src is emerging as a promising target for anticancer therapy [148, 149]. Src inhibition also results in a reduction of cancer progression in several cancer types [150–152], thus suggesting a potential clinical usefulness to inhibiting Src. There are several small molecule inhibitors for Src kinase that are undergoing clinical trials after promishing preclinical studies, such as the ATP-binding competitive inhibitors dasatinib (BMS-354825, Sprycel), bosutinib (SKI-606), saracatinib (AZD530), ponatinib (AP24534), bafetinib (INNO-406), and the substrate binding-site inhibitor Kxo-I (KX2-391) [153–157]. Preliminary data suggest that the agents are well tolerated at doses that achieve clinically meaningful plasma drug concentrations. Recent clinical studies with Src inhibitors as single agents or in combination are shown in Table 1.
Table 1.
Src inhibitors with other agents in clinical trials.
| Drug | Phase | Tumor type | Combination agent |
|---|---|---|---|
| Dasatinib | II | Advanced-NSCLC/Colorectal/Pancreatic/HNSCC/Breast/SCLC/Melanoma | — |
| II | Resectable NSCLC/HNSCC | Erlotinib | |
| I-II | Advanced NSCLC | Erlotinib | |
| I | Breast | Capecitabine | |
| I | Breast | Paclitaxel | |
| I-II | Prostate/castration resistant prostate cancer | Docetaxel | |
| I | Colon | FOLFOX6/Cetuximab | |
|
| |||
| Saracatinib | II | Prostate/Pancreatic/Osteosarcoma/Soft tissue sarcoma/Melanoma/Gastration-resistant prostate cancer/Thymoma/Colorectal/HNSCC | — |
| II | Advanced NSCLC/SCLC | Carboplatin/Paclitaxel | |
| I | Advanced solid tumor | Cediranib | |
| I-II | Pancreatic | Gemcitabine | |
| II | Ovarian | Carboplatin | |
| II | Prostate/Breast with bone metastasis | Zoledronic acid | |
|
| |||
| Bosutinib | II | Breast | — |
| II | Breast | Exemestane | |
| II | Breast | Letrozole/Capecitabine | |
| I-II | Advanced solid tumor | Capecitabine | |
|
| |||
| XL228 | I | Advanced solid tumor | — |
|
| |||
| KX2-391 | I | Advanced solid tumor/Lymphoma | — |
|
| |||
| AZM475271 | I-II | Pancreatic | — |
|
| |||
| XL999 | I | Advanced solid tumor | — |
Dasatinib suppressed invasion and induced cell cycle arrest in HNSCC cells in vitro [158], affected the mechanisms of prostate tumor progression [159], and greatly inhibited the development of liver metastasis in an orthotopic murine model of pancreatic carcinoma. Studies of dasatinib in prostate [160] and colon cancer cell lines [161] showed inhibition of cellular adhesion, migration, and invasion. Breast cancer cell lines belonging to the basal/“triple-negative” subtype were particularly sensitive to dasatinib. Breast cancers within this subgroup express basal cell cytokeratins (CK5 and CK17), with ER, PR and Her2 negative phenotype [162, 163], and are well known for poor prognosis [164]. Interestingly, in EGFR-overexpressing breast cancer cell lines, dasatinib inhibited cell growth, invasion, and angiogenesis, and stimulated apoptosis by activating caspase 8 and 9 [165].
Bosutinib showed activity against colon cancer in a murine model and was well tolerated. In cellular assays, bosutinib treatment resulted in a dose-dependent reduction in proliferation, invasion, and migration of breast cancer cells [166, 167]. Furthermore, in a murine model of breast carcinoma, bosutinib inhibited tumor growth and significantly reduced the number of liver, spleen, and lung metastases. Clinical trials with bosutinib for breast cancer, other solid tumors, and leukemia are ongoing [168].
Saracatinib (formerly AZD0530; AstraZeneca) is another ATP-competitive inhibitor of SFKs, with activity against ABL and activated mutant forms of EGFR (L858R and L861Q) [169]. In a panel of 13 human cancer cell lines treated with saracatinib, there was growth inhibition in four different cell lines (derived from colon, prostate, and lung tumors) and inhibitory effects on migration and invasion [170, 171].
In a recent phase II trial with dasatinib as a first line of treatment for metastatic NSCLC several patients had prolonged stable disease and one patient had a near complete response that persisted 2 years after the start of therapy, suggesting that there was a subset of patients with NSCLC who benefited from Src inhibition [172]. Another independent phase I/II study in NSCLC using the combination of Src and EGFR inhibitors also demonstrated clinical responses [173, 174]. These observations further validate the preclinical findings that suggest there is cooperation between EGFR kinase activity and Src in NSCLC [158, 175–177].
In a phase II trial in 2008, Yu et al. demonstrated that dasatinib improves the overall survival in castration resistant prostate cancer [178]. Based on promising results from phase I/II clinical trials of combination treatment with dasatinib and docetaxel in prostate cancer patients, this combination is now being tested in a phase III clinical trials [118, 119].
M475271 is an oral inhibitor of Src and vascular endothelial growth factor receptor (VEGFR) that has shown preclinical activity in lung adenocarcinoma cell lines [179]. Another SFK inhibitor, KX2-391, targets the peptide substrate-binding site rather than the ATP-binding site. Based on the promising results from phase I study, a phase II study has been initiated with Castration-Resistant Prostate Cancer Bone-Metastatic patients [180], (http://www.clinicaltrial.gov/). All these therapeutic agents appear to be well tolerated and we eagerly await their detailed clinical results.
10. Conclusions
Our understanding of Src structure and function, regulation, and localization has increased dramatically since its discovery. One-hundred years after the original description of Src, this protein continues to attract keen interest because of its multiplicity of actions in the molecular signaling pathways underlying developmental as well as oncogenic events. Many studies have addressed the molecular mechanisms of Src regulation in cells and tumor tissues. In order to clarify and fully elucidate the normal physiologic function of Src and other SFKs and to fully comprehend Src signaling networks in various cancers, Src interactions with specific targets or binding partners in different subcellular localization studies should be characterized in as much detail as possible. Special focus should be placed on the role of Src in bone metastasis because of the protein's role in osteoclast and osteoblast function. Moreover, preclinical reports of combination treatments involving chemotherapy, radiation therapy, and targeted therapies with a Src inhibitor warrant further investigation [181, 182].
Acknowledgments
The authors thank Ms. M. E. Veech for editorial assistance. F. M. Johnson is supported by R01CA14336901, and by the University of Texas MD Anderson Head and Neck SPORE Grant no. (P50 CA097007).
References
- 1.Rous P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. The Journal of Experimental Medicine. 1911;13:397–411. doi: 10.1084/jem.13.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stehelin D, Fujita DJ, Padgett T. Detection and enumeration of transformation defective strains of avian sarcoma virus with molecular hybridization. Virology. 1977;76(2):675–684. doi: 10.1016/0042-6822(77)90250-1. [DOI] [PubMed] [Google Scholar]
- 3.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260(5547):170–173. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 4.Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(3 I):1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collett MS, Purchio AF, Erikson RL. Avian sarcoma virus-transforming protein, pp60(src) shows protein kinase activity specific for tyrosine. Nature. 1980;285(5761):167–169. doi: 10.1038/285167a0. [DOI] [PubMed] [Google Scholar]
- 6.Levinson AD, Oppermann H, Varmus HE, Bishop JM. The purified product of the transforming gene of avian sarcoma virus phosphorylates tyrosine. Journal of Biological Chemistry. 1980;255(24):11973–11980. [PubMed] [Google Scholar]
- 7.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 8.Thomas SM, Brugge JS. Cellular functions regulated by SRC family kinases. Annual Review of Cell and Developmental Biology. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 9.Cance WG, Craven RJ, Bergman M, Xu L, Alitalo K, Liu ET. Rak, a novel nuclear tyrosine kinase expressed in epithelial cells. Cell Growth and Differentiation. 1994;5(12):1347–1355. [PubMed] [Google Scholar]
- 10.Serfas MS, Tyner AL. Brk, Srm, Frk, and Src42A form a distinct family of intracellular Src-like tyrosine kinases. Oncology Research. 2002;13(6–10):409–419. doi: 10.3727/096504003108748438. [DOI] [PubMed] [Google Scholar]
- 11.Annerén C, Lindholm CK, Kriz V, Welsh M. The FRK/RAK-SHB signaling cascade: a versatile signal-transduction pathway that regulates cell survival, differentiation and proliferation. Current Molecular Medicine. 2003;3(4):313–324. doi: 10.2174/1566524033479744. [DOI] [PubMed] [Google Scholar]
- 12.Meyer T, Xu L, Chang J, Lui ET, Craven RJ, Cance WG. Breast cancer cell line proliferation blocked by the Src-related Rak tyrosine kinase. International Journal of Cancer. 2003;104(2):139–146. doi: 10.1002/ijc.10925. [DOI] [PubMed] [Google Scholar]
- 13.Roskoski R., Jr. Src protein-tyrosine kinase structure and regulation. Biochemical and Biophysical Research Communications. 2004;324(4):1155–1164. doi: 10.1016/j.bbrc.2004.09.171. [DOI] [PubMed] [Google Scholar]
- 14.Martin GS. The hunting of the Src. Nature Reviews Molecular Cell Biology. 2001;2(6):467–474. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 15.Toyoshima K, Yamamoto T, Kawai S, Yoshida M. Viral oncogenes, v-yes and v-erbB, and their cellular counterparts. Advances in Virus Research. 1987;32:97–127. doi: 10.1016/s0065-3527(08)60475-3. [DOI] [PubMed] [Google Scholar]
- 16.Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochimica et Biophysica Acta. 2002;1602(2):114–130. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 17.Parsons JT, Weber MJ. Genetics of src: structure and functional organization of a protein tyrosine kinase. Current Topics in Microbiology and Immunology. 1989;147:79–127. doi: 10.1007/978-3-642-74697-0_3. [DOI] [PubMed] [Google Scholar]
- 18.Yeatman TJ. A renaissance for SRC. Nature Reviews Cancer. 2004;4(6):470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 19.Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochimica et Biophysica Acta. 1996;1287(2-3):121–149. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- 20.Towler DA, Gordon JI, Adams SP, Glaser L. The biology and enzymology of eukaryotic protein acylation. Annual Review of Biochemistry. 1988;57:69–99. doi: 10.1146/annurev.bi.57.070188.000441. [DOI] [PubMed] [Google Scholar]
- 21.Sato I, Obata Y, Kasahara K, et al. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. Journal of Cell Science. 2009;122(7):965–975. doi: 10.1242/jcs.034843. [DOI] [PubMed] [Google Scholar]
- 22.McCabe JB, Berthiaume LG. Functional roles for fatty acylated amino-terminal domains in subcellular localization. Molecular Biology of the Cell. 1999;10(11):3771–3786. doi: 10.1091/mbc.10.11.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Resh MD. Fatty acylation of proteins: New insights into membrane targeting of myristoylated and palmitoylated proteins. Biochimica et Biophysica Acta. 1999;1451(1):1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 24.Cohen GB, Ren R, Baltimore D. Modular binding domains in signal transduction proteins. Cell. 1995;80(2):237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 25.Songyang Z, Cantley LC. Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends in Biochemical Sciences. 1995;20(11):470–475. doi: 10.1016/s0968-0004(00)89103-3. [DOI] [PubMed] [Google Scholar]
- 26.Waksman G, Kuriyan J. Structure and specificity of the SH2 domain. Cell. 2004;116(2):S45–S48. doi: 10.1016/s0092-8674(04)00043-1. [DOI] [PubMed] [Google Scholar]
- 27.Gonfloni S, Weijland A, Kretzschmar J, Superti-Furga G. Crosstalk between the catalytic and regulatory domains allows bidirectional regulation of Src. Nature Structural Biology. 2000;7(4):281–286. doi: 10.1038/74041. [DOI] [PubMed] [Google Scholar]
- 28.Okada M, Nakagawa H. A protein tyrosine kinase involved in regulation of pp60(c-src) function. Journal of Biological Chemistry. 1989;264(35):20886–20893. [PubMed] [Google Scholar]
- 29.Cooper JA, Gould KL, Cartwright CA, Hunter T. Tyr is phosphorylated in pp60(c-src): implications for regulation. Science. 1986;231(4744):1431–1434. doi: 10.1126/science.2420005. [DOI] [PubMed] [Google Scholar]
- 30.Zrihan-Licht S, Lim J, Keydar I, Sliwkowski MX, Groopman JE, Avraham H. Association of Csk-homologous kinase (CHK) (formerly MATK) with HER- 2/ErbB-2 in breast cancer cells. Journal of Biological Chemistry. 1997;272(3):1856–1863. doi: 10.1074/jbc.272.3.1856. [DOI] [PubMed] [Google Scholar]
- 31.Masaki T, Okada M, Tokuda M, et al. Reduced C-terminal Src kinase (Csk) activities in hepatocellular carcinoma. Hepatology. 1999;29(2):379–384. doi: 10.1002/hep.510290239. [DOI] [PubMed] [Google Scholar]
- 32.Nakagawa T, Tanaka S, Suzuki H, et al. Overexpression of the csk gene suppresses tumor metastasis in vivo. International Journal of Cancer. 2000;88(3):384–391. [PubMed] [Google Scholar]
- 33.Nada S, Yagi T, Takeda H, et al. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell. 1993;73(6):1125–1135. doi: 10.1016/0092-8674(93)90642-4. [DOI] [PubMed] [Google Scholar]
- 34.Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochimica et Biophysica Acta. 2008;1784(1):56–65. doi: 10.1016/j.bbapap.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Oneyama C, Hikita T, Enya K, et al. The lipid raft-anchored adaptor protein Cbp controls the oncogenic potential of c-Src. Molecular Cell. 2008;30(4):426–436. doi: 10.1016/j.molcel.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 36.Oneyama C, Hikita T, Nada S, Okada M. Functional dissection of transformation by c-Src and v-Src. Genes to Cells. 2008;13(1):1–12. doi: 10.1111/j.1365-2443.2007.01145.x. [DOI] [PubMed] [Google Scholar]
- 37.Resh MD. Trafficking and signaling by fatty-acylated and prenylated proteins. Nature Chemical Biology. 2006;2(11):584–590. doi: 10.1038/nchembio834. [DOI] [PubMed] [Google Scholar]
- 38.Solheim SA, Torgersen KM, Taskén K, Berge T. Regulation of FynT function by dual domain docking on PAG/Cbp. Journal of Biological Chemistry. 2008;283(5):2773–2783. doi: 10.1074/jbc.M705215200. [DOI] [PubMed] [Google Scholar]
- 39.Tauzin S, Ding H, Khatib K, et al. Oncogenic association of the Cbp/PAG adaptor protein with the Lyn tyrosine kinase in human B-NHL rafts. Blood. 2008;111(4):2310–2320. doi: 10.1182/blood-2007-05-090985. [DOI] [PubMed] [Google Scholar]
- 40.Zheng XM, Wang Y, Pallen CJ. Cell transformation and activation of pp60(c-src) by overexpression of a protein tyrosine phosphatase. Nature. 1992;359(6393):336–339. doi: 10.1038/359336a0. [DOI] [PubMed] [Google Scholar]
- 41.Krueger NX, Streuli M, Saito H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO Journal. 1990;9(10):3241–3252. doi: 10.1002/j.1460-2075.1990.tb07523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaplan R, Morse B, Huebner K, et al. Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(18):7000–7004. doi: 10.1073/pnas.87.18.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang KS, Barker K, Sudol M, Hanafusa H. A transmembrane protein-tyrosine phosphatase contains spectrin-like repeats in its extracellular domain. Journal of Biological Chemistry. 1994;269(19):14056–14063. [PubMed] [Google Scholar]
- 44.Harder KW, Moller NPH, Peacock JW, Jirik FR. Protein-tyrosine phosphatase a regulates Src family kinases and alters cell-substratum adhesion. Journal of Biological Chemistry. 1999;273(48):31890–31900. doi: 10.1074/jbc.273.48.31890. [DOI] [PubMed] [Google Scholar]
- 45.Bhandari V, Lim KL, Pallen CJ. Physical and functional interactions between receptor-like protein- tyrosine phosphatase α and p59(fyn) Journal of Biological Chemistry. 1998;273(15):8691–8698. doi: 10.1074/jbc.273.15.8691. [DOI] [PubMed] [Google Scholar]
- 46.Arnott CH, Sale EM, Miller J, Sale GJ. Use of an antisense strategy to dissect the signaling role of protein-tyrosine phosphatase α . Journal of Biological Chemistry. 1999;274(37):26105–26112. doi: 10.1074/jbc.274.37.26105. [DOI] [PubMed] [Google Scholar]
- 47.Ponniah S, Wang DZM, Kah Leong L, Pallen CJ. Targeted disruption of the tyrosine phosphatase PTPα leads to constitutive downregulation of the kinases Src and Fyn. Current Biology. 1999;9(10):535–538. doi: 10.1016/s0960-9822(99)80238-3. [DOI] [PubMed] [Google Scholar]
- 48.Su J, Muranjan M, Sap J. Receptor protein tyrosine phosphatase α activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Current Biology. 1999;9(10):505–511. doi: 10.1016/s0960-9822(99)80234-6. [DOI] [PubMed] [Google Scholar]
- 49.Fang KS, Sabe H, Saito H, Hanafusa H. Comparative study of three protein-tyrosine phosphatases. Chicken protein- tyrosine phosphatase λ dephosphorylates c-Src tyrosine 527. Journal of Biological Chemistry. 1994;269(31):20194–20200. [PubMed] [Google Scholar]
- 50.Chappel J, Ross FP, Abu-Amer Y, Shaw A, Teitelbaum SL. 1,25-dihydroxyvitamin D regulates pp60(c-src) activity and expression of a pp60(c-src) activating phosphatase. Journal of Cellular Biochemistry. 1997;67(4):432–438. doi: 10.1002/(sici)1097-4644(19971215)67:4<432::aid-jcb2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 51.Falet H, Ramos-Morales F, Bachelot C, Fischer S, Rendu F. Association of the protein tyrosine phosphatase PTP1C with the protein tyrosine kinase c-Src in human platelets. FEBS Letters. 1996;383(3):165–169. doi: 10.1016/0014-5793(96)00232-3. [DOI] [PubMed] [Google Scholar]
- 52.Somani AK, Bignon JS, Mills GB, Siminovitch KA, Branch DR. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. Journal of Biological Chemistry. 1997;272(34):21113–21119. doi: 10.1074/jbc.272.34.21113. [DOI] [PubMed] [Google Scholar]
- 53.Peng ZY, Cartwright CA. Regulation of the Src tyrosine kinase and Syp tyrosine phosphatase by their cellular association. Oncogene. 1995;11(10):1955–1962. [PubMed] [Google Scholar]
- 54.Walter AO, Peng ZY, Cartwright CA. The Shp-2 tyrosine phosphatase activates the Src tyrosine kinase by a non-enzymatic mechanism. Oncogene. 1999;18(11):1911–1920. doi: 10.1038/sj.onc.1202513. [DOI] [PubMed] [Google Scholar]
- 55.Charbonneau H, Tonks NK, Kumar S, et al. Human placenta protein-tyrosine-phosphatase: amino acid sequence and relationship to a family of receptor-like proteins. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(14):5252–5256. doi: 10.1073/pnas.86.14.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tonks NK, Diltz CD, Fischer EH. Purification of the major protein-tyrosine-phosphatases of human placenta. Journal of Biological Chemistry. 1988;263(14):6722–6730. [PubMed] [Google Scholar]
- 57.Chernoff J, Schievella AR, Jost CA, Erikson RL, Neel RG. Cloning of a cDNA for a major human protein-tyrosine-phosphatase. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(7):2735–2739. doi: 10.1073/pnas.87.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guan K, Haun RS, Watson SJ, Geahlen RL, Dixon JE. Cloning and expression of a protein-tyrosine-phosphatase. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(4):1501–1505. doi: 10.1073/pnas.87.4.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown-Shimer S, Johnson KA, Lawrence JB, et al. Molecular cloning and chromosome mapping of the human gene encoding protein phosphotyrosyl phosphatase 1B. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(13):5148–5152. doi: 10.1073/pnas.87.13.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. Journal of Biological Chemistry. 2000;275(52):41439–41446. doi: 10.1074/jbc.M004852200. [DOI] [PubMed] [Google Scholar]
- 61.O’Connor TJ, Neufeld E, Bechberger J, Fujita DJ. pp60c-src in human melanocytes and melanoma cells exhibits elevated specific activity and reduced tyrosine 530 phosphorylation compared to human fibroblast pp60c-src. Cell Growth & Differentiation. 1992;3(7):435–442. [PubMed] [Google Scholar]
- 62.Egan C, Pang A, Durda D, Cheng HC, Wang JH, Fujita DJ. Activation of Src in human breast tumor cell lines: elevated levels of phosphotyrosine phosphatase activity that preferentially recognizes the Src carboxy terminal negative regulatory tyrosine 530. Oncogene. 1999;18(5):1227–1237. doi: 10.1038/sj.onc.1202233. [DOI] [PubMed] [Google Scholar]
- 63.Arregui CO, Balsamo J, Lilien J. Impaired integrin-mediated adhesion and signaling in fibroblasts expressing a dominant, negative mutant PTP1B. Journal of Cell Biology. 1998;143(3):861–873. doi: 10.1083/jcb.143.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Irby RB, Mao W, Coppola D, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nature Genetics. 1999;21(2):187–190. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- 65.Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23(48):7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- 66.Abram CL, Courtneidge SA. Src family tyrosine kinases and growth factor signaling. Experimental Cell Research. 2000;254(1):1–13. doi: 10.1006/excr.1999.4732. [DOI] [PubMed] [Google Scholar]
- 67.Ishizawar R, Parsons SJ. C-Src and cooperating partners in human cancer. Cancer Cell. 2004;6(3):209–214. doi: 10.1016/j.ccr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 68.Lu Y, Yu Q, Liu JH, et al. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. Journal of Biological Chemistry. 2003;278(41):40057–40066. doi: 10.1074/jbc.M303621200. [DOI] [PubMed] [Google Scholar]
- 69.Belsches AP, Haskell MD, Parsons SJ. Role of c-Src tyrosine kinase in EGF-induced mitogenesis. Frontiers in Bioscience. 1997;2:d501–518. doi: 10.2741/a208. [DOI] [PubMed] [Google Scholar]
- 70.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 71.Guarino M. Src signaling in cancer invasion. Journal of Cellular Physiology. 2010;223(1):14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
- 72.Lai SY, Johnson FM. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resistance Updates. 2010;13(3):67–78. doi: 10.1016/j.drup.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 73.Rucci N, Šuša M, Teti A. Inhibition of protein kinase c-Src as a therapeutic approach for cancer and bone metastases. Anti-Cancer Agents in Medicinal Chemistry. 2008;8(3):342–349. doi: 10.2174/187152008783961905. [DOI] [PubMed] [Google Scholar]
- 74.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer and Metastasis Reviews. 2003;22(4):337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 75.Mori S, Ronnstrand L, Yokote K, et al. Identification of two juxtamembrane autophosphorylation sites in the PGDF β-receptor; involvement in the interaction with Src family tyrosine kinases. EMBO Journal. 1993;12(6):2257–2264. doi: 10.1002/j.1460-2075.1993.tb05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Broome MA, Hunter T. The PDGF receptor phosphorylates Tyr 138 in the c-Src SH3 domain in vivo reducing peptide ligand binding. Oncogene. 1997;14(1):17–34. doi: 10.1038/sj.onc.1200798. [DOI] [PubMed] [Google Scholar]
- 77.Stover DR, Furet P, Lydon NB. Modulation of the SH2 binding specificity and kinase activity of Src by tyrosine phosphorylation within its SH2 domain. Journal of Biological Chemistry. 1996;271(21):12481–12487. doi: 10.1074/jbc.271.21.12481. [DOI] [PubMed] [Google Scholar]
- 78.Kypta RM, Goldberg Y, Ulug ET, Courtneidge SA. Association between the PDGF receptor and members of the src family of tyrosine kinases. Cell. 1990;62(3):481–492. doi: 10.1016/0092-8674(90)90013-5. [DOI] [PubMed] [Google Scholar]
- 79.Alonso G, Koegl M, Mazurenko N, Courtneidge SA. Sequence requirements for binding of Src family tyrosine kinases to activated growth factor receptors. Journal of Biological Chemistry. 1995;270(17):9840–9848. doi: 10.1074/jbc.270.17.9840. [DOI] [PubMed] [Google Scholar]
- 80.Cobb BS, Schaller MD, Leu TH, Parsons JT. Stable association of pp60(src) and pp59(fyn) with the focal adhesion- associated protein tyrosine kinase, pp125(FAK) Molecular and Cellular Biology. 1994;14(1):147–155. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eide BL, Turck CW, Escobedo JA. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60(src) association in the focal adhesion kinase, pp125(FAK) Molecular and Cellular Biology. 1995;15(5):2819–2827. doi: 10.1128/mcb.15.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125(FAK), directs SH2- dependent binding of pp60(src) Molecular and Cellular Biology. 1994;14(3):1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kanner SB, Reynolds AB, Vines RR, Parsons JT. Monoclonal antibodies to individual tyrosine-phosphorylated protein substrates of oncogene-encoded tyrosine kinases. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(9):3328–3332. doi: 10.1073/pnas.87.9.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burnham MR, Bruce-Staskal PJ, Harte MT, et al. Regulation of c-SRC activity and function by the adapter protein CAS. Molecular and Cellular Biology. 2000;20(16):5865–5878. doi: 10.1128/mcb.20.16.5865-5878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPα . EMBO Journal. 2000;19(5):964–978. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moarefi I, LaFevre-Bernt M, Sicheri F, et al. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385(6617):650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- 87.Alexandropoulos K, Baltimore D. Coordinate activation of c-Src by SH3-and SH2-binding sites on a novel, p130(Cas)-related protein, Sin. Genes and Development. 1996;10(11):1341–1355. doi: 10.1101/gad.10.11.1341. [DOI] [PubMed] [Google Scholar]
- 88.Roche S, Koegl M, Barone MV, Roussel MF, Courtneidge SA. DNA synthesis induced by some but not all growth factors requires Src family protein tyrosine kinases. Molecular and Cellular Biology. 1995;15(2):1102–1109. doi: 10.1128/mcb.15.2.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wilson LK, Luttrell DK, Parsons JT, Parsons SJ. pp60(c-src) tyrosine kinase, myristylation, and modulatory domains are required for enhanced mitogenic responsiveness to epidermal growth factor seen in cells overexpressing c-src. Molecular and Cellular Biology. 1989;9(4):1536–1544. doi: 10.1128/mcb.9.4.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(4):1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maa MC, Leu TH, Mccarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(15):6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johns TG, Perera RM, Vernes SC, et al. The efficacy of epidermal growth factor receptor-specific antibodies against glioma xenografts is influenced by receptor levels, activation status, and heterodimerization. Clinical Cancer Research. 2007;13(6):1911–1925. doi: 10.1158/1078-0432.CCR-06-1453. [DOI] [PubMed] [Google Scholar]
- 93.Lu KV, Zhu S, Cvrljevic A, et al. Fyn and Src are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Research. 2009;69(17):6889–6898. doi: 10.1158/0008-5472.CAN-09-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Olayioye MA, Beuvink I, Horsch K, Daly JM, Hynes NE. ErbB receptor-induced activation of Stat transcription factors is mediated by Src tyrosine kinases. Journal of Biological Chemistry. 1999;274(24):17209–17218. doi: 10.1074/jbc.274.24.17209. [DOI] [PubMed] [Google Scholar]
- 95.Sen B, Peng S, Saigal B, Williams MD, Johnson FM. Distinct interactions between c-Src and c-Met in mediating resistance to c-Src inhibition in head and neck cancer. Clinical Cancer Research. 2011;17(3):514–524. doi: 10.1158/1078-0432.CCR-10-1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang LQ, Feng X, Zhou W, Knyazev PG, Ullrich A, Chen Z. Csk-binding protein (Cbp) negatively regulates epidermal growth factor-induced cell transformation by controlling Src activation. Oncogene. 2006;25(40):5495–5506. doi: 10.1038/sj.onc.1209554. [DOI] [PubMed] [Google Scholar]
- 97.Biscardi JS, Tice DA, Parsons SJ. C-Src, receptor tyrosine kinases, and human cancer. Advances in Cancer Research. 1999;76:117–119. doi: 10.1016/s0065-230x(08)60774-5. [DOI] [PubMed] [Google Scholar]
- 98.Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of Src kinase. Oncogene. 2000;19(49):5620–5635. doi: 10.1038/sj.onc.1203923. [DOI] [PubMed] [Google Scholar]
- 99.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116(2):191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 100.Lo SH. Tensin. International Journal of Biochemistry and Cell Biology. 2004;36(1):31–34. doi: 10.1016/s1357-2725(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 101.Chen H, Lo SUH. Regulation of tensin-promoted cell migration by its focal adhesion binding and Src homology domain 2. Biochemical Journal. 2003;370(3):1039–1045. doi: 10.1042/BJ20021308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qian X, Li G, Vass WC, et al. The Tensin-3 protein, including its SH2 domain, is phosphorylated by Src and contributes to tumorigenesis and metastasis. Cancer Cell. 2009;16(3):246–258. doi: 10.1016/j.ccr.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davis S, Lu ML, Lo SH, et al. Presence of an SH2 domain in the actin-binding protein tensin. Science. 1991;252(5006):712–715. doi: 10.1126/science.1708917. [DOI] [PubMed] [Google Scholar]
- 104.Qian X, Li G, Asmussen HK, et al. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):9012–9017. doi: 10.1073/pnas.0703033104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Willingham MC, Jay G, Pastan I. Localization of the ASV src gene product to the plasma membrane of transformed cells by electron microscopic immunocytochemistry. Cell. 1979;18(1):125–134. doi: 10.1016/0092-8674(79)90361-1. [DOI] [PubMed] [Google Scholar]
- 106.Courtneidge SA, Levinson AD, Bishop JM. The protein encoded by the transforming gene of avian sarcoma virus (pp60(src)) and a homologous protein in normal cells (pp60(proto-src)) are associated with the plasma membrane. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(7):3783–3787. doi: 10.1073/pnas.77.7.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Resh MD, Erikson RL. Highly specific antibody to Rous sarcoma virus src gene product recognizes a novel population of pp60(v-src) and pp60(c-src) molecules. Journal of Cell Biology. 1985;100(2):409–417. doi: 10.1083/jcb.100.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.David-Pfeuty T, Nouvian-Dooghe Y. Highly specific antibody to Rous sarcoma virus src gene product recognizes nuclear and nucleolar antigens in humans cells. Journal of Virology. 1995;69(3):1699–1713. doi: 10.1128/jvi.69.3.1699-1713.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaplan KB, Swedlow JR, Varmus HE, Morgan DO. Association of p60(c-src) with endosomal membranes in mammalian fibroblasts. Journal of Cell Biology. 1992;118(2):321–333. doi: 10.1083/jcb.118.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.David-Pfeuty T, Nouvian-Dooghe Y. Immunolocalization of the cellular src protein in interphase and mitotic NIH c-src overexpresser cells. Journal of Cell Biology. 1990;111(6):3097–3116. doi: 10.1083/jcb.111.6.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Parsons SJ, Creutz CE. p60(c-src) activity detected in the chromaffin granule membrane. Biochemical and Biophysical Research Communications. 1986;134(2):736–742. doi: 10.1016/s0006-291x(86)80482-x. [DOI] [PubMed] [Google Scholar]
- 112.Rendu F, Lebret M, Danielian S, Fagard R, Levy-Toledano S, Fischer S. High pp60(c-src) level in human platelet dense bodies. Blood. 1989;73(6):1545–1551. [PubMed] [Google Scholar]
- 113.Linstedt AD, Vetter ML, Bishop JM, Kelly RB. Specific association of the proto-oncogene product pp60(c-src) with an intracellular organelle, the PC12 synaptic vesicle. Journal of Cell Biology. 1992;117(5):1077–1084. doi: 10.1083/jcb.117.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64(4):693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 115.Verbeek BS, Vroom TM, Adriaansen-Slot SS, et al. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. Journal of Pathology. 1996;180(4):383–388. doi: 10.1002/(SICI)1096-9896(199612)180:4<383::AID-PATH686>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 116.Chackalaparampil I, Shalloway D. Altered phosphorylation and activation of PP60 during fibroblast mitosis. Cell. 1988;52(6):801–810. doi: 10.1016/0092-8674(88)90422-9. [DOI] [PubMed] [Google Scholar]
- 117.Wyke AW, Cushley W, Wyke JA. Mitogenesis by v-Src: a need for active oncoprotein both in leaving G0 and in completing G1 phases of the cell cycle. Cell Growth & Differentiation. 1993;4(8):671–678. [PubMed] [Google Scholar]
- 118.Barlat I, Maurier F, Duchesne M, Guitard E, Tocque B, Schweighoffer F. A role for Sam68 in cell cycle progression antagonized by a spliced variant within the KH domain. Journal of Biological Chemistry. 1997;272(6):3129–3132. doi: 10.1074/jbc.272.6.3129. [DOI] [PubMed] [Google Scholar]
- 119.Neel H, Gondran P, Weil D, Dautry F. Regulation of pre-mRNA processing by src. Current Biology. 1995;5(4):413–422. doi: 10.1016/s0960-9822(95)00082-0. [DOI] [PubMed] [Google Scholar]
- 120.Hartmann AM, Nayler O, Schwaiger FW, Obermeier A, Stamm S. The interaction and colocalization of Sam68 with the splicing-associated factor YT521-B in nuclear dots is regulated by the Src family kinase p59(fyn) Molecular Biology of the Cell. 1999;10(11):3909–3926. doi: 10.1091/mbc.10.11.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gondran P, Dautry F. Regulation of mRNA splicing and transport by the tyrosine kinase activity of src. Oncogene. 1999;18(16):2547–2555. doi: 10.1038/sj.onc.1202598. [DOI] [PubMed] [Google Scholar]
- 122.Richard S, Yu D, Blumer KJ, et al. Association of p62, a multifunctional SH2- and SH3-domain-binding protein, with src family tyrosine kinases, Grb2, and phospholipase Cγ-1. Molecular and Cellular Biology. 1995;15(1):186–197. doi: 10.1128/mcb.15.1.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Finan PM, Hall A, Kellie S. Sam68 from an immortalised B-cell line associates with a subset of SH3 domains. FEBS Letters. 1996;389(2):141–144. doi: 10.1016/0014-5793(96)00552-2. [DOI] [PubMed] [Google Scholar]
- 124.Bunnell SC, Henry PA, Kolluri R, Kirchhausen T, Rickles RJ, Berg LJ. Identification of Itk/Tsk Src homology 3 domain ligands. Journal of Biological Chemistry. 1996;271(41):25646–25656. doi: 10.1074/jbc.271.41.25646. [DOI] [PubMed] [Google Scholar]
- 125.Fusaki N, Iwamatsu A, Iwashima M, Fujisawa J-I. Interaction between Sam68 and Src family tyrosine kinases, Fyn and Lck, in T cell receptor signaling. Journal of Biological Chemistry. 1997;272(10):6214–6219. doi: 10.1074/jbc.272.10.6214. [DOI] [PubMed] [Google Scholar]
- 126.Jabado N, Pallier A, Le Deist F, Bernard F, Fischer A, Hivroz C. CD4 ligands inhibit the formation of multifunctional transduction complexes involved in T cell activation. Journal of Immunology. 1997;158(1):94–103. [PubMed] [Google Scholar]
- 127.Jabado N, Jauliac S, Pallier A, Bernard F, Fischer A, Hivroz C. Sam68 association with p120GAP in CD4 T cells is dependent on CD4 molecule expression. Journal of Immunology. 1998;161(6):2798–2803. [PubMed] [Google Scholar]
- 128.Lawe DC, Hahn C, Wong AJ. The Nck SH2/SH3 adaptor protein is present in the nucleus and associates with the nuclear protein SAM68. Oncogene. 1997;14(2):223–231. doi: 10.1038/sj.onc.1200821. [DOI] [PubMed] [Google Scholar]
- 129.Trüb T, Frantz JD, Miyazaki M, Band H, Shoelson SE. The role of a lymphoid-restricted, Grb2-like SH3-SH2-SH3 protein in T cell receptor signaling. Journal of Biological Chemistry. 1997;272(2):894–902. doi: 10.1074/jbc.272.2.894. [DOI] [PubMed] [Google Scholar]
- 130.Guitard E, Barlat I, Maurier F, Schweighoffer F, Tocque B. Sam68 is a Ras-GAP-associated protein in mitosis. Biochemical and Biophysical Research Communications. 1998;245(2):562–566. doi: 10.1006/bbrc.1998.8374. [DOI] [PubMed] [Google Scholar]
- 131.Brown MT, Andrade J, Radhakrishna H, Donaldson JG, Cooper JA, Randazzo PA. ASAP1, a phospholipid-dependent Arf GTPase-activating protein that associates with and is phosphorylated by Src. Molecular and Cellular Biology. 1998;18(12):7038–7051. doi: 10.1128/mcb.18.12.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Randazzo PA, Andrade J, Miura K, et al. The Arf GTPase-activating protein ASAP1 regulates the actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(8):4011–4016. doi: 10.1073/pnas.070552297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Barnekow A, Jahn R, Schartl M. Synaptophysin: a substrate for the protein tyrosine kinase pp60(c-src) in intact synaptic vesicles. Oncogene. 1990;5(7):1019–1024. [PubMed] [Google Scholar]
- 134.Onofri F, Giovedì S, Vaccaro P, et al. Synapsin I interacts with c-Src and stimulates its tyrosine kinase activity. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(22):12168–12173. doi: 10.1073/pnas.94.22.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Foster-Barber A, Bishop JM. Src interacts with dynamin and synapsin in neuronal cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(8):4673–4677. doi: 10.1073/pnas.95.8.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jakymiw A, Raharjo E, Rattner JB, Eystathioy T, Chan EKL, Fujita DJ. Identification and characterization of a novel Golgi protein, golgin-67. Journal of Biological Chemistry. 2000;275(6):4137–4144. doi: 10.1074/jbc.275.6.4137. [DOI] [PubMed] [Google Scholar]
- 137.Abu-Amer Y, Ross FP, Schlesinger P, Tondravi MM, Teitelbaum SL. Substrate recognition by osteoclast precursors induces C- src/microtubule association. Journal of Cell Biology. 1997;137(1):247–254. doi: 10.1083/jcb.137.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19(49):5636–5642. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 139.Bolen JB, Veillette A, Schwartz AM. Activation of pp60(c-src) protein kinase activity in human colon carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(8):2251–2255. doi: 10.1073/pnas.84.8.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cartwright CA, Kamps MP, Meisler AI, Pipas JM, Eckhart W. pp60(c-src) activation in human colon carcinoma. Journal of Clinical Investigation. 1989;83(6):2025–2033. doi: 10.1172/JCI114113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ottenhoff-Kalff AE, Rijksen G, Van Beurden EACM, Hennipman A, Michels AA, Staal GEJ. Characterization of protein tyrosine kinases from human breast cancer: involvement of the c-src oncogene product. Cancer Research. 1992;52(17):4773–4778. [PubMed] [Google Scholar]
- 142.Byers LA, Sen B, Saigal B, et al. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clinical Cancer Research. 2009;15(22):6852–6861. doi: 10.1158/1078-0432.CCR-09-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sen B, Salgai B, Parikh N, Gallick G, Johnson FM. Sustained src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered jan us-Activated kinase-STAT3 binding. Cancer Research. 2009;69(5):1958–1965. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang XHF, Wang Q, Gerald W, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16(1):67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yim EK, Peng G, Dai H, et al. Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell. 2009;15(4):304–314. doi: 10.1016/j.ccr.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Carretero J, Shimamura T, Rikova K, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17(6):547–559. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sgroi DC. Breast cancer SRC activity: bad to the bone. Cancer Cell. 2009;16(1):1–2. doi: 10.1016/j.ccr.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 148.Johnson FM, Gallick GE. Src family nonreceptor tyrosine kinases as molecular targets for cancer therapy. Anti-Cancer Agents in Medicinal Chemistry. 2007;7(6):651–659. doi: 10.2174/187152007784111278. [DOI] [PubMed] [Google Scholar]
- 149.Gallick GE, Johnson FM. Src family kinase inhibitors in cancer therapy. In: Georgiev B, Markovski S, editors. Serpins and Protein Kinase Inhibitors: Novel Functions, Structural Features and Molecular Mechanisms. Hauppauge, NY, USA: Nova Science Publishers; 2010. [Google Scholar]
- 150.Nam JS, Ino Y, Sakamoto M, Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clinical Cancer Research. 2002;8(7):2430–2436. [PubMed] [Google Scholar]
- 151.Coluccia AML, Benati D, Dekhil H, De Filippo A, Lan C, Gambacorti-Passerini C. SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of β-catenin and its nuclear signaling. Cancer Research. 2006;66(4):2279–2286. doi: 10.1158/0008-5472.CAN-05-2057. [DOI] [PubMed] [Google Scholar]
- 152.González L, Agulló-Ortuño MT, García-Martínez JM, et al. Role of c-Src in human MCF7 breast cancer cell tumorigenesis. Journal of Biological Chemistry. 2006;281(30):20851–20864. doi: 10.1074/jbc.M601570200. [DOI] [PubMed] [Google Scholar]
- 153.Summy JM, Gallick GE. Treatment for advanced tumors: Src reclaims center stage. Clinical Cancer Research. 2006;12(5):1398–1401. doi: 10.1158/1078-0432.CCR-05-2692. [DOI] [PubMed] [Google Scholar]
- 154.Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clinical Cancer Research. 2007;13(24):7232–7236. doi: 10.1158/1078-0432.CCR-07-1902. [DOI] [PubMed] [Google Scholar]
- 155.Rothschild S, Gautschi O, Haura E, Johnson F. Src inhibitors in lung cancer: current status and future directions. Clinical Lung Cancer. 2010;11(4):238–242. doi: 10.3816/CLC.2010.n.030. [DOI] [PubMed] [Google Scholar]
- 156.Aleshin A, Finn RS. SRC: a century of science brought to the clinic. Neoplasia. 2010;12(8):599–607. doi: 10.1593/neo.10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mayer EL, Krop IE. Advances in targeting Src in the treatment of breast cancer and other solid malignancies. Clinical Cancer Research. 2010;16(14):3526–3532. doi: 10.1158/1078-0432.CCR-09-1834. [DOI] [PubMed] [Google Scholar]
- 158.Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clinical Cancer Research. 2005;11(19):6924–6932. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 159.Im HI, Joo WS, Nam E, Lee ES, Hwang YJ, Kim YS. Baicalein prevents 6-hydroxydopamine-induced dopaminergic dysfunction and lipid peroxidation in mice. Journal of Pharmacological Sciences. 2005;98(2):185–189. doi: 10.1254/jphs.sc0050014. [DOI] [PubMed] [Google Scholar]
- 160.Nam S, Kim D, Cheng JQ, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Research. 2005;65(20):9185–9189. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- 161.Serrels A, Macpherson IRJ, Evans TRJ, et al. Identification of potential biomarkers for measuring inhibition of Src kinase activity in colon cancer cells following treatment with dasatinib. Molecular Cancer Therapeutics. 2006;5(12):3014–3022. doi: 10.1158/1535-7163.MCT-06-0382. [DOI] [PubMed] [Google Scholar]
- 162.Finn RS, Dering J, Ginther C, et al. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“triple- negative” breast cancer cell lines growing in vitro. Breast Cancer Research and Treatment. 2007;105(3):319–326. doi: 10.1007/s10549-006-9463-x. [DOI] [PubMed] [Google Scholar]
- 163.Huang F, Reeves K, Han X, et al. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selection. Cancer Research. 2007;67(5):2226–2238. doi: 10.1158/0008-5472.CAN-06-3633. [DOI] [PubMed] [Google Scholar]
- 164.Kaklamani VG, Gradishar WJ. Gene expression in breast cancer. Current Treatment Options in Oncology. 2006;7(2):123–128. doi: 10.1007/s11864-006-0047-0. [DOI] [PubMed] [Google Scholar]
- 165.Nautiyal J, Majumder P, Patel BB, Lee FY, Majumdar APN. Src inhibitor dasatinib inhibits growth of breast cancer cells by modulating EGFR signaling. Cancer Letters. 2009;283(2):143–151. doi: 10.1016/j.canlet.2009.03.035. [DOI] [PubMed] [Google Scholar]
- 166.Jallal H, Valentino ML, Chen G, Boschelli F, Ali S, Rabbani SA. A Src/Abl kinase inhibitor, SKI-606, blocks breast cancer invasion, growth, and metastasis in vitro and in vivo. Cancer Research. 2007;67(4):1580–1588. doi: 10.1158/0008-5472.CAN-06-2027. [DOI] [PubMed] [Google Scholar]
- 167.Vultur A, Buettner R, Kowolik C, et al. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Molecular Cancer Therapeutics. 2008;7(5):1185–1194. doi: 10.1158/1535-7163.MCT-08-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Golas JM, Lucas J, Etienne C, et al. SKI-606, a Src/Abl inhibitor with in vivo activity in colon tumor xenograft models. Cancer Research. 2005;65(12):5358–5364. doi: 10.1158/0008-5472.CAN-04-2484. [DOI] [PubMed] [Google Scholar]
- 169.Hennequin LF, Allen J, Breed J, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy] -5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. Journal of Medicinal Chemistry. 2006;49(22):6465–6488. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 170.Purnell PR, Mack PC, Tepper CG, et al. The src inhibitor AZD0530 blocks invasion and may act as a radiosensitizer in lung cancer cells. Journal of Thoracic Oncology. 2009;4(4):448–454. doi: 10.1097/JTO.0b013e31819c78fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008;27(49):6365–6375. doi: 10.1038/onc.2008.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Johnson FM, Bekele BN, Feng L, et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. Journal of Clinical Oncology. 2010;28(30):4609–4615. doi: 10.1200/JCO.2010.30.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Haura EB, Tanvetyanon T, Chiappori A, et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. Journal of Clinical Oncology. 2010;28(8):1387–1394. doi: 10.1200/JCO.2009.25.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chiappori AA, Tanvetyanon T, Williams CA, et al. Phase I trial evaluating the epidermal growth factor receptor inhibitor erlotinib in combination with the SRC kinase inhibitor dasatinib for patients with recurrent non-small-cell lung cancer (NSCLC) Journal of Clinical Oncology. 2008;26(supplement 15):p. 635s. abstract 14605. [Google Scholar]
- 175.Leung ELH, Tam IYS, Tin VPC, et al. Src promotes survival and invasion of lung cancers with epidermal growth factor receptor abnormalities and is a potential candidate for molecular-targeted therapy. Molecular Cancer Research. 2009;7(6):923–932. doi: 10.1158/1541-7786.MCR-09-0003. [DOI] [PubMed] [Google Scholar]
- 176.Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Research. 2006;66(11):5542–5548. doi: 10.1158/0008-5472.CAN-05-4620. [DOI] [PubMed] [Google Scholar]
- 177.Chung BM, Dimri M, George M, et al. The role of cooperativity with Src in oncogenic transformation mediated by non-small cell lung cancer-associated EGF receptor mutants. Oncogene. 2009;28(16):1821–1832. doi: 10.1038/onc.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yu EY, Wilding G, Posadas E, et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clinical Cancer Research. 2009;15(23):7421–7428. doi: 10.1158/1078-0432.CCR-09-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zheng R, Yano S, Matsumori Y, et al. Src tyrosine kinase inhibitor, M475271, suppresses subcutaneous growth and production of lung metastasis via inhibition of proliferation, invasion, and vascularization of human lung adenocarcinoma cells. Clinical and Experimental Metastasis. 2005;22(3):195–204. doi: 10.1007/s10585-005-7768-5. [DOI] [PubMed] [Google Scholar]
- 180.Adjei AA, Cohen RB, Kurzrock R, et al. Results of a phase I trial of KX2-391, a novel non-ATP competitive substrate-pocket directed SRC inhibitor, in patients with advanced malignancies. Journal of Clinical Oncology. 2009;27(supplement 15):p. 148s. doi: 10.1007/s10637-013-9929-8. abstract 3511. [DOI] [PubMed] [Google Scholar]
- 181.Kopetz S, Lesslie DP, Dallas NA, et al. Synergistic activity of the Src family kinase inhibitor dasatinib and oxaliplatin in colon carcinoma cells is mediated by oxidative stress. Cancer Research. 2009;69(9):3842–3849. doi: 10.1158/0008-5472.CAN-08-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ceppi P, Papotti M, Monica V, et al. Effects of Src kinase inhibition induced by dasatinib in non-small cell lung cancer cell lines treated with cisplatin. Molecular Cancer Therapeutics. 2009;8(11):3066–3074. doi: 10.1158/1535-7163.MCT-09-0151. [DOI] [PubMed] [Google Scholar]