

Abstract
The in vitro and in vivo evidence compatible with a role for oxidative stress in OTA carcinogenicity has been collected and described. Several potential oxido-reduction mechanisms have been identified in the past. More recently, the possibility of a reduction of cellular antioxidant defense has been raised as an indirect source of oxidative stress. Consequences resulting from the production of oxidative stress are observed at different levels. First, OTA exposure has been associated with increased levels of oxidative DNA, lipid, and protein damage. Second, various biological processes known to be mobilized under oxidative stress were shown to be altered by OTA. These effects have been observed in both in vitro and in vivo test systems. In vivo, active doses were often within doses documented to induce renal tumors in rats. In conclusion, the evidence for the induction of an oxidative stress response resulting from OTA exposure can be considered strong. Because the contribution of the oxidative stress response in the development of cancers is well established, a role in OTA carcinogenicity is plausible. Altogether, the data reviewed above support the application of a threshold-based approach to establish safe level of dietary human exposure to OTA.
1. Introduction
Ochratoxin A (OTA) is a mycotoxin produced by several food-borne species of Aspergillus and Penicillium fungi. OTA occurs in various food materials and therefore humans are continuously exposed to relatively small amounts of it. Because of its wide occurrence and consequent exposure, together with a potent carcinogenic potential in animal models, OTA has attracted significant public health attention over the last few years. Several national and international food safety organizations and expert groups have conducted a thorough review of the situation as well as risk assessments in order to provide an insight on the health significance of OTA in food.
Human epidemiology has been inconclusive: a number of studies have suggested a correlation between exposure to OTA and Balkan Endemic Nephropathy (BEN) and mortality from urinary tract tumors [1, 2]. Epidemiological data were recently reviewed by several expert groups [3–6]. All concluded that causality between intake of OTA and human nephropathy could not be established. Therefore, the IARC statement that there is inadequate evidence for carcinogenicity in humans (group 2B) [7] appears still valid. Recently, other nephrotoxic agents have been put forward as the primary cause of BEN [6, 8, 9].
In absence of adequate human data, risk assessments have relied on animal data. Kidney has been considered as the key target organ of OTA toxicity. In all animal species studied, OTA was found to produce renal toxicity, while in rodents renal carcinogenicity was clearly established. Recently, OTA renal and hepatic carcinogenicity was also observed in chicks [10]. Using a LOAEL of 8 mcg/kg bw/day based on early markers of renal toxicity in pig (the most sensitive animal species) and applying an uncertainty factor of 450, EFSA [4] allocated a Tolerable Weekly Intake (TWI) of 120 ng/kg bw. Analysis of dietary exposure throughout Europe revealed that the current average OTA exposure (50–60 ng/kg bw/week) is well within the TWI [4]. The joint FAO/WHO Expert Committee on Food Additives (JECFA) first evaluated OTA at their 37th meeting [11]. Based on the LOAEL in pig, and applying an uncertainty factor of 500, JECFA allocated a Provisional Tolerable Weekly Intake (PTWI) of 112 ng/kg bw [11]. This value was rounded to 100 ng/kg bw/week and confirmed in several subsequent meetings [12, 13].
In 2008, JECFA applied a benchmark dose (BMD) modeling approach using carcinogenicity data [14]. The BMD is the dose estimated to cause a predefined increase (e.g., 10% for the BMD10) in tumor incidence over background. The BMDL is the lower limit of a one-sided 95% confidence interval of the BMD. The occurrence of combined adenomas and carcinomas in the kidneys of male rats was considered by JECFA to be the most appropriate data for modeling. Values of 18–33 mcg/kg bw/d and 15–25 mcg/kg bw/day were calculated for, respectively, the BMD10 and BMDL10. Because the BMD approach did not provide a lower point of departure than the LOAEL in pig, JECFA decided to reconfirm the PTWI of 100 ng/kg bw/day [14].
A recent health risk assessment, performed by Health Canada [15] recommends to regulate OTA as a nonthreshold carcinogen, because of the uncertainties regarding the mode of action. The authors defined a negligible cancer risk intake (NCRI, risk level 1 : 100 000) using the tumorigenic dose at which 5% of rats are likely to develop tumors (TD05, derived through modeling) as point of departure. Importantly, there is considerable convergence between the NCRI established in this assessment and the TDI derived by EFSA.
In the risk assessment of carcinogenic substances, consideration of the mode of action (MOA) is essential, determining the method to be applied in order to define levels of exposure below which a low safety concern is expected. The key events analysis framework of the MOA has not yet been formally applied to OTA. However, the approach used by most expert groups (EFSA, JECFA, ILSI) to establish the safe level of exposure of OTA (based on uncertainty factors) implies the consideration of key events compatible with a threshold effect. For these groups, amongst the mechanisms of action highlighted as possible, oxidative stress has been presented as one of the most probable [5, 14].
1.1. Scope of the Paper
Over the last decades, studies aimed at elucidating the modes of action implicated in OTA toxicity and carcinogenicity have been published [16]. There has been considerable debate for many years over the genotoxicity of OTA and its actual role in carcinogenicity [3–5, 11–15, 17, 18].
Although genotoxicity is likely to play a role in OTA carcinogenicity [3, 4], the actual molecular mechanism involved, either through covalent adduct formation, through other indirect modifications, or both is still unclear. The potential of OTA to form covalent DNA adducts has been subjected to debate due to conflicting data in the literature. Using 32P-postlabelling analysis, large numbers of putative OTA-derived DNA adducts have been reported to be present in a wide range of tissues from OTA-treated rats, mice as well as pigs [2, 17, 19–22]. However, so far, these adducts have never been observed by any other highly specific techniques such as radioactivity measurements using 3H-labelled OTA (3H-OTA) [23], accelerator mass spectrometry (AMS) [24], or liquid chromatography-tandem mass spectrometry (LC-MS/MS) [25].
The present paper is not intended to provide a thorough review of the complex and controversial scientific literature on DNA-adduct formation. However, it is important to keep in mind that DNA adducts are increasingly considered as markers of exposures and not only of effects [26, 27] and that DNA-covalent binding does not necessarily determine the shape of the dose-response at low level of exposure [28, 29]. According to Mantle and coworkers [21, 30], experimental dose-response data for OTA's renal carcinogenesis makes a compelling case for OTA being a thresholded carcinogen in male rat. In this context, it appears important to also consider other potential modes of action, which could potentially contribute to OTA carcinogenicity. In the near future, the application of the mode of action framework [28] will likely help to understand the individual contribution of all mechanisms described up to now for OTA.
The focus of the present short paper was to collect and highlight the evidence associated with a role for oxidative stress as a plausible mechanism to consider for OTA. A list of the studies used to illustrate the main messages of the present paper is provided in Table 1. Although not exhaustive, the list shows that over the last two decades, numerous investigators have documented the generation of oxidative stress as a result of OTA treatment in both in vitro and in vivo model systems.
Table 1.
OTA oxidative stress-related studies.
| Model | Gender | Via | Time treatment | Dose | Aim | Results/Conclusion | Ref. |
|---|---|---|---|---|---|---|---|
| BALB/c macrophage J774a cell line | 24, 48, 72 h | 30 nM–100 μM | OTA immunotoxicity and modulation inflammatory process | Induction of iNOs, COX-2 and NF-κb expression by OTA. OTA is an immunotoxic compound | [51] | ||
| Porcine kidney tubuli cells LL-PK1 | 6–24 h | 1–100 μmol/L | Characterization effect of OTA on Nrf2 response | Nrf2 potential signal transduction pathway by which OTA impairs its own detoxification | [45] | ||
| Porcine kidney tubuli cells LL-PK1 | 24 h | 1–100 μmol/L | Impact of OTA on Nrf2, AP-1 activity, antioxidant enzymes and GST | Enhanced production of ROS, GST impairment. Nrf2 and AP-1 disruption by OTA. Impairment of the detoxification machinery | [44] | ||
| Rat Sprague-Dawley | male | diet | 15 days | 3.0 mg/kg bw | Oxidative stress protection study | OTA-induced oxidative stress chemoprotection by Inula crithmoides | [102] |
| Rat F344 | male | gavage | 7 and 21 days | 0.5 mg/kg bw | Mechanism of action study-microarrays | Oxidative stress, calcium homeostasis, cytoskeleton structure | [61] |
| Human hepatocytes HepG2; Monkey kidney Vero cells | 0–100 μM | Decrease GSH | No induction of heat shock protein (HSP) | [103] | |||
| Rat Wistar | male | diet | 15 days | 5 ng/g bw 50 ng/kg bw | Oxidative damage study (proteins and lipids) | Malondialdehyde (MDA) and protein carbonylation (PC) increase in kidney > liver | [76] |
| Chinese Hamster lung V79 cells; Lymphoma mouse LY5178 cells | 0–438 μM | OTA mutagenicity | OTA is mutagenic at cytotoxic doses in mammalian cells via oxidative DNA damage induction. | [104] | |||
| Rats Sprague-Dawley | male | diet | 4 weeks | 200 ppb | Oxidative stress protection study | OTA-induced oxidative stress and DNA damage chemoprotection by cyanidin 3-O-β-D-glucoside | [105] |
| Pig kidney cell line LLC-PK1 | 24 h | 0, 10, 15, 20 μM | Oxidative stress protection study | OTA-induced ROS. Scavenging by cathechins (epigallocathechin gallate (EGCG) and epicatechin gallate (ECG)) | [106] | ||
| Human epithelial colorectal adenocarcinoma Caco-2 cells | 100 μM | Effect of OTA on barrier function impairment | Loss of microdomains associated with tight junctions maybe due to oxidative events | [107] | |||
| Neural stem/progenitor cells (NSCs) | 0.01–100 μg/mL | Vulnerability of brains mouse cells to OTA | Robust increased in total and mitochondrial SOD activity. OTA impaired hippocampus neurogenesis | [108] | |||
| Rats | Male/liver and kidney | Diet (drinking water) | 4 weeks | 289 μg/kg | Oxidative stress protection study | Melatonin protection against OTA-induced oxidative damage in liver and kidney. CoQ protective in liver. | [79] |
| Human renal cell line HK-2 | 6 and 24 h | 50 μM | Mechanism of action study-microarrays | Significant increase in ROS level and oxidative DNA damage. | [61] | ||
| Human renal proximal tubular epithelial cell line HK-2 | 50–800 μM | Evaluate single-strand DNA breaks and oxidative damage induction by OTA | Oxidative stress precedes cytotoxicity and genotoxicity | [57] | |||
| Male Fischer 344; Primary hepatocytes; adherent proximal tubules epithelial NRK cells; rat liver RL-34 | Rats 2 years; in vitro culture 24–48 h; | 300 μg–100/kg bw; 1.5–6.0 μM | Demonstration of cellular defense reduction by OTA | OTA induces depletion of antioxidant defense by inhibition of Nrf2 responsible of oxidative stress response | [46] | ||
| Eker and wild type rats | male | gavage | 1–14 days | 210 μg/kg bw | Early carcinogen-specific gene expression study | Oxidative DNA damage response genes, general stress response, and cell proliferation | [109] |
| Wistar rats | gavage | 90 days | 289 μg/kg bw | Early effects of chronic OTA administration in liver | Reduction in the ability to counteract oxidative stress in liver | [63] | |
| Swiss ICR | male | i. p | 6, 24, 72 hours | 0–6 mg/kg bw | Oxidative stress and OTA neurotoxicity | Acute depletion of striate DA on a background of globally increased oxidative stress and transient inhibition of oxidative DNA repair | [110] |
| Swiss mice | male | I.p; infusion | 2 weeks | Acute 3.5 mg/kg; chronic 4, 8, 16 mg/kg | Effect of chronic low dose OTA exposure on regional brain oxidative stress and stratial DA metabolism | Low doses exposure caused global oxidative stress | [111] |
| F344 rats | male | diet | 7 and 21 d; 4, 7 and 12 months | 300 mg/kg bw | OTA mechanism of action-microarrays study in liver and kidney | Oxidative stress and metabolic response modulated involving mainly Nrf2 and HNF4α pathway disruption | [47] |
| Swiss mice | male | oral | 24 hours | 10 mg/kg | Immune cells response after acute OTA exposure | OTA-induced oxidative stress response responsible of its own toxicity. | [112] |
| Wistar rats | female | Intraperitoneally | 7, 14 and 21 days | 0.5 mg/kg bw/day | Genotoxic potential of OTA measuring DNA strand breaks (comet assay) in the kidney | OTA-induced DNA strand breaks were detected, OTA concentration in the kidney and duration of the treatment correlated with severity of the DNA damage | [62] |
| Wistar rats | male | Oral | 15 days | 5 ng; 0.05 mg; 0.5 mg/bw | Effect of OTA on DNA damage | Oxidative stress responsible for OTA-DNA damage as shown by Fpg-modified comet assay | [113] |
| Pig kidney microsomes, human bronchial epithelial cells, human kidney cells | Cells: 2, 7, 24 hours | 0.5, 1.0, 2.5 μM | Genotoxicity of the hydroquinone (OTHQ) metabolite of OTA | OTQ-mediated adduct spots form in a dose-and-time-dependent manner | [114] | ||
| Wistar rats | female | oral | 7, 14 and 21 days | 0.5 mg/kg bw | Effect of OTA on protein oxidation in kidney and liver | Increased protein carbonyls in the kidney and liver | [68] |
| F344 rats | male | gavage | 0.03, 0.10, 0.30 mg/kg bw | Evaluate relevance of OTA-induced oxidative damage on nephrotoxicity and carcinogenicity | Tumours in rat kidney may be attributable to oxidative DNA damage in combination with cell-specific cytotoxic and proliferation-stimulating effects as cell-signaling response | [69] | |
| V79 (Chinese hamster lung fibroblasts) cells, CV-1 (African green monkey, kidney) cells, primary rat kidney cells | 1–24 hours | 2.5, 100 μmol/L OTA | Relevance of OTA-induced oxidative damage in nephrotoxicity and carcinogenicity | Cytotoxicity and oxidative DNA damage already at low doses could be a relevant factor for the nephrocarcinogenicity | [58] | ||
| Rat lymphoid cells | 1 hour | 0.5, 2, 20 μM | OTA immune function modification | Protein synthesis inhibition, oxidative metabolism of OTA, prostaglandin synthesis implicated in NK cells toxicity | [115] | ||
| Human hepatoma—derived cell lines (HepG2), human colonic adenocarcinoma cell line (Caco-2) | 24, 48, 72 hours | 0–100 μM | Oxidative stress protection study | OTA-induced oxidative stress damage. Protective effect by Cyanidin-3-O-β glucopyranoside (C-3-G) | [116] | ||
| F344 Fischer rats | male | gavage | 2 weeks | 0–2000 μg/kg bw | Genotoxicity of OTA | DNA strand breaks in target and nontarget tissues probably involving oxidative stress mechanism | [60] |
| Human hepatoma—derived cell line (HepG2) | 48–72 hours | 35–10 mM | Oxidative damage protection study | No cytotoxicity protection observed with Vitamine E, polyphenols | [117] | ||
| Sprague-Dawley | male | diet | 15 days | 3 mg/kg | Oxidative stress protection in vivo study | Preventive effect against OTA-induced oxidative stress and lipid peroxidation by melatonin | [75] |
| Human fibroblast cells | 48–72 hours | 0–50 μM | Oxidative stress protection study | Reduction of free radical species production and DNA damage prevention by cyanin 3-O-β-D-glucoside (C3G) | [118] | ||
| Fetal rat telencephalon aggregating cells | 24–48 hours, 9 days | 0–20 nM | Adverse effect of OTA in brain | Brain inflammatory response induction of HO-1, iNOs, PPARγ, cytoskeletal damage | [50] | ||
| Human hepatoma-derived cell line (HepG2) | 24 hours | 0–40 μg/mL | Genotoxicity of OTA | Dose-dependent induction of DNA single strand breaks (comet assay) and micronuclei (MN) | [119] | ||
| Primary proximal tubules renal (PT) cells, proximal tubular cell line (LLC-PK1) | 0–24 hours | 0–100 μM | OTA mediated oxidative stress response in proximal tubular cells, oxidative stress protection | Oxidative stress contributes to tubular toxicity. Antioxidants (α-tocopherol, N-acetyl-Lcysteine (NAC) treatment prevents OTA toxicity | [59] | ||
| Wistar rats | male | gavage | 10, 30, 60 days | 120 μg/kg bw | Kidney low dose OTA response: sequence of events leading to cell death | Low dose induces oxidative stress, apoptosis in proximal, and distal tubule kidney cells | [78] |
| Human hepatoma—derived cell line (HepG2) | 1 hour, 24 hours | 0–50 μg/mL | Genotoxicity of OTA | No inductions of mutations in the Ames assay, a dose-dependent induction of micronuclei in the MN assay, and DNA migration (comet assay) were detected | [120] | ||
| Proximal tubular cells (PTC), Wistar rats | male | gavage | 24–72 hours (in vivo and in vitro) | 5.0 μM, 12.5 μM in vitro; 1 and 10 mg/kg bw | In vivo and in vitro gene expression comparative study | In vitro and in vivo gene expression data were comparable. Response to oxidative stress-related genes hypoxia-inducible factor 1 and catalase was observed | [121] |
| Dark Agouti (DA), Lewis rats | male | Intragastric intubation | 0.4 mg/kg bw | Life-time | Life-time study to evaluate if MESNA leads to a more effective reduction of OTA-induced tumour development or urinary tract damage | Lack of effect of mesna on OTA-induced urinary tract damage or renal tumor development | [122] |
| Dark Agouti (DA), Lewis rats | male | Intragastric intubation | 0.4 mg/kg bw | 2 years | Life-time study to evaluate the potential protective effect of 2 mercaptoethane sulfonate (MESNA) and N-acetyl cysteine (NAC ) | MESNA decreased karyomegalies in kidney, but had no beneficial effect on renal tumour incidence | [123] |
| Fischer rats | male | gavage | 4, 8, 24, 48 hours | 0–2.0 mg/kg bw | Chemical and biological markers induced by OTA exposure associated with oxidative stress | Oxidative stress may contribute to mechanism of OTA renal toxicity and carcinogenicity in rats over long term exposure | [77] |
| Bronchial epithelial cells incubated with microsomes of seminal vesicles of pig | 4 hours | 10 μM | Roles of cyclooxygenase and lipoxygenase in ochratoxin A genotoxicity in human epithelial lung cells | OTA is biotransformed into genotoxic metabolite via a lipoxygenase, whereas prostaglandin—H-synthetase (PGHS) decreases OTA genotoxicity | [124] | ||
| Sprague-Dawley liver microsomes, liver mitochondria and hepatocytes cells | female | 2.5 mM | Free radical generation by OTA in hepatocytes, mitochondria, and microsomes using electron paramagnetic resonance (EPR) | Oxidative damage may be one of the manifestations of cellular damage in the toxicity of OTA | [125] | ||
| Bacillus brevis | 10 min | 1 mg/mL | Study free radical generation in bacteria as model system | OTA induces free radical production, enhancing permeability of the cellular membrane to Ca2+ | [37] | ||
| Swiss mice | Male | Gastric intubation | 48 hours | 2 mg/kg bw | Effects of vitamins on genotoxicity of OTA | Vitamins E, A, and C also reduced OTA-DNA adduct formation in mice kidney | [126] |
| Wistar rat | male | Gastric intubation | Every 48 hours/3 weeks | 289 μg/kg body weight | Protective effect of superoxide dismutase (SOD) and catalase | SOD + catalase prevents the nephrotoxicity induced by OTA in rats | [127] |
| Wistar rat liver microsomes, kidney microsomes | male | 6 mg/kg bw | Lipid peroxidation induction by OTA | lipid peroxidation may play a role in the observed toxicity of ochratoxin A | [73] |
2. Sources of OTA-Mediated Oxygen-Species Generation
Production of reactive oxygen species (ROS) leading to oxidative stress and macromolecular damage is known to contribute to the pathogenesis of age-related as well as chronic diseases including cancer [31–35]. A number of studies are available documenting that OTA is associated with the production of reactive oxygen species and resulting oxidative stress through various direct and indirect mechanisms.
2.1. Oxido-Reduction Mechanisms
Several oxido-reduction mechanisms elicited by OTA have been proposed. In a reconstituted system consisting of phospholipid vesicles, the flavoprotein NADPH-cytochrome P450 reductase and Fe3+, OTA was found to chelate ferric ions (Fe3+), facilitating their reduction to ferrous ions (Fe2+), which in the presence of oxygen, provided the active species initiating lipid peroxidation [36]. Results indicated that the hydroxyl radical was not involved in the process. A role for cytochrome P450 in this reaction was also suggested [36]. In contrast, others found that OTA induced oxidative damage through the generation of hydroxyl radicals. This reaction conducted with microsomes, in presence of NADPH and O2 did not require exogenous Fe [20]. Structure-activity studies have also suggested that the toxicity of OTA may be attributable to its isocoumarin moiety and that the lactone carbonyl group may be involved in its toxicity. Using a Bacillus brevis model, Hoehler's et al. showed that OTA behaved as a cell pro-oxidant through mobilization of Fe2+ and Ca2+ pathway leading to uncoupling oxidative phosphorylation and increased production of hydroxyl radical via the Fenton reaction [37]. However, in other studies using OTA and structural analogs, a direct correlation between toxicity and iron chelating capacity was only partially supported [38].
The generation of an OTA hydroquinone/quinone couple from the oxidation of OTA (phenol oxidation) by electrochemical, photochemical, and chemical processes was reported [39, 40]. The quinone is thought to undergo reductions to form hydroquinone, postulated to be responsible for the formation of the glutathione conjugate of OTA. Such events are likely to result in redox cycling and in the generation of reactive oxygen species [20, 40, 41]. The formation of OTA-derived quinones has been observed in cell cultures in vitro [41] as well as in vivo [42, 43].
2.2. Reduction of Antioxidant Defenses
OTA was found to reduce the expression of several genes regulated by nuclear factor-erythroid 2 p45-relatetd factor (Nrf2) [44–47]. This was observed at the RNA and protein levels, both in vitro and in vivo test systems. Nrf2 is involved in both the basal expression as well as in the induction of genes encoding detoxification, cytoprotective, and antioxidant enzymes [48, 49]. A reduction of the expression of these genes is likely to result in decrease in antioxidant defenses leading to oxidative stress and macromolecular damage. This was confirmed with OTA. Through in vitro and in vivo studies, a correlation was observed between the OTA-dependent reduction of the Nrf2 pathway and an increased production of oxidative damage [46]. In this context, it is interesting to note that OTA was found to increase the expression of inducible nitric oxide synthase (iNOs) [50–52], an enzyme responsible for the production of nitric oxide (NO). An association between iNOs expression and the development of cancers was suggested in humans and animals in vivo [53]. In excess, NO may behave as a toxic radical producing nitrosative stress. NO is known to react with oxygen anion radical superoxide to form the pro-oxidant peroxynitrite. Under physiological conditions, peroxynitrite rapidly decomposes to generate a nitro radical intermediate leading to protein and DNA nitration. OTA was shown to stimulate protein and possibly DNA nitration [52, 53] indicating that OTA exposure may be considered as a source of both oxygen and also reactive nitrogen radicals/species (RNS).
3. OTA-Mediated Oxidative Damage
3.1. DNA-Damage
ROS, such as hydroxyl radicals and nitric oxide, are capable of forming oxidized DNA bases that directly produce diverse types of DNA damage [54–56]. The oxidized DNA bases appear to be capable of inducing mutations that are commonly observed in neoplasia [33]. As illustrated in Table 1, diverse biomarkers have been analyzed showing that OTA induces DNA damage. In cell cultures, the OTA-dependent production of ROS was correlated with an increased formation of 8-oxoguanine [57–59]. Moreover, OTA was shown to induce DNA strand breaks as assessed by comet assay in liver, kidney, and spleen of F344 rats given 0, 0.25, 0.50, 1.0, and 2.0 mg/kg bw/day by gavage for 2 weeks [60]. In liver and kidney, the extent of DNA damage analyzed by comet assay was further enhanced in the presence of formamidopyrimidine glycosylase (Fpg), an enzyme involved in excision repair of oxidized DNA bases [57, 58, 61]. Another study [62] detected DNA-strand breaks (using the comet assay) in the kidney of female Wistar rats treated intraperitoneally with OTA (0.5 mg/kg bw/day for 7, 14, and 21 days; n = 5 per group). The severity of the DNA lesions in the kidney increased according to the OTA dose and was at maximum after 21 days of treatment. Other authors have observed oxidative DNA-damage in various tissues of animals treated with a wide range of OTA doses and treatment durations [46, 60, 63, 64].
3.2. Protein Damage
Carbonylation of proteins occurs through a variety of oxidative pathways [54, 65–67]. Carbonylation is an important protein modification associated with alterations of protein (enzymes) function, protein misfolding, protein fate, and proteolysis. An increase in protein carbonyl content of tissues has been associated with a number of pathological disorders. Due to their abundance in mammalian cells, cytoskeletal proteins like actin are common targets for a variety of ROS and low-molecular weight reactive carbonyl species [67]. Inconsistent data have been reported on the potential impact of OTA on protein carbonylation [52, 59, 63, 68, 69]. No increase in protein oxidation was observed in liver and kidney of F344 rats treated with OTA at 0.3 mg/kg bw per day for 4 weeks [69]. The same finding was reported in liver of Wistar rats treated with OTA at 289 μg/kg bw for 90 days [63]. In contrast, in another in vivo study measuring protein oxidation in Wistar rats treated with OTA (0.5 mg/kg bw/day) for 24 h, 7, 14 and 21 days, a significant increase in protein carbonyls was found after 14 and 21 days of treatment in, respectively, the kidney and the liver [68]. Oxidative protein modification was observed in vitro [52].
3.3. Lipid Damage
Lipid peroxidation is among the most extensively investigated processes induced by free radicals. Of these, the by-products, 4-hydroxy-2-nonenal (HNE), the tautomer malondialdehyde (MDA), acrolein and crotonaldehyde have been widely studied. The ability of these reactive electrophiles to modify DNA bases, yielding promutagenic lesions, is considered to contribute to the mutagenic and carcinogenic effects associated with oxidative stress-induced lipid peroxidation. HNE and MDA have increasingly been implicated in carcinogenesis [33, 70–72]. OTA has been reported to increase MDA formation. Initially, Rahimtula's group [73] observed that OTA was able to stimulate lipid peroxidation when added to liver or kidney microsomes or when administered to rats in vivo. Stimulation of lipid peroxidation by OTA-iron complex facilitating the reduction of iron was further reported [36]. Moreover, additional studies [74] indicated that OTA induced lipid peroxidation accompanied by leakage of calcium from calcium-loaded microsomes. Increased formation of MDA was observed in animal models treated orally with different doses of OTA [75–79]. HNE-protein adducts were measured in cell cultures treated with OTA [52].
4. OTA-Mediated Biological Response
It is widely acknowledged that reactive oxygen and nitrosative species can trigger biological responses such as stimulation or inhibition of signal transduction and gene expression. Such biological responses are considered to contribute to the expression of the carcinogenic potential of the reactive chemicals. A number of in vitro and in vivo studies are consistent with a role of oxidative and probably nitrosative stress as messengers involved in the adverse biological effects of OTA (Table 1).
5. Cell Signaling
ROS induces release of calcium from intracellular stores, resulting in the activation of kinases, such as protein kinase C (PKC). ROS species play also a critical role in the selective mobilization of other cell signaling responses. Cell signaling phosphoproteins of mitogen-activated protein (MAP) kinases including ERK, c-Jun N-terminal kinases (JNK), and p38 kinases are involved in proliferation, differentiation, and apoptosis. Activation of these molecules has been observed in response to changes in the cellular redox balance and are considered as vectors of ROS biological effects [33]. In vitro experiments provided evidence of an effect of OTA on intracellular calcium (Ca2+) homeostasis [74, 80–82]. Ca2+ homeostasis modulation by OTA was also observed in vivo [83]. An increased rate of microsomal Ca2+ uptake was observed after OTA administration in vivo and in vitro [84]. Gene profiling analysis suggested a modulation of genes involved in calcium homeostasis by OTA [47, 85]. These data indicate that Ca2+ dependent signal transduction pathways may be affected by OTA treatment. Interferences were observed with other cell signaling pathways. OTA was shown to stimulate phosphorylation of ERK1/2, SAP/JNK, and p38 using in vitro models [82, 86, 87]. Using an in vivo model ERK1/2 a specific signaling response was also observed with mobilization of the atypical protein kinase C (PKCζ) and the insulin-like growth factor-1 (IGF-1) system [88]. Gene profiling data supported a potential role of the (IGF)-PI3K-PKB pathway in OTA-mediated renal toxicity in male rats [89].
5.1. Redox Modulation of Transcription Factors
Numerous reports have characterized interactions of ROS and RNS with activity of transcription factors [90–92]. Transcription factors contain a conserved redox sensitive cysteine residue; the oxidation of this residue inactivates the DNA-binding domain of the factor. Several studies have observed an inactivation of transcription factors resulting from increased concentrations of ROS [93–95]. For example, it was shown that the complex AP-1 is a basic leucine zipper protein, which is highly sensitive to changes in redox environment due to a cysteine residue in the DNA-binding domain [96]. Gene expression profile studies performed using in vivo models have shown that OTA impairs the antioxidant defense system regulated by Nrf2 [47]. This effect was supported by further studies [44–46]. Using in vitro models together with electrophoretic mobility shift assay, an inhibition of Nrf2 and AP-1 activity was shown as a result of OTA treatment [46]. Interestingly, Nrf2 binding site represents a bZip domain that interacts with the Antioxidant Response Element (ARE) DNA-binding site triggering the transcription of Nrf2 regulated genes. The ARE motif (5′-A/GTGAC/TnnnGCA/G-3′) shares structural similarity with AP-1 binding site (5′-TGACTCA) involving both cysteine rich residues, the target of ROS oxidation [97, 98]. These data strongly suggest a correlation between the generation of ROS by OTA and the subsequent inactivation of Nrf2 as previously described for AP-1. This molecular mechanism appears as a likely molecular component explaining the reduction of the defense response observed under OTA treatment.
In vivo experiments provided direct evidence that S-nitrosylation can interfere with transcription [91, 92]. Nitric oxide (NO) induces the nitrosylation of cysteine residues (thiol groups) within or near the DNA-binding domain and/or insertion of the zinc finger, which is a DNA-binding motif, resulting in the inhibition of the DNA-binding activity of transcription factors [99–101]. This is illustrated by the example of the suppression of P450 gene expression by NO. NO-donors were found to suppress CYP2D6 promoter activity through inhibition of the transcription factor called hepatocyte nuclear factor 4 (HNF4) [94, 95]. Interestingly, in a gene expression study performed by our group, OTA was associated with a significant reduction of the expression of genes regulated by HNF4 suggesting an indirect evidence of the role oxidative stress and the transcription factors regulation by OTA [47].
5.2. Alterations of Gap-Junction Intercellular Communication (GJIC)
A strong correlation between the ability of a compound to block cell-to-cell communication in cultured cells and its ability to induce rodent tumors through nongenotoxic mechanisms has been documented [33, 128–131]. Disruption of gap junction intercellular communication was specifically reported in human renal cancer cell lines [132]. For example, Connexin 32 (Cx32) was discovered to be generally downregulated in human renal cell carcinoma (RCC) cell lines and in cancerous regions of the kidney [133]. ROS such as H2O2, an established tumor promoter, is known to modulate cell-to-cell communication. Likewise, certain chemicals inducing ROS were shown to inhibit intercellular communication in a variety of cells in culture systems [33]. Data on the potential effects of OTA on GJIC are inconsistent. Kidney epithelial cells treated with OTA resulted in modulation of gap junction-mediated intercellular communication, through a reduced expression of the gap-junction protein CX43 [134]. In addition, OTA strongly reduced the expression of other gap-junction proteins, CX26, CX32, and CX43, in liver of rats treated with OTA [63]. In another study, even though a strong reduction of CX43 was found in renal cells in vitro, OTA inhibited GJIC only in liver but not in kidney [135].
6. Prevention of OTA-Induced Oxidative Stress
6.1. Counteracting OTA-Mediated Reduction of Nrf2 Activity
As mentioned earlier, OTA was found to reduce the expression of antioxidant enzymes through inhibition of Nrf2 activity [46]. This reduction in antioxidant gene expression was correlated with increased oxidative damage of protein and DNA [46]. To further confirm the actual role of the reduction of defense mechanisms in the induction of cellular oxidative damage, inducers of Nrf2 were applied in cell cultures in vitro. All reported OTA effects at the levels of Nrf2 activity, Nrf2-regulated gene expression, and DNA damage were prevented in cell pretreated with Nrf2 inducers [46], strongly indicating a causal relationship between Nrf2 effects and oxidative damage.
6.2. Application of Radical Scavengers
Several studies have been performed to try to counteract the adverse effects of oxygen radicals generated under OTA-treatment. A number of molecules with various antioxidant properties were tested, including inula crithmoides, cyanidin 3-O-b-D-glucoside, catechins, melatonin, superoxide dismutase, catalase, and N-acetyl-L-cysteine (NAC), using in vivo or in vitro models. Protection against OTA-induced DNA damage, lipid peroxidation as well as cytotoxicity was observed [75, 79, 102, 105, 106, 116, 118, 127] further confirming the link between OTA exposure and oxidative macromolecular damage. However, up to now, the application of chemicals known to possess antioxidant properties failed to prevent the development of OTA-induced tumors in animal models. Authors reported protection against nephrotoxicity but not carcinogenicity induced by ochratoxin A, implicating two separate pathways [122, 123].
6.3. Application of Peroxidase Inhibitors
Indomethacin and aspirin were found to prevent OTA genotoxicity in the urinary bladder and kidney of mice [136]. These data suggested the possible co-oxidation of OTA by enzymes involved in arachidonic acid biotransformation, especially prostaglandin-H-synthase (PGHS) and/or lipoxygenase [124]. Such reactions thought to produce activated OTA metabolites are also known to generate ROS which then may induce oxidation [124].
7. Conclusion
The carcinogenic mycotoxin OTA has been reviewed by a number of expert groups [3–5, 14, 137]. These expert groups identified the production of oxidative stress as an important event in the mode of action of OTA-induced nephrocarcinogenicity. In the present paper, the actual evidence available supporting such hypothesis was collected and reviewed.
It has been clearly shown that OTA generates oxidative stress predominantly in kidney through potentially direct (redox cycling) and indirect (reduction of cellular antioxidant defenses) mechanisms. Interestingly, these two mechanisms may interact with each other. The reduction of defense may amplify the impact of the direct production of radicals. Consequences resulting from the production of oxidative stress were observed at different levels. High kidney susceptibility to oxidative stress conditions may explain the target-specific toxicity of OTA. Oxidative stress has been incriminated in a number of kidney pathological pathways [138–140].
As depicted in Figure 1, first, OTA exposure was associated with increased levels of oxidative DNA, lipid, and protein damage. Second, various biological pathways known to be mobilized under oxidative stress were shown to be altered by OTA. Importantly, these effects were observed in both in vitro and in vivo test systems. Active in vivo doses were within doses known to induce tumors in kidney. These mechanisms are likely to be relevant for humans.
Figure 1.
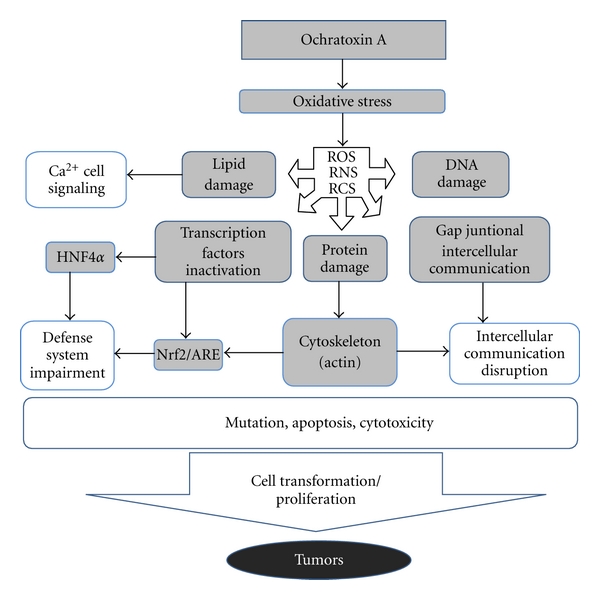
Scheme to illustrate the oxidative stress-mediated mode of action proposed for OTA. Increased production of ROS, RNS, and RCS is likely to originate either from direct redox reactions involving OTA or through the inhibition of cellular defenses such as through the inhibition of transcription factors as Nrf2 which regulates enzymes with antioxidant properties. The generation of radicals will induce macromolecular oxidative damage such as oxidized DNA bases which may be converted into mutation resulting into generation of transformed cells. In addition, radicals will trigger biological responses which may impair intercellular communication and induce cell proliferation as well as reduction in cellular defense in oxidative stress. This last effect is likely to amplify the oxidative stress-mediated effects of OTA. Altogether, these molecular mechanisms will result in cancer development.
In conclusion, the evidence for the induction of an oxidative stress response resulting from OTA exposure can be considered strong. Because the contribution of the oxidative stress response in the development of cancers is well established, a role in OTA carcinogenicity is plausible. Altogether, the data reviewed above support the application of a threshold-based approach to establish safe level of dietary human exposure to OTA.
References
- 1.Tatu CA, Drugarin D, Paunescu V, Stanescu DI, Schneider F. Balkan endemic nephropathy, the haematopoietic system and the environmental connection. Food and Chemical Toxicology. 1998;36(3):245–247. doi: 10.1016/s0278-6915(97)00137-3. [DOI] [PubMed] [Google Scholar]
- 2.Pfohl-Leszkowicz A, Manderville RA. Ochratoxin A: an overview on toxicity and carcinogenicity in animals and humans. Molecular Nutrition and Food Research. 2007;51(1):61–99. doi: 10.1002/mnfr.200600137. [DOI] [PubMed] [Google Scholar]
- 3.Fink-Gremmels J. Conclusions from the workshops on Ochratoxin A in Food: recent developments and significance, organized by ILSI Europe in Baden (Austria), 29 June-1 July 2005. Food Additives and Contaminants. 2005;22(supplement 1):1–5. [Google Scholar]
- 4.EFSA. Opinion of the Scientific Panel on Contaminants in the food chain on a request from the commission related to Ochratoxin A in Food. 365, 1-56, 2006.
- 5.JECFA. Ochratoxin A. WHO Technical Report Series 947. IPCS, WHO, Geneva, Switzerland, pp. 169–180. 2007.
- 6. Symposium on Recent Advances in Endemic Nephropathy: the role of toxins in an environmental disease, Croatia, 2006. [PubMed]
- 7.IARC. Some Naturally Occuring Substances: Food Items and Constituents, Hetrocyclic Aromatic Amines and Mycotoxins. Lyon, France: IARC; 1993. (IARC Monographs on the Evaluation of Carcinogenic Risk to Humans). [Google Scholar]
- 8.Grollman AP, Jelaković B. Role of environmental toxins in endemic (Balkan) nephropathy. Journal of the American Society of Nephrology. 2007;18(11):2817–2823. doi: 10.1681/ASN.2007050537. [DOI] [PubMed] [Google Scholar]
- 9.Grollman AP, Shibutani S, Moriya M, et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(29):12129–12134. doi: 10.1073/pnas.0701248104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoev SD. Studies on some feed additives and materials giving partial protection against the suppressive effect of ochratoxin A on egg production of laying hens. Research in Veterinary Science. 2010;88:486–491. doi: 10.1016/j.rvsc.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 11.JECFA. WHO Technical Report Series. 806. Geneva, Switzerland: WHO; 1991. Evaluation of certain food additives and contaminants: ochratoxin A. [PubMed] [Google Scholar]
- 12.JECFA. WHO Technical Report Series. 859. Geneva, Switzerland: WHO; 1995. Evaluation of certain food additives and contaminants: ochratoxin A. [PubMed] [Google Scholar]
- 13.JECFA. WHO Technical Report Series. 906. Geneva, Switzerland: WHO; 2002. Evaluation of certain mycotoxins in food: ochratoxin A. [PubMed] [Google Scholar]
- 14.JECFA. Ochratoxin A (addendum) WHO Food Additive Series 59: Safety Evaluations of certain food additives and contaminants. IPCS, WHO, Geneva, Switzerland, 2008.
- 15.Kuiper-Goodman T, Hilts C, Billiard SM, Kiparissis Y, Richard IDK, Hayward S. Health risk assessment of ochratoxin a for all age-sex strata in a market economy. Food Additives and Contaminants A. 2010;27(2):212–240. doi: 10.1080/02652030903013278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schilter B, Marin-Kuan M, Delatour T, Nestler S, Mantle P, Cavin C. Ochratoxin A: potential epigenetic mechanisms of toxicity and carcinogenicity. Food Additives and Contaminants. 2005;22(supplement 1):88–93. doi: 10.1080/02652030500309319. [DOI] [PubMed] [Google Scholar]
- 17.Manderville RA. A case for the genotoxicity of ochratoxin a by bioactivation and covalent DNA adduction. Chemical Research in Toxicology. 2005;18(7):1091–1097. doi: 10.1021/tx050070p. [DOI] [PubMed] [Google Scholar]
- 18.Turesky RJ. Perspective: ochratoxin A is not a genotoxic carcinogen. Chemical Research in Toxicology. 2005;18(7):1082–1090. doi: 10.1021/tx050076e. [DOI] [PubMed] [Google Scholar]
- 19.Faucet V, Pfohl-Leszkowicz A, Dai J, Castegnaro M, Manderville RA. Evidence for covalent DNA adduction by ochratoxin A following chronic exposure to rat and subacute exposure to pig. Chemical Research in Toxicology. 2004;17(9):1289–1296. doi: 10.1021/tx049877s. [DOI] [PubMed] [Google Scholar]
- 20.Manderville RA, Pfohl-Leszkowicz A. Chapter 4 genotoxicity of chlorophenols and ochratoxin A. Advances in Molecular Toxicology. 2006;1:85–138. [Google Scholar]
- 21.Mantle PG, Faucet-Marquis V, Manderville RA, Squillaci B, Pfohl-Leszkowicz A. Structures of covalent adducts between DNA and ochratoxin a: a new factor in debate about genotoxicity and human risk assessment. Chemical Research in Toxicology. 2010;23(1):89–98. doi: 10.1021/tx900295a. [DOI] [PubMed] [Google Scholar]
- 22.Pfohl-Leszkowicz A, Castegnaro M. Further arguments in favour of direct covalent binding of Ochratoxin A (OTA) after metabolic biotransformation. Food Additives and Contaminants. 2005;22(supplement 1):75–87. doi: 10.1080/02652030500309400. [DOI] [PubMed] [Google Scholar]
- 23.Schlatter C, Studer-Rohr J, Rásonyi T. Carcinogenicity and kinetic aspects of ochratoxin A. Food Additives and Contaminants. 1996;13, supplement:43–44. [PubMed] [Google Scholar]
- 24.Mally A, Zepnik H, Wanek P, et al. Ochratoxin A: lack of formation of covalent DNA adducts. Chemical Research in Toxicology. 2004;17(2):234–242. doi: 10.1021/tx034188m. [DOI] [PubMed] [Google Scholar]
- 25.Delatour T, Mally A, Richoz J, et al. Absence of 2′-deoxyguanosine-carbon 8-bound ochratoxin A adduct in rat kidney DNA monitored by isotope dilution LC-MS/MS. Molecular Nutrition and Food Research. 2008;52(4):472–482. doi: 10.1002/mnfr.200700276. [DOI] [PubMed] [Google Scholar]
- 26.Jarabek AM, Pottenger LH, Andrews LS, et al. Creating context for the use of DNA adduct data in cancer risk assessment: I. Data organization. Critical Reviews in Toxicology. 2009;39(8):659–678. doi: 10.1080/10408440903164155. [DOI] [PubMed] [Google Scholar]
- 27.Swenberg JA, Fryar-Tita E, Jeong YC, et al. Biomarkers in toxicology and risk assessment: informing critical dose-response relationships. Chemical Research in Toxicology. 2008;21(1):253–265. doi: 10.1021/tx700408t. [DOI] [PubMed] [Google Scholar]
- 28.Boobis AR, Daston GP, Preston RJ, Olin SS. Application of key events analysis to chemical carcinogens and noncarcinogens. Critical Reviews in Food Science and Nutrition. 2009;49(8):690–707. doi: 10.1080/10408390903098673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson GE, Doak SH, Griffiths SM, et al. Non-linear dose-response of DNA-reactive genotoxins: recommendations for data analysis. Mutation Research. 2009;678(2):95–100. doi: 10.1016/j.mrgentox.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Mantle PG. Minimum tolerable exposure period and maximum threshold dietary intake of ochratoxin A for causing renal cancer in male Dark Agouti rats. Food and Chemical Toxicology. 2009;47(10):2419–2424. doi: 10.1016/j.fct.2009.05.043. [DOI] [PubMed] [Google Scholar]
- 31.Loft S, Poulsen HE. Cancer risk and oxidative DNA damage in man. Journal of Molecular Medicine. 1996;74(6):297–312. doi: 10.1007/BF00207507. [DOI] [PubMed] [Google Scholar]
- 32.Klaunig JE, Xu Y, Isenberg JS, et al. The role of oxidative stress in chemical carcinogenesis. Environmental Health Perspectives. 1998;106(supplement 1):289–295. doi: 10.1289/ehp.98106s1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annual Review of Pharmacology and Toxicology. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 34.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 35.Goetz ME, Luch A. Reactive species: a cell damaging rout assisting to chemical carcinogens. Cancer Letters. 2008;266(1):73–83. doi: 10.1016/j.canlet.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 36.Omar RF, Hasinoff BB, Mejilla F, Rahimtula AD. Mechanism of ochratoxin a stimulated lipid peroxidation. Biochemical Pharmacology. 1990;40(6):1183–1191. doi: 10.1016/0006-2952(90)90382-u. [DOI] [PubMed] [Google Scholar]
- 37.Hoehler D, Marquardt RR, Mclntosh AR, Xiao H. Free radical generation as induced by ochratoxin A and its analogs in bacteria (Bacillus brevis) Journal of Biological Chemistry. 1996;271(44):27388–27394. doi: 10.1074/jbc.271.44.27388. [DOI] [PubMed] [Google Scholar]
- 38.Xiao H, Madhyastha S, Marquardt RR, et al. Toxicity of ochratoxin A, its opened lactone form and several of its analogs: structure-activity relationships. Toxicology and Applied Pharmacology. 1996;137(2):182–192. doi: 10.1006/taap.1996.0071. [DOI] [PubMed] [Google Scholar]
- 39.Dai J, Park G, Wright MW, Adams M, Akman SA, Manderville RA. Detection and characterization of a glutathione conjugate of ochratoxin A. Chemical Research in Toxicology. 2002;15(12):1581–1588. doi: 10.1021/tx0255929. [DOI] [PubMed] [Google Scholar]
- 40.Gillman IG, Clark TN, Manderville RA. Oxidation of ochratoxin A by an Fe-porphyrin system: model for enzymatic activation and DNA cleavage. Chemical Research in Toxicology. 1999;12(11):1066–1076. doi: 10.1021/tx9901074. [DOI] [PubMed] [Google Scholar]
- 41.Faucet-Marquis V, Pont F, Størmer FC, Rizk T, Castegnaro M, Pfohl-Leszkowicz A. Evidence of a new dechlorinated ochratoxin A derivative formed in opossum kidney cell cultures after pretreatment by modulators of glutathione pathways: correlation with DNA-adduct formation. Molecular Nutrition and Food Research. 2006;50(6):530–542. doi: 10.1002/mnfr.200500219. [DOI] [PubMed] [Google Scholar]
- 42.Manderville RA. Pfohl-Leszkowicz bioactivation and DNA adduction as a rationale for ochratoxin A carcinogenesis. World Mycotoxin Journal. 2008;1:357–367. [Google Scholar]
- 43.Pfohl-Leszkowicz A. Ochratoxin A and aristolochic acid involvement in nephropathies and associated urothelial tract tumours. Arhiv za Higijenu Rada i Toksikologiju. 2009;60(4):465–483. doi: 10.2478/10004-1254-60-2009-2000. [DOI] [PubMed] [Google Scholar]
- 44.Boesch-Saadatmandi C, Loboda A, Jozkowicz A, et al. Effect of ochratoxin A on redox-regulated transcription factors, antioxidant enzymes and glutathione-S-transferase in cultured kidney tubulus cells. Food and Chemical Toxicology. 2008;46(8):2665–2671. doi: 10.1016/j.fct.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 45.Boesch-Saadatmandi C, Wagner AE, Graeser AC, Hundhausen C, Wolffram S, Rimbach G. Ochratoxin A impairs Nrf2-dependent gene expression in porcine kidney tubulus cells. Journal of Animal Physiology and Animal Nutrition. 2009;93(5):547–554. doi: 10.1111/j.1439-0396.2008.00838.x. [DOI] [PubMed] [Google Scholar]
- 46.Cavin C, Delatour T, Marin-Kuan M, et al. Reduction in antioxidant defenses may contribute to ochratoxin A toxicity and carcinogenicity. Toxicological Sciences. 2007;96(1):30–39. doi: 10.1093/toxsci/kfl169. [DOI] [PubMed] [Google Scholar]
- 47.Marin-Kuan M, Nestler S, Verguet C, et al. A toxicogenomics approach to identify new plausible epigenetic mechanisms of ochratoxin a carcinogenicity in rat. Toxicological Sciences. 2006;89(1):120–134. doi: 10.1093/toxsci/kfj017. [DOI] [PubMed] [Google Scholar]
- 48.Hayes JD, Chanas SA, Henderson CJ, et al. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochemical Society Transactions. 2000;28(2):33–41. doi: 10.1042/bst0280033. [DOI] [PubMed] [Google Scholar]
- 49.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annual Review of Pharmacology and Toxicology. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 50.Zurich MG, Lengacher S, Braissant O, Monnet-Tschudi F, Pellerin L, Honegger P. Unusual astrocyte reactivity caused by the food mycotoxin ochratoxin a in aggregating rat brain cell cultures. Neuroscience. 2005;134(3):771–782. doi: 10.1016/j.neuroscience.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 51.Ferrante MC, Raso GM, Bilancione M, Esposito E, Iacono A, Meli R. Differential modification of inflammatory enzymes in J774A.1 macrophages by ochratoxin A alone or in combination with lipopolysaccharide. Toxicology Letters. 2008;181(1):40–46. doi: 10.1016/j.toxlet.2008.06.866. [DOI] [PubMed] [Google Scholar]
- 52.Cavin C, Delatour T, Marin-Kuan M, et al. Ochratoxin A—mediated DNA and protein damage: roles of nitrosative and oxidative stresses. Toxicological Sciences. 2009;110(1):84–94. doi: 10.1093/toxsci/kfp090. [DOI] [PubMed] [Google Scholar]
- 53.Crowell JA, Steele VE, Sigman CC, Fay JR. Is inducible nitric oxide synthase a target for chemoprevention? Molecular Cancer Therapeutics. 2003;2(8):815–823. [PubMed] [Google Scholar]
- 54.De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19(3):169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 55.Hiraku Y, Kawanishi S. Immunohistochemical analysis of 8-nitroguanine, a nitrative DNA lesion, in relation to inflammation-associated carcinogenesis. Methods in Molecular Biology. 2009;512:3–13. doi: 10.1007/978-1-60327-530-9_1. [DOI] [PubMed] [Google Scholar]
- 56.Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Formation of 8-oxo-7,8-dihydroguanine-radicals in γ-irradiated DNA by multiple one-electron oxidations. Nucleic Acids Research. 2004;32(22):6565–6574. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arbillaga L, Azqueta A, Ezpeleta O, De Cerain AL. Oxidative DNA damage induced by ochratoxin A in the HK-2 human kidney cell line: evidence of the relationship with cytotoxicity. Mutagenesis. 2007;22(1):35–42. doi: 10.1093/mutage/gel049. [DOI] [PubMed] [Google Scholar]
- 58.Kamp HG, Eisenbrand G, Schlatter J, Würth K, Janzowski C. Ochratoxin A: induction of (oxidative) DNA damage, cytotoxicity and apoptosis in mammalian cell lines and primary cells. Toxicology. 2005;206(3):413–425. doi: 10.1016/j.tox.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 59.Schaaf GJ, Nijmeijer SM, Maas RFM, Roestenberg P, De Groene EM, Fink-Gremmels J. The role of oxidative stress in the ochratoxin A-mediated toxicity in proximal tubular cells. Biochimica et Biophysica Acta. 2002;1588(2):149–158. doi: 10.1016/s0925-4439(02)00159-x. [DOI] [PubMed] [Google Scholar]
- 60.Mally A, Pepe G, Ravoori S, et al. Ochratoxin A causes DNA damage and cytogenetic effects but no DNA adducts in rats. Chemical Research in Toxicology. 2005;18(8):1253–1261. doi: 10.1021/tx049650x. [DOI] [PubMed] [Google Scholar]
- 61.Arbillaga L, Azqueta A, van Delft JHM, López de Cerain A. In vitro gene expression data supporting a DNA non-reactive genotoxic mechanism for ochratoxin A. Toxicology and Applied Pharmacology. 2007;220(2):216–224. doi: 10.1016/j.taap.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 62.Zeljezic D, Domijan AM, Peraica M. DNA damage by ochratoxin A in rat kidney assessed by the alkaline comet assay. Brazilian Journal of Medical and Biological Research. 2006;39(12):1563–1568. doi: 10.1590/s0100-879x2006001200006. [DOI] [PubMed] [Google Scholar]
- 63.Gagliano N, Donne ID, Torri C, et al. Early cytotoxic effects of ochratoxin A in rat liver: a morphological, biochemical and molecular study. Toxicology. 2006;225(2-3):214–224. doi: 10.1016/j.tox.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 64.Kamp HG, Eisenbrand G, Janzowski C, et al. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Molecular Nutrition and Food Research. 2005;49(12):1160–1167. doi: 10.1002/mnfr.200500124. [DOI] [PubMed] [Google Scholar]
- 65.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clinica Chimica Acta. 2003;329(1-2):23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 66.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends in Molecular Medicine. 2003;9(4):169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 67.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clinical Chemistry. 2006;52(4):601–623. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- 68.Domijan AM, Rudeš K, Peraica M. The effect of ochratoxin A on the concentration of protein carbonyls in rats. Arhiv za Higijenu Rada i Toksikologiju. 2005;56(4):311–315. [PubMed] [Google Scholar]
- 69.Kamp HG, Eisenbrand G, Janzowski C, et al. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Molecular Nutrition and Food Research. 2005;49(12):1160–1167. doi: 10.1002/mnfr.200500124. [DOI] [PubMed] [Google Scholar]
- 70.Marnett LJ. Lipid peroxidation—DNA damage by malondialdehyde. Mutation Research. 1999;424(1-2):83–95. doi: 10.1016/s0027-5107(99)00010-x. [DOI] [PubMed] [Google Scholar]
- 71.Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181-182:219–222. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
- 72.Nair U, Bartsch H, Nair J. Lipid peroxidation-induced DNA damage in cancer-prone inflammatory diseases: a review of published adduct types and levels in humans. Free Radical Biology and Medicine. 2007;43(8):1109–1120. doi: 10.1016/j.freeradbiomed.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 73.Rahimtula AD, Bereziat JC, Bussacchini-Griot V, Bartsch H. Lipid peroxidation as a possible cause of ochratoxin a toxicity. Biochemical Pharmacology. 1988;37(23):4469–4477. doi: 10.1016/0006-2952(88)90662-4. [DOI] [PubMed] [Google Scholar]
- 74.Khan S, Martin M, Bartsch H, Rahimtula AD. Perturbation of liver microsomal calcium homeostasis by ochratoxin A. Biochemical Pharmacology. 1989;38(1):67–72. doi: 10.1016/0006-2952(89)90150-0. [DOI] [PubMed] [Google Scholar]
- 75.Abdel-Wahhab MA, Abdel-Galil MM, El-Lithey M. Melatonin counteracts oxidative stress in rats fed an ochratoxin A contaminated diet. Journal of Pineal Research. 2005;38(2):130–135. doi: 10.1111/j.1600-079X.2004.00184.x. [DOI] [PubMed] [Google Scholar]
- 76.Domijan AM, Peraica M, Vrdoljak AL, Radić B, Žlender V, Fuchs R. The involvement of oxidative stress in ochratoxin A and fumonisin B toxicity in rats. Molecular Nutrition and Food Research. 2007;51(9):1147–1151. doi: 10.1002/mnfr.200700079. [DOI] [PubMed] [Google Scholar]
- 77.Gautier JC, Holzhaeuser D, Markovic J, Gremaud E, Schilter B, Turesky RJ. Oxidative damage and stress response from ochratoxin A exposure in rats. Free Radical Biology and Medicine. 2001;30(10):1089–1098. doi: 10.1016/s0891-5849(01)00507-x. [DOI] [PubMed] [Google Scholar]
- 78.Petrik J, Žanić-Grubišić T, Barišić K, et al. Apoptosis and oxidative stress induced by ochratoxin A in rat kidney. Archives of Toxicology. 2003;77(12):685–693. doi: 10.1007/s00204-003-0501-8. [DOI] [PubMed] [Google Scholar]
- 79.Sutken E, Aral E, Ozdemir F, Uslu S, Alatas O, Colak O. Protective role of melatonin and coenzyme Q in ochratoxin A toxicity in rat liver and kidney. International Journal of Toxicology. 2007;26(1):81–87. doi: 10.1080/10915810601122893. [DOI] [PubMed] [Google Scholar]
- 80.Benesic A, Mildenberger S, Gekle M. Nephritogenic ochratoxin A interferes with hormonal signalling in immortalized human kidney epithelial cells. Pflugers Archiv European Journal of Physiology. 2000;439(3):278–287. doi: 10.1007/s004249900172. [DOI] [PubMed] [Google Scholar]
- 81.Chong X, Rahimtula AD. Alterations in ATP-dependent calcium uptake by rat renal cortex microsomes following ochratoxin A administration in vivo or addition in vitro. Biochemical Pharmacology. 1992;44(7):1401–1409. doi: 10.1016/0006-2952(92)90542-q. [DOI] [PubMed] [Google Scholar]
- 82.Gekle M, Sauvant C, Schwerdt G. Ochratoxin A at nanomolar concentrations: a signal modulator in renal cells. Molecular Nutrition and Food Research. 2005;49(2):118–130. doi: 10.1002/mnfr.200400062. [DOI] [PubMed] [Google Scholar]
- 83.Rached E, Hard GC, Blumbach K, et al. Ochratoxin A: 13-week oral toxicity and cell proliferation in male F344/ N rats. Toxicological Sciences. 2007;97(2):288–298. doi: 10.1093/toxsci/kfm042. [DOI] [PubMed] [Google Scholar]
- 84.Rahimtula AD, Chong X. Alterations in calcium homeostasis as a possible cause of ochratoxin A nephrotoxicity. IARC Scientific Publications. 1991;(115):207–214. [PubMed] [Google Scholar]
- 85.Arbillaga L, Vettorazzi A, Gil AG, van Delft JH, García-Jalón JA, López de Cerain A. Gene expression changes induced by ochratoxin A in renal and hepatic tissues of male F344 rat after oral repeated administration. Toxicology and Applied Pharmacology. 2008;230(2):197–207. doi: 10.1016/j.taap.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 86.Gekle M, Schwerdt G, Freudinger R, et al. Ochratoxin A induces JNK activation and apoptosis in MDCK-C7 cells at nanomolar concentrations. Journal of Pharmacology and Experimental Therapeutics. 2000;293(3):837–844. [PubMed] [Google Scholar]
- 87.Sauvant C, Holzinger H, Gekle M. Proximal tubular toxicity of ochratoxin A is amplified by simultaneous inhibition of the extracellular signal-regulated kinases 1/2. Journal of Pharmacology and Experimental Therapeutics. 2005;313(1):234–241. doi: 10.1124/jpet.104.079475. [DOI] [PubMed] [Google Scholar]
- 88.Marin-Kuan M, Nestler S, Verguet C, et al. MAPK-ERK activation in kidney of male rats chronically fed ochratoxin A at a dose causing a significant incidence of renal carcinoma. Toxicology and Applied Pharmacology. 2007;224(2):174–181. doi: 10.1016/j.taap.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 89.Stemmer K, Ellinger-Ziegelbauer H, Ahr HJ, Dietrich DR. Carcinogen-specific gene expression profiles in short-term treated Eker and wild-type rats indicative of pathways involved in renal tumorigenesis. Cancer Research. 2007;67(9):4052–4068. doi: 10.1158/0008-5472.CAN-06-3587. [DOI] [PubMed] [Google Scholar]
- 90.Arrigo AP. Gene expression and the thiol redox state. Free Radical Biology and Medicine. 1999;27(9-10):936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 91.Marshall HE, Merchant K, Stamler JS. Nitrosation and oxidation in the regulation of gene expression. FASEB Journal. 2000;14(13):1889–1900. doi: 10.1096/fj.00.011rev. [DOI] [PubMed] [Google Scholar]
- 92.Vossen C, Erard M. Down-regulation of nuclear receptor DNA-binding activity by nitric oxide—HNF4 as a model system. Medical Science Monitor. 2002;8(10):RA217–RA220. [PubMed] [Google Scholar]
- 93.Giles GI. The redox regulation of thiol dependent signaling pathways in cancer. Current Pharmaceutical Design. 2006;12(34):4427–4443. doi: 10.2174/138161206779010549. [DOI] [PubMed] [Google Scholar]
- 94.Hara H, Adachi T. Contribution of hepatocyte nuclear factor-4 to down-regulation of CYP2D6 gene expression by nitric oxide. Molecular Pharmacology. 2002;61(1):194–200. doi: 10.1124/mol.61.1.194. [DOI] [PubMed] [Google Scholar]
- 95.Vuppugalla R, Mehvar R. Hepatic disposition and effects of nitric oxide donors: rapid and concentration-dependent reduction in the cytochrome P450-mediated drug metabolism in isolated perfused rat livers. Journal of Pharmacology and Experimental Therapeutics. 2004;310(2):718–727. doi: 10.1124/jpet.104.065557. [DOI] [PubMed] [Google Scholar]
- 96.Karimpour S, Lou J, Lin LL, et al. Thioredoxin reductase regulates AP-1 activity as well as thioredoxin nuclear localization via active cysteines in response to ionizing radiation. Oncogene. 2002;21(41):6317–6327. doi: 10.1038/sj.onc.1205749. [DOI] [PubMed] [Google Scholar]
- 97.Iwasaki K, MacKenzie EL, Hailemariam K, Sakamoto K, Tsuji Y. Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of ref-1 during erythroid differentiation of K562 cells. Molecular and Cellular Biology. 2006;26(7):2845–2856. doi: 10.1128/MCB.26.7.2845-2856.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tsuji Y. JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene. 2005;24(51):7567–7578. doi: 10.1038/sj.onc.1208901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kröncke KD, Klotz LO, Suschek CV, Sies H. Comparing nitrosative versus oxidative stress toward zinc finger-dependent transcription. Unique role for NO. Journal of Biological Chemistry. 2002;277(15):13294–13301. doi: 10.1074/jbc.M111216200. [DOI] [PubMed] [Google Scholar]
- 100.Kröncke KD. Nitrosative stress and transcription. Biological Chemistry. 2003;384(10-11):1365–1377. doi: 10.1515/BC.2003.153. [DOI] [PubMed] [Google Scholar]
- 101.Nikitovic D, Holmgren A, Spyrou G. Inhibition of AP-1 DNA binding by nitric oxide involving conserved cysteine residues in Jun and Fos. Biochemical and Biophysical Research Communications. 1998;242(1):109–112. doi: 10.1006/bbrc.1997.7930. [DOI] [PubMed] [Google Scholar]
- 102.Abdel-Wahhab MA, Abdel-Azim SH, El-Nekeety AA. Inula crithmoides extract protects against ochratoxin A-induced oxidative stress, clastogenic and mutagenic alterations in male rats. Toxicon. 2008;52(4):566–573. doi: 10.1016/j.toxicon.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 103.Hassen W, Ayed-Boussema I, Bouslimi A, Bacha H. Heat shock proteins (Hsp 70) response is not systematic to cell stress. Case of the mycotoxin ochratoxin A. Toxicology. 2007;242(1–3):63–70. doi: 10.1016/j.tox.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 104.Palma N, Cinelli S, Sapora O, Wilson SH, Dogliotti E. Ochratoxin A-induced mutagenesis in mammalian cells is consistent with the production of oxidative stress. Chemical Research in Toxicology. 2007;20(7):1031–1037. doi: 10.1021/tx700027j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Di Giacomo C, Acquaviva R, Piva A, et al. Protective effect of cyanidin 3-O-β-d-glucoside on ochratoxin A-mediated damage in the rat. British Journal of Nutrition. 2007;98(5):937–943. doi: 10.1017/S0007114507756908. [DOI] [PubMed] [Google Scholar]
- 106.Costa S, Utan A, Cervellati R, Speroni E, Guerra MC. Catechins: natural free-radical scavengers against ochratoxin A-induced cell damage in a pig kidney cell line (LLC-PK1) Food and Chemical Toxicology. 2007;45(10):1910–1917. doi: 10.1016/j.fct.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 107.Lambert D, Padfield PJ, McLaughlin J, Cannell S, O’Neill CA. Ochratoxin A displaces claudins from detergent resistant membrane microdomains. Biochemical and Biophysical Research Communications. 2007;358(2):632–636. doi: 10.1016/j.bbrc.2007.04.180. [DOI] [PubMed] [Google Scholar]
- 108.Sava V, Velasquez A, Song S, Sanchez-Ramos J. Adult hippocampal neural stem/progenitor cells in vitro are vulnerable to the mycotoxin ochratoxin-A. Toxicological Sciences. 2007;98(1):187–197. doi: 10.1093/toxsci/kfm093. [DOI] [PubMed] [Google Scholar]
- 109.Stemmer K, Ellinger-Ziegelbauer H, Ahr HJ, Dietrich DR. Carcinogen-specific gene expression profiles in short-term treated Eker and wild-type rats indicative of pathways involved in renal tumorigenesis. Cancer Research. 2007;67(9):4052–4068. doi: 10.1158/0008-5472.CAN-06-3587. [DOI] [PubMed] [Google Scholar]
- 110.Sava V, Reunova O, Velasquez A, Harbison R, Sánchez-Ramos J. Acute neurotoxic effects of the fungal metabolite ochratoxin-A. NeuroToxicology. 2006;27(1):82–92. doi: 10.1016/j.neuro.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 111.Sava V, Reunova O, Velasquez A, Sanchez-Ramos J. Can low level exposure to ochratoxin-A cause parkinsonism? Journal of the Neurological Sciences. 2006;249(1):68–75. doi: 10.1016/j.jns.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 112.Ferrante MC, Bilancione M, Raso GM, et al. Expression of COX-2 and hsp72 in peritoneal macrophages after an acute ochratoxin A treatment in mice. Life Sciences. 2006;79(13):1242–1247. doi: 10.1016/j.lfs.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 113.Domijan AM, Želježić D, Kopjar N, Peraica M. Standard and Fpg-modified comet assay in kidney cells of ochratoxin A- and fumonisin B-treated rats. Toxicology. 2006;222(1-2):53–59. doi: 10.1016/j.tox.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 114.Tozlovanu M, Faucet-Marquis V, Pfohl-Leszkowicz A, Manderville RA. Genotoxicity of the hydroquinone metabolite of ochratoxin A: structure-activity relationships for covalent DNA adduction. Chemical Research in Toxicology. 2006;19(9):1241–1247. doi: 10.1021/tx060138g. [DOI] [PubMed] [Google Scholar]
- 115.Alvarez-Erviti L, Leache C, González-Peñas E, López De Cerain A. Alterations induced in vitro by ochratoxin A in rat lymphoid cells. Human and Experimental Toxicology. 2005;24(9):459–466. doi: 10.1191/0960327105ht554oa. [DOI] [PubMed] [Google Scholar]
- 116.Guerra MC, Galvano F, Bonsi L, et al. Cyanidin-3-O-β-glucopyranoside, a natural fee-radical scavenger against aflatoxin B1-and ochratoxin A-induced cell damage in a human hepatoma cell line (Hep G2) and a human colonic adenocarcinoma cell line (CaCo-2) British Journal of Nutrition. 2005;94(2):211–220. doi: 10.1079/bjn20051425. [DOI] [PubMed] [Google Scholar]
- 117.Hundhausen C, Bösch-Saadatmandi C, Augustin K, Blank R, Wolffram S, Rimbach G. Effect of vitamin E and polyphenols on ochratoxin A-induced cytotoxicity in liver (HepG2) cells. Journal of Plant Physiology. 2005;162(7):818–822. doi: 10.1016/j.jplph.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 118.Russo A, La Fauci L, Acquaviva R, et al. Ochratoxin A-induced DNA damage in human fibroblast: protective effect of cyanidin 3-O-β-D-glucoside. Journal of Nutritional Biochemistry. 2005;16(1):31–37. doi: 10.1016/j.jnutbio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 119.Knasmüller S, Cavin C, Chakraborty A, et al. Structurally related mycotoxins ochratoxin A, ochratoxin B, and citrinin differ in their genotoxic activities and in their mode of action in human-derived liver (HepG2) cells: implications for risk assessment. Nutrition and Cancer. 2004;50(2):190–197. doi: 10.1207/s15327914nc5002_9. [DOI] [PubMed] [Google Scholar]
- 120.Ehrlich V, Darroudi F, Uhl M, et al. Genotoxic effects of ochratoxin A in human-derived hepatoma (HepG2) cells. Food and Chemical Toxicology. 2002;40(8):1085–1090. doi: 10.1016/s0278-6915(02)00045-5. [DOI] [PubMed] [Google Scholar]
- 121.Lühe A, Hildebrand H, Bach U, Dingermann T, Ahr HJ. A new approach to studying ochratoxin A (OTA)-induced nephrotoxicity: expression profiling in vivo and in vitro employing cDNA microarrays. Toxicological Sciences. 2003;73(2):315–328. doi: 10.1093/toxsci/kfg073. [DOI] [PubMed] [Google Scholar]
- 122.Son WC, Kamino K, Lee YS, Kang KS. Lack of effects of sodium 2-mercaptoethane sulfonate (mesna) on Ochratoxin A induced renal tumorigenicity following life-time oral administration of Ochratoxin A in DA and Lewis rats. Toxicology Letters. 2003;142(1-2):19–27. doi: 10.1016/s0378-4274(02)00471-x. [DOI] [PubMed] [Google Scholar]
- 123.Pfohl-Leszkowicz A, Bartsch H, Azémar B, Mohr U, Estève J, Castegnaro M. MESNA protects rats against nephrotoxicity but not carcinogenicity induced by ochratoxin A, implicating two separate pathways. Facta Universitatis Series Medicine and Biology. 2002;9:57–63. [Google Scholar]
- 124.Pinelli E, El Adlouni C, Pipy B, Quartulli F, Pfohl-Leszkowicz A. Roles of cyclooxygenase and lipoxygenases in ochratoxin A genotoxicity in human epithelial lung cells. Environmental Toxicology and Pharmacology. 1999;7(2):95–107. doi: 10.1016/s1382-6689(99)00008-3. [DOI] [PubMed] [Google Scholar]
- 125.Hoehler D, Marquardt RR, McIntosh AR, Hatch GM. Induction of free radicals in hepatocytes, mitochondria and microsomes of rats by ochratoxin A and its analogs. Biochimica et Biophysica Acta. 1997;1357(2):225–233. doi: 10.1016/s0167-4889(97)00026-8. [DOI] [PubMed] [Google Scholar]
- 126.Grosse Y, Chekir-Ghedira L, Huc A, et al. Retinol, ascorbic acid and α-tocopherol prevent DNA adduct formation in mice treated with the mycotoxins ochratoxin A and zearalenone. Cancer Letters. 1997;114(1-2):225–229. doi: 10.1016/s0304-3835(97)04669-7. [DOI] [PubMed] [Google Scholar]
- 127.Baudrimont I, Betbeder AM, Gharbi A, Pfohl-Leszkowicz A, Dirheimer G, Creppy EE. Effect of superoxide dismutase and catalase on the nephrotoxicity induced by subchronical administration of ochratoxin A in rats. Toxicology. 1994;89(2):101–111. doi: 10.1016/0300-483x(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 128.Klaunig JE, Kamendulis LM, Xu Y. Epigenetic mechanisms of chemical carcinogenesis. Human and Experimental Toxicology. 2000;19:543–555. doi: 10.1191/096032700701546442. [DOI] [PubMed] [Google Scholar]
- 129.Trosko JE, Ruch RJ. Cell-cell communication in carcinogenesis. Frontiers in Bioscience. 1998;3:d208–d236. doi: 10.2741/a275. [DOI] [PubMed] [Google Scholar]
- 130.Trosko JE, Chang CC, Upham B, Wilson M. Epigenetic toxicology as toxicant-induced changes in intracellular signalling leading to altered gap junctional intercellular communication. Toxicology Letters. 1998;102-103:71–78. doi: 10.1016/s0378-4274(98)00288-4. [DOI] [PubMed] [Google Scholar]
- 131.Yamasaki H, Krutovskikh V, Mesnil M, Columbano A, Tsuda H, Ito N. Gap junctional intercellular communication and cell proliferation during rat liver carcinogenesis. Environmental Health Perspectives. 1993;101(supplement 5):191–197. doi: 10.1289/ehp.93101s5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Noguchi M, Nomata K, Watanabe JI, Sato H, Kanetake H, Saito Y. Disruption of gap junctional intercellular communication in human renal cancer cell lines. Urology. 1999;53(1):218–222. doi: 10.1016/s0090-4295(98)00412-9. [DOI] [PubMed] [Google Scholar]
- 133.Sato H, Hagiwara H, Ohde Y, Senba H, Virgona N, Yano T. Regulation of renal cell carcinoma cell proliferation, invasion and metastasis by connexin 32 gene. Journal of Membrane Biology. 2007;216(1):17–21. doi: 10.1007/s00232-007-9020-5. [DOI] [PubMed] [Google Scholar]
- 134.Mally A, Decker M, Bekteshi M, Dekant W. Ochratoxin A alters cell adhesion and gap junction intercellular communication in MDCK cells. Toxicology. 2006;223(1-2):15–25. doi: 10.1016/j.tox.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 135.Horvath A, Upham BL, Ganev V, Trosko JE. Determination of the epigenetic effects of ochratoxin in a human kidney and a rat liver epithelial cell line. Toxicon. 2002;40(3):273–282. doi: 10.1016/s0041-0101(01)00219-7. [DOI] [PubMed] [Google Scholar]
- 136.Obrecht-Pflumio S, Grosse Y, Pfohl-Leszkowicz A, Dirheimer G. Protection by indomethacin and aspirin against genotoxicity of ochratoxin A, particularly in the urinary bladder and kidney. Archives of Toxicology. 1996;70(3-4):244–248. doi: 10.1007/s002040050267. [DOI] [PubMed] [Google Scholar]
- 137.Centre for Food Safety (CFS) Ochratoxin A in Food. Food and Environmental Hygiene Department. Risk Assessment Studies Report no. 23. The Government of the Hong Kong Special Administrative Region, Hong Kong, 2006.
- 138.Beisswenger PJ, Drummond KS, Nelson RG, Howell SK, Szwergold BS, Mauer M. Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes. 2005;54(11):3274–3281. doi: 10.2337/diabetes.54.11.3274. [DOI] [PubMed] [Google Scholar]
- 139.Coşkun C, Kural A, Döventaş Y, et al. Hemodialysis and protein oxidation products. Annals of the New York Academy of Sciences. 2007;1100:404–408. doi: 10.1196/annals.1395.045. [DOI] [PubMed] [Google Scholar]
- 140.Descamps-Latscha B, Witko-Sarsat V. Importance of oxidatively modified proteins in chronic renal failure. Kidney International, Supplement. 2001;59(78):S108–S113. doi: 10.1046/j.1523-1755.2001.59780108.x. [DOI] [PubMed] [Google Scholar]