

Abstract
CCN5 is a member of the CCN (connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed) family and was identified as an estrogen-inducible gene in estrogen receptor-positive cell lines. However, the role of CCN5 in breast carcinogenesis remains unclear. We report here that the CCN5 protein is localized mostly in the cytoplasm and in part in the nucleus of human tumor breast tissue. Using a heterologous transcription assay, we demonstrate that CCN5 can act as a transcriptional repressor presumably through association with histone deacetylase 1 (HDAC1). Microarray gene expression analysis showed that CCN5 represses expression of genes associated with epithelial-mesenchymal transition (EMT) as well as expression of key components of the transforming growth factor β (TGF-β) signaling pathway, prominent among them TGF-βRII receptor. We show that CCN5 is recruited to the TGF-βRII promoter, thereby providing a mechanism by which CCN5 restricts transcription of the TGF-βRII gene. Consistent with this finding, CCN5, we found, functions to suppress TGF-β-induced transcriptional responses and invasion that is concomitant with EMT. Thus, our data uncovered CCN5 as a novel transcriptional repressor that plays an important role in regulating tumor progression functioning, at least in part, by inhibiting the expression of genes involved in the TGF-β signaling cascade that is known to promote EMT.
INTRODUCTION
CCN5 (previously known as WISP-2) is a 29-kDa protein member of the connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family (2, 5, 23). The CCN family is composed of six members grouped on the basis of similar structural analogies (7). CCN proteins appear to play important roles in several biological processes, including cell growth, adhesion, and migration as well as numerous endocrine-regulated functions (9, 39, 41). CCN proteins encompass four structural domains: an insulin-like growth factor-binding protein (IGF-BP) domain, a von Willebrand factor type C (VWC) domain, a thrombospondin (TSP-1) domain, and a cysteine knot (CT) domain reported to act as a potential proliferation module (29). Although the physiological function of CCN5 is not well defined, its domain structure suggests that its function may be different from that of other members of the CCN family. CCN5 contains only three structural domains and lacks the CT domain (7, 8, 35). It has been shown that CCN5 suppresses proliferation and its expression is reduced in cancers (14, 31, 35). Previously, we showed that upon hormone binding, the estrogen receptor (ER) directly regulates the ccn5 gene in all ER-positive breast cancer cell lines tested (17). Moreover, we found that CCN5 knockdown not only induced estradiol-independent growth of these cells, owing to a loss of estrogen receptor α (ERα) expression, but also promoted epithelial-mesenchymal transition (EMT) (18), a process involved in tumor invasiveness and metastasis (13, 43, 46, 51).
Consistent with its role in tumor progression, our recent studies have shown that CCN5 is strongly expressed in less-aggressive breast cancer cell lines, such as MCF-7, while it is undetectable in highly aggressive breast cancer cell lines, such as MDA-MB-231 (18). Untransformed cells express low levels of CCN5 (18). Furthermore, in vivo studies have shown that CCN5 expression is detected mainly in preneoplastic disorders, such as noninvasive ductal carcinoma in situ (DCIS) and atypical ductal hyperplasia, whereas expression levels were either very low or undetectable in invasive breast tumors (4).
There is strong evidence that a number of secreted factors and cell surface receptors can be internalized by endocytosis and translocated to the cytoplasm and sometimes to the nucleus (26, 34, 37). Their nuclear localization is often transient, appearing only during certain phases of the cell cycle. It has been reported that CCN2/CTGF and the amino-truncated CCN3/Nov protein are addressed to the nucleus (38, 48). Furthermore, it has also been reported that CCN6/WISP-3 can localize to the nuclei of breast cancer cells (22). Finally, it has been reported that CCN2 and CCN5 are detected in the cytoplasmic compartment as well as the nuclear compartment in many rodent embryonic and adult tissues as well as in fetal human tissues (20, 27, 50). Collectively, these data are suggestive of diverse functions for the CCN family, including nuclear function for several of its members.
Here we show that CCN5 is localized both in the cytoplasm and in the nucleus of a human breast cancer cell line and noninvasive breast tumors. We demonstrate that CCN5 interacts with histone deacetylases (HDACs) and exhibits strong transcriptional repressor activity. We also show that the loss of CCN5 expression leads to increased expression of important components of the transforming growth factor β (TGF-β) signaling system, in turn favoring EMT and associated cellular invasion. Our data reveal a new role for CCN5 in transcriptional repression.
MATERIALS AND METHODS
Tissue samples, histological analyses, and immunohistochemistry.
Clinical data and primary breast carcinoma samples were collected at Ghent University Hospital and Saint-Antoine Hospital (Paris). Written informed consent was obtained from each patient according to the recommendations of the local ethics committee. Immunochemical staining was performed on paraformaldehyde-fixed, paraffin-embedded ductal carcinoma in situ and invasive ductal carcinoma specimens (n = 20). Sections were deparaffinized in xylene and hydrated with a graded series of ethanol. Subsequently, slides were loaded in the Ventana autostainer (NexES; Ventana Medical Systems, Inc., Tucson, AZ) and stained with the iView DAB detection system (Ventana Medical Systems, Inc., Tucson, AZ) according to the manufacturer's instructions. Antigen retrieval was performed using protease pretreatment (Ventana). Sections were then incubated with anti-WISP2 rabbit polyclonal IgG1 antibody (Abcam Laboratories, Inc., Cambridge, United Kingdom) for 30 min at 37°C (1:100 dilution).
Plasmids.
The expression vector for full-length human CCN5 (hCCN5) was previously described (18). S. Khochbin provided the L8G5E1b-Luc, LexA-VP16, hemagglutinin (HA)-histone deacetylase 5 (HDAC5), and HA-HDAC6 expression vectors. A. Atfi and A. Sharrocks provided the pG5-E1B-Luc and GAL4-tk-Luc reporter plasmids, respectively. To generate GAL4-DNA binding domain (DBD) and CCN5 fusion protein constructs, fragments encoding various portions of human CCN5 were generated by PCR and subcloned into the XbaI/EcoRI sites of the pM vector (Clontech) to fuse CCN5 in frame to the C terminus of GAL4-DBD. Flag-HDAC1 and Flag-HDAC3 expression vectors were a gift of E. Seto. The TβRII promoter luciferase reporter plasmids were provided by J. W. Freeman; ARE3-Lux, FAST1, and CAGA9-Lux are described in references 36 and 40.
Cell culture and transfection.
Human embryonic kidney 293T cells and HeLa cells were maintained in Dulbecco modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). MCF-7, MCF-7-sh-scrambled, and MCF-7-sh-CCN5 were cultured as described previously (18). For luciferase reporter gene assays, cells were seeded in 6-well plates and transfected with plasmids by using the Lipofectamine reagent (Invitrogen). After 36 h, cells were treated with or without TGF-β1 (2 ng/ml) for 16 h. Luciferase activity was measured (Promega) and was normalized for transfection efficiency using a β-galactosidase-expressing vector and the Galacto-Star system (PerkinElmer).
Confocal microscopy.
MCF-7 cells were seeded in 2-well Lab-Teck glass chamber slides for 24 h. The cells were fixed with 4% paraformaldehyde in phosphate-buffered saline and then treated briefly with 0.1% Triton X-100 in phosphate-buffered saline (PBS). After three rinses with PBS, cells were incubated for 1 h at room temperature with primary antibodies to CCN5 (Santa Cruz Biotechnology), rinsed, and incubated for 1 h at room temperature with fluorescein isothiocyanate (FITC)-conjugated secondary antibody (goat anti-rabbit; Invitrogen). Nuclei were stained with 4′,6′-diamino-2-phenylindole (DAPI) at a concentration of 1 μg/ml. Confocal microscopy was conducted with Leica SP2 biphoton laser scanning.
Biochemical cell fractionation.
Cells were harvested at 80% confluence through trypsination. Isolation of nuclei and cytosol was carried out using NE-PER nuclear and cytoplasmic extraction reagents (Pierce) according to the manufacturer's instructions.
GST pulldown.
BL21(DE3) Escherichia coli (Invitrogen) bacteria were used for expression of glutathione S-transferase (GST) and GST-CCN5. The CCN5 open reading frame (ORF) was subcloned into the pGEX4T-1 (GE Healthcare Life Sciences). After adding IPTG (isopropyl-β-d-thiogalactopyranoside) (final concentration of 1 mM), the bacteria were cultured for 4 h at 30°C. The fusion proteins were purified as inclusion bodies and were denatured and renatured using the Rapid GST inclusion body solubilization and renaturation kit (Cell Biolabs, Inc.) according to the manufacturer's instructions. Purified GST or GST-CCN5 was incubated with glutathione-Sepharose 4B transferase (GE Healthcare Life Sciences) for 2 h at 4°C with gentle agitation. The suspensions were then centrifuged, and pellets were washed four times with ice-cold NTEN buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.1 mM dithiothreitol [DTT], supplemented with complete protease inhibitor cocktail [Sigma]). Epitope-tagged proteins were expressed in 293T cells and extracted in NTEN supplemented with protease inhibitors. Sepharose-bound GST fusion proteins were incubated with cell lysates for 2 h at 4°C, washed five times with extraction buffer, fractionated on 10% SDS-PAGE, and detected by Western blotting using horseradish peroxidase-conjugated anti-Flag antibody (Sigma-Aldrich). Membranes were washed extensively and developed with an enhanced chemiluminescence kit (ECL; GE Healthcare).
Immunoblotting.
Cell lysates were made in TNMG lysis buffer (0.5% NP-40, 20 mM Tris-HCl [pH 8], 25 mM MgCl2, 10% glycerol, and 150 mM NaCl) containing 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1 μM leupeptin, and 1 μM aprotinin and cleared by centrifugation. Fifty micrograms of protein was separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with saturating buffer for 1 h at room temperature. The membranes were probed with specific antibodies: anti-GAL4 (DBD), anti-TGF-βRII, anti-plasminogen activator inhibitor 1 (PAI1), anti-HA, anti-CCN5, anti-actin (Santa Cruz Biotechnology), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), anti-lamin A plus C (Millipore), and anti-Flag (Sigma-Aldrich) overnight at 4°C. Membranes were then washed and incubated with horseradish peroxidase-conjugated secondary antibodies for 2 h. Membranes were washed extensively and developed with an enhanced chemiluminescence kit (ECL; GE Healthcare).
Microrrays.
Microarray analysis has been performed in the genomic and microgenomic core facility profileXpert (Bron, France). Total RNA was extracted from frozen cells with the RNeasy minikit (Qiagen, Hilden, Germany) and quantified with NanoDrop. The quality of total RNA was verified by microchips on an Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA). Total RNA (100 ng) was amplified and biotin labeled by in vitro transcription (IVT) with a Message Amp antisense RNA (aRNA) kit (Ambion, Austin, Texas). Before amplification, spikes of synthetic mRNA at different concentrations were added to all samples; these positive controls were used to ascertain the quality of the process. The aRNA yield was measured with a UV spectrophotometer, and the quality on nanochips with the Agilent 2100 bioanalyzer (Agilent).
Ten micrograms of biotin-labeled aRNA was fragmented using 5 μl of fragmentation buffer in a final volume of 20 μl, mixed with 240 μl of Amersham hybridization solution (GE Healthcare Europe GmbH, Freiburg, Germany), and injected onto CodeLink human whole-genome bioarrays containing human oligonucleotide gene probes targeting 57,000 transcripts and expressed sequence tags (EST) (both from GE Healthcare Europe GmbH, Freiburg, Germany). Arrays were hybridized overnight at 37°C at 300 rpm in an incubator. The slides were washed in stringent TNT buffer (0.01 M Tris-HCl [pH 8], 0.15 N NaCl, 0.05% Tween 20) at 46°C for 1 h, and then a streptavidin-cy5 (GE Healthcare) detection step was performed. Each slide was incubated for 30 min in 3.4 ml of streptavidin-cy5 solution as described previously (16), washed 4 times in 240 ml of TNT buffer, rinsed twice in 240 ml of water containing 0.2% Triton X-100, and dried by centrifugation at 600 rpm. The slides were scanned using a Genepix 4000B scanner (Axon, Union City) and Genepix software, with the laser set at 635 mm, the laser power at 100%, and the photomultiplier tube voltage at 60%. The scanned image files were analyzed using CodeLink expression software, version 4.2 (GE Healthcare), which produces both a raw and normalized hybridization signal for each spot on the array.
Microarray data analysis.
The relative intensity of the raw hybridization signal on arrays varies in different experiments. CodeLink software was therefore used to normalize the raw hybridization signal on each array to the median of the array (median intensity is 1 after normalization) for better cross-array comparison. The threshold of detection was calculated using the normalized signal intensity of the 100 negative-control samples in the array; spots with signal intensities below this threshold are referred to as “absent.” The quality of processing was evaluated by generating scatter plots of positive-signal distribution. Signal intensities were then converted to log base 2 values.
SYBR green real-time RT-PCR.
Reverse transcription-PCR (RT-PCR) analysis was carried out as described previously (19). Primers for the amplification of the TGF-βRII gene were as follows: upper, 5′-GGGGAAACAATACTGGCTGA-3′; and lower, 5′-GAGCTCTTGAGGTCCCTGTG-3′.
ChIP assay.
Chromatin immunoprecipitation (ChIP) assays were performed largely as previously described (18). Briefly, a small portion (1%) of the cross-linked, sheared chromatin solution was saved as input DNA, and the remainder was used for immunoprecipitation by specific antibodies against CCN5 (Santa Cruz), acetylated H3, acetylated H4, HDAC1, or Sp1 (Millipore). Immunoprecipitated DNA was deproteinized, precipitated by ethanol, and resuspended in 30 μl of water. Two microliters of DNA were then subjected to PCR (30 cycles) using the following primer pairs for hTGF-βRII gene promoter amplification: upper, 5′-GGAGCAATCTGAAGAAAGCTGA-3′; and lower, 5′-GGGGAAACAGGAAACTCCTC-3′.
Collagen invasion assay.
Acid-extracted type I collagen was commercially available (BD Biosciences). Collagen gels were prepared in 6-well plates with 1.25 ml of type I collagen used at a final concentration of 1 mg/ml (12). After gelling (45 min at 37°C), 1 × 105 to 2 × 105 MCF-7-sh-scrambled and MCF-7-sh-CCN5 cells in media supplemented with 10% heat-inactivated fetal calf serum (FCS) were added with vehicle only (0.1% dimethyl sulfoxide [DMSO]) or SB431542 (10 μM, dissolved in DMSO). After a 14-day culture period (with medium refreshments every 2 days), samples were formalin fixed, cross-sectioned, and hematoxylin and eosin (H&E) stained, and the number of deeply invasive cells was measured in 6 randomly selected fields. The experiments were performed 3 times.
Data analysis.
Data are shown as the average ± the standard deviation (SD) of results of at least three independent experiments. Differences between test and control conditions were assessed by Student's t test analysis or the χ2 test (collagen type I invasion assay).
Microarray data accession number.
Microarray data sets are available at http://www.ncbi.nlm.nih.gov/geo/ under accession code GSE25431.
RESULTS
Subcellular localization of CCN5.
To gain insight into the mechanisms by which CCN5 regulates tumor progression, we analyzed whether CCN5 could display intracellular localization in human breast cancer cells. We examined the subcellular distribution of CCN5 in the MCF-7 cells by confocal immunofluorescence microscopy using anti-CCN5 antibodies. The results revealed the presence of CCN5 in discrete nuclear structures in MCF-7 cells, in addition to a cytoplasmic staining (Fig. 1A). We then used biochemical methods of cell fractionation followed by Western blot analysis with anti-CCN5 antibodies. As shown in Fig. 1B, CCN5 is present in both the cytosolic and nuclear fractions. Control antibodies against the cytoplasmic protein GAPDH and the nuclear proteins lamin A and C confirmed the absence of cross-contamination between the cytoplasmic and nuclear fractions. Moreover, we examined CCN5 subcellular distribution in samples of human breast tumor tissue. We found that noninvasive lesions, such as ductal carcinoma in situ (DCIS), had a marked expression of CCN5. These tumors were estrogen receptor and progesterone receptor positive and ErB-2/Neu negative, features characteristic of well-differentiated tumors. CCN5 is observed with the cytoplasm and also with many nuclei of these cells (Fig. 1C). In contrast, CCN5 expression is weak or undetectable in poorly differentiated invasive ductal carcinoma with lymph node metastasis (Fig. 1C).
Fig. 1.

Subcellular localization of CCN5. (A) Immunochemical detection. MCF-7 cells were fixed, permeabilized, and then immunostained with anti-CCN5 antibodies as described in Materials and Methods. Nuclear DNA was stained with DAPI. Images were obtained by confocal microscopy (magnification, ×63). The results were confirmed by three independent experiments. Arrows indicate nuclear structure stained with anti-CCN5 antibodies. (B) Biochemical evidence. MCF-7 cells were fractionated into cytosolic and nuclear fractions, and equal amounts of protein were analyzed by Western blotting. GAPDH was used as a cytoplasmic marker, and lamin A and C were used as nuclear markers to exclude cross-contamination. (C) Immunohistochemistry analysis of human DCIS and invasive carcinoma sections using anti-CCN5 antibodies. Arrows indicate marked nuclei. Scale bars, 100 μm.
Mapping of the transcriptional repression domain in CCN5.
Since CCN5 localizes in the nucleus, we decided to test whether CCN5 might regulate transcription. To address this possibility, we analyzed the effect of GAL4-DBD full-length CCN5 fusion protein (GAL4-CCN5) on the activity of a GAL4-dependent reporter in a transient transfection assay in HeLa cells. GAL4-CCN5 strongly inhibited the transcription of a GAL4-driven E1B-luciferase reporter construct in a dose-dependent manner (Fig. 2A). Repression by GAL4-CCN5 requires binding to the heterologous promoter, as the transcriptional activity of a reporter lacking the GAL4-binding sites was practically insensitive to the expression of GAL4-CCN5 (Fig. 2B). To map the CCN5 domain responsible for the transcriptional repression activity, we tested the ability of a series of truncated GAL4-CCN5 fusion proteins to regulate the activity of two GAL4-driven luciferase reporter genes in two different cell lines, 293T and HeLa cells (Fig. 2C). In both cell lines, GAL4-CCN5(24-250), deleted of the peptide signal, and GAL4-CCN5(24-150), deleted of the peptide signal and of the C-terminal part of the protein, displayed the most efficient transcriptional repressor activity. However, deletions of any of the three structural domains resulted in a significant decrease in repression, suggesting that multiple domains of CCN5 are important for basal transcriptional repression (Fig. 2C). At the same time, we noted that none of the three domains by themselves produced notable transcriptional repression (compare fragments from positions 1 to 94, 98 to 164, and 194 to 238), and the VWC domain alone (positions 98 to 164) actually stimulated GAL4-dependent transcription. As judged by Western blot analysis with whole-cell extracts of 293T transfected cells, all of the fusion proteins were expressed at comparable levels, with the exception of GAL4-CCN5(194-238), which displayed low abundance (Fig. 2D).
Fig. 2.

CCN5 contains multiple transcriptional repression domains. (A) CCN5 represses basal transcription. Increasing amounts of GAL4-CCN5 (0.25, 0.5, and 1 μg) were transfected into HeLa cells along with 0.5 μg of pG5-E1B-Luc and 0.1 μg of an RSV-β-galactosidase construct as an internal control. Cells were harvested 48 h later. Extracts were assayed for luciferase and β-galactosidase activities. Fold repression was determined relative to the activity of GAL4-DBD and represents an average of triplicate assays. (B) Requirement of GAL4 binding sites for transcriptional repression by GAL4-CCN5. HeLa cells were transfected with 1 μg of GAL4-CCN5 or GAL4-DBD along with 0.5 μg of GAL4-tk-Luc and tk-Luc reporters and 0.1 μg of RSV-β-galactosidase construct as an internal control. Cells were harvested 48 h later. Extracts were assayed for luciferase and β-galactosidase activities. Fold repression was determined relative to the activity of GAL4-DBD and represents an average of triplicate assays. (C) Mapping of the CCN5 sequences required for repression. Schematic representation of GAL4-CCN5 deletion mutants is shown, and their effects on promoter activity in 293T and HeLa cells are summarized. 293T cells and HeLa cells were cotransfected with 1 μg of constructs encoding the indicated CCN5 fragments fused to GAL4 and 0.5 μg of GAL4-driven luciferase reporter plasmids containing the minimal E1B or TK promoter and 0.1 μg of RSV-β-galactosidase construct as an internal control. Cells were harvested 48 h later. Extracts were assayed for luciferase and β-galactosidase activities. The graph shows the fold repression of the basal GAL4-E1B promoter activity in the presence of GAL4-CCN5 fusion proteins relative to GAL4-DBD alone in 293T cells. The results shown represent the average of three independent experiments assayed in duplicate. Significant differences: *, P < 0,05; **, P < 0.01, versus controls. (D) Expression of GAL4-CCN5 mutants (fragments indicated) in 293T transfected cells. The transfected lysates were analyzed by SDS-PAGE and immunoblotting by using mouse anti-GAL4-DBD monoclonal antibody.
The minimal E1B promoter-driven reporter construct displays a low basal transcriptional activity promoter, indicating that CCN5 can repress basal transcription. To establish whether the ability of CCN5 to repress transcription could occur in an inducible setting, we analyzed the effect of expressing CCN5 on an E1B promoter preceded by LexA and GAL4 binding sites (Fig. 3A). In the presence of the highly active LexA-VP16 fusion protein, this reporter is strongly activated (Fig. 3B) (30). A series of GAL4-CCN5 fusion proteins were then added in trans to investigate their potential to repress LexA-VP16-dependent transcription. As shown in Fig. 3B, GAL4-CCN5(1-250) as well as GAL4-CCN5(24-150) repressed the LexA-VP16-induced transcription from the LexA-GAL4-E1B-Luc reporter (Fig. 3B). We therefore conclude that GAL4-CCN5(1-250) acts as a repressor of both basal and induced transcription and that multiple regions located essentially between amino acids 24 to 150 are important for transcriptional repression.
Fig. 3.

CCN5 represses activated transcription. (A) Scheme of LexA-GAL4-driven E1B promoter-reporter construct. LexA-VP16 is an activator and GAL4-CCN5 a repressor. (B) Activities of the indicated GAL4-CCN5 fusion proteins relative to the reporter alone in 293T cells. Cells were cotransfected with 0.1 μg of LexA-VP16 alone or in combination with 1 μg of constructs encoding the indicated GAL4-CCN5 fragments and 0.5 μg of GAL4-driven luciferase reporter and 0.1 μg of RSV-β-galactosidase construct as an internal control. Cells were harvested 48 h later. Extracts were assayed for luciferase and β-galactosidase activities. The presence of LexA-VP16 is indicated. The results shown represent the average of three independent experiments assayed in duplicate. Significant differences: *, P < 0.05 versus control.
It is important to note that GAL4-CCN5(98-164) corresponding to the VWC module, although expressed at a low level relative to that of wild-type CCN5, behaved as a transcriptional activator not only in the case of basal (Fig. 2C) but also VP16-stimulated transcription (Fig. 3B). In fact, in the presence of LexA-VP16, GAL4-CCN5(98-164) increased the transcription mediated by LexA-VP16 from the LexA-GAL4-E1B-Luc reporter approximately 2-fold (Fig. 3B). In association with the IGFBP (positions 1 to 164) and/or TSP motifs (positions 124 to 250), the transcription-activating character of VWC is too weak or absent, possibly due to the overall conformation of the resulting protein (compare fragments from positions 98 to 164, 1 to 164, and 124 to 250). Interestingly, a similar transcriptional activator effect has been reported when the VWC module of CCN3 was fused to GAL4-DBD (37).
CCN5 interacts with HDAC.
Histone deacetylases (HDACs) catalyze the removal of the acetyl groups of lysine residues on histone tails, causing chromatin compaction and transcriptional repression (25). Numerous studies have shown that many transcriptional repressors exert their action through recruitment of histone deacetylases (28, 33). In order to test whether the transcriptional repression activity of GAL4-CCN5 is mediated by HDACs, we assayed the effect of trichostatine A (TSA), a specific inhibitor of HDAC activity, on transcriptional repression mediated by GAL4-CCN5 in a transfection assay. Addition of TSA significantly reduces the ability of CCN5 to repress transcription, while it has no effect on Gal4-DBD alone, suggesting that the full transcriptional repression effect of CCN5 requires histone deacetylase activity (Fig. 4A). Other authors also observed that histone deacetylase inhibitors are not able to relieve all of the silencing activity of corepressors (49, 52).
Fig. 4.
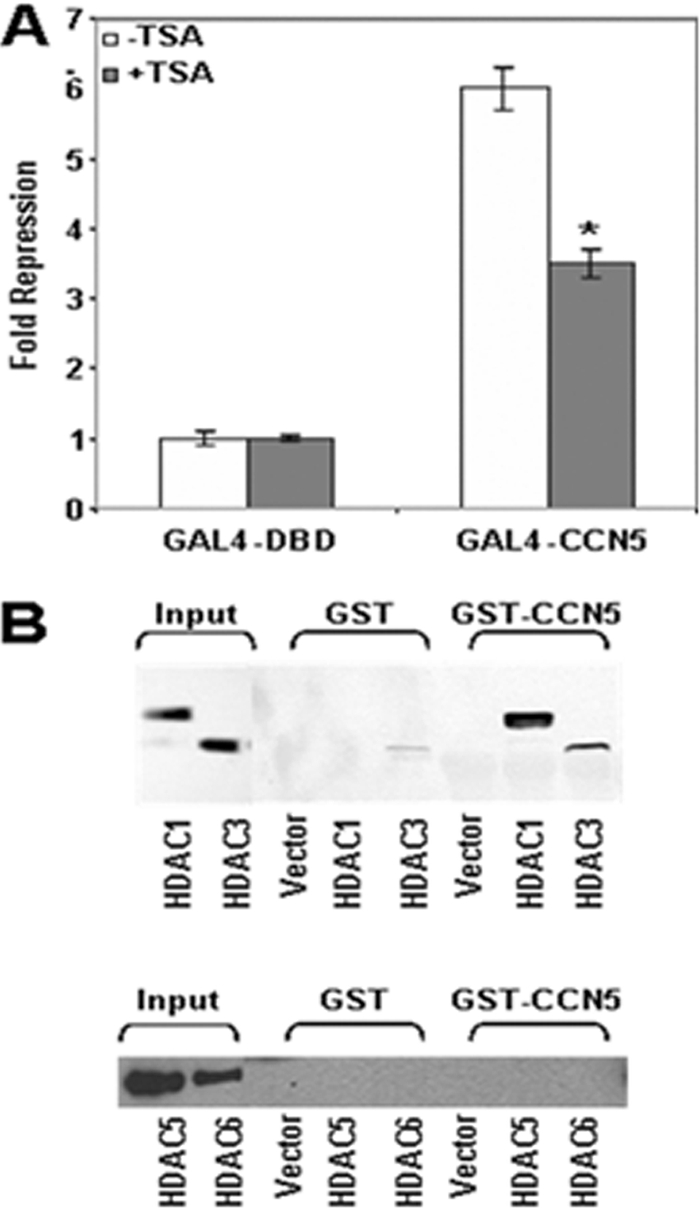
Interaction of CCN5 with HDAC. (A) TSA partially reverses transcriptional repression by GAL4-CCN5. 293T cells were transfected with 1 μg of GAL4-DBD or GAL4-CCN5 expression vector together with 0.5 μg of GAL4-E1B luciferase reporter. Transfected cells were treated with 200 nM TSA for 16 h before harvesting. The repression by GAL4-CCN5 was determined relative to GAL4-DBD alone. The results shown represent the average of three independent experiments assayed in duplicate. Significant differences: *, P < 0.05 versus control. (B) HDAC and CCN5 form a complex. 293T cells were transfected with pCEP4-HDAC1-Flag, pCEP4-HDAC3-Flag, pcDNA3-HA-HDAC5, or pcDNA3-HA-HDAC6. Equal amounts of cellular extracts were incubated with bacterially expressed GST or GST-CCN5 fusion proteins immobilized on glutathione-Sepharose beads. Following incubation at 4°C for 2 h, complexes were washed five times with binding buffer, resolved by SDS-PAGE, and detected by immunoblotting using anti-Flag antibody to detect HDAC1 or HDAC3 or anti-HA antibody to detect HDAC5 or HDAC6. The expression levels of each protein in total lysates were monitored by immunoblotting with anti-Flag antibody or anti-HA antibody (Input).
To approach the question of how HDACs contribute to the ability of CCN5 to repress transcription, we looked for their possible association. FLAG-HDAC1, FLAG-HDAC3, HA-HDAC5, and HA-HDAC6, members of two distinct HDAC families, were transfected into 293T cells. Whole-cell extracts prepared from transfected cells were then incubated with GST-CCN5 fusion protein immobilized on glutathione agarose beads. After several washes and elution, specific complexes were analyzed by SDS-PAGE and quantified by immunoblotting analysis with specific antibodies. As seen in Fig. 4B, only HDACs of class I, HDAC1 and to a lesser extent HDAC3, were captured by CCN5. There was no HDAC retained with GST alone. This result indicates that CCN5 interacts with HDACs, which provides a potential mechanism by which CCN5 exerts its repressive function.
Gene expression profiling identifies global changes resulting from CCN5 loss.
To investigate in more details the role of CCN5 in transcriptional repression, we performed microarray gene expression profiling of MCF-7-sh-CCN5 and MCF-7-sh-scrambled cells (control cells). Among the genes that are significantly induced in MCF-7-sh-CCN5 cells relative to the control were a number of mesenchymal markers known to be associated with passage through an EMT, including fibronectin and vimentin. Conversely, a number of epithelial markers were downregulated relative to the controls (Table 1), which is consistent with our previous results indicating that depletion of CCN5 in MCF-7 cells can culminate in EMT. Consistent with their dedifferentiated phenotype, MCF-7-sh-CCN5 cells show a reduction of cell surface marker CD24 (20.4-fold) and elevation of CD44 (14.7-fold) (Table 1).
Table 1.
Microarray gene expression profiling of MCF-7-sh-CCN5 and MCF-7-sh-scrambled cells
| Gene symbol | Gene descriptiona | Fold change in MCF-sh-CCN5 expression relative to MCF7-sh-scrambled |
|---|---|---|
| Mesenchymal markers | ||
| CD44 | CD44 molecule | +14.7 |
| IGFBP7 | Insulin-like growth factor binding protein 7 | +8.5 |
| ITGA5 | Integrin α5 | +5.1 |
| LAMB3 | Laminin beta 3 | +37 |
| LAMC2 | Laminin gamma 2 | +19.6 |
| PROCR | Protein C receptor, endothelial | +8.2 |
| SNAI2/SLUG | Snail homolog 2 | +72.2 |
| VIM | Vimentin | +188.3 |
| ZEB1 | Zinc finger E-box binding homeobox 1 | +6.6 |
| Epithelial markers | ||
| CD24 | CD24 molecule | −20.4 |
| CDH1 | E-cadherin | −110.2 |
| CLDN1 | Claudin 1 | −2 |
| DSP | Desmoplakin | −3 |
| ERBB2 | v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 | −4 |
| ESR1 | Estrogen receptor 1 | −148 |
| FOXA1 | Forkhead box A1 | −65 |
| GATA3 | GATA binding protein 3 | −32 |
| JUP | Junction plakoglobin | −25 |
| KRT8 | Keratin 8 | −11 |
| KRT18 | Keratin 18 | −6 |
| MUC1 | Mucin 1, cell surface associated | −5 |
| TFF1 | Trefoil factor 1 | −427 |
| TFF3 | Trefoil factor 3 | −2 |
| XBP1 | X-box binding protein 1 | −12 |
| TGF-β pathway markers | ||
| CTGF | Connective tissue growth factor | +12.1 |
| FN1 | Fibronectin 1 | +8.9 |
| JUN | v-jun sarcoma virus 17 oncogene homolog | +2.6 |
| SERPINE1 | Serpin peptidase inhibitor member 1 | +116.3 |
| SMAD1 | SMAD, mothers against DPP homolog 1 | −5.7 |
| SMAD3 | SMAD, mothers against DPP homolog 3 | +2.1 |
| SMAD6 | SMAD, mothers against DPP homolog 6 | −27.9 |
| SPARC | Secreted protein acidic cysteine rich | +2 |
| TGFBR1 | TGF-β receptor 1 | +3.2 |
| TGFBR2 | TGF-β receptor 2 | +416.5 |
| TGFBR3 | TGF-β receptor 3 | +44.6 |
| TGFB1 | TGF-β 1 | +2.6 |
| TGFB3 | TGF-β 3 | −37.6 |
DPP, decapentaplegic homologs.
We show that suppression of CCN5 in MCF-7-sh-CCN5 cells was accompanied by, in addition to EMT markers, deregulated expression of several components of the TGF-β signaling pathway. Notably, we identified 13 genes that were differentially expressed (Table 1), with the TGF-β type II receptor, TGF-βRII, being the most markedly affected. Analysis of TGF-βRII mRNA levels by real-time RT-PCR and protein expression by immunoblotting validated the expression array data, indicating that TGF-βRII was specifically upregulated in MCF-7-sh-CCN5 cells versus control cells (Fig. 5A and B). Taken together, these results provide conclusive evidence that CCN5 functions as a transcriptional repressor and further suggest that CCN5 might play an important role in TGF-β signaling.
Fig. 5.

Silencing of CCN5 increases TGF-β signaling in MCF-7 cells. (A) mRNA was isolated from MCF-7-sh-scrambled and MCF-7-sh-CCN5 cells, and TGF-βRII expression was analyzed by real-time RT-PCR. The results, after normalization, represent the relative hTGF-βRII mRNA transcript levels among these different cell lines and are the means ± SD of triplicate experiments. (B) Protein extracts were prepared and tested by Western blotting for TGF-βRII expression. The levels of β-actin in cell lysates were measured by Western blotting and included as a loading control. (C) MCF-7-sh-scrambled and MCF-7-sh-CCN5 cells were transfected with 0.5 μg of CAGA9-Lux or ARE3-Lux and FAST1 and 0.1 μg of RSV-β-galactosidase construct as an internal control. Cells were treated with TGF-β for 16 h and analyzed for luciferase and β-galactosidase activities. Luciferase was expressed as mean ± SD of triplicates from a representative experiment performed at least three times. Significant differences: *, P < 0.05 versus control; **, P < 0.01 versus control. (D) MCF-7-sh-scrambled and MCF-7-sh-CCN5 cells were treated with TGF-β for 16 h. The expression of PAI1 was analyzed by immunoblotting using specific antibodies.
Silencing of CCN5 increases TGF-β signaling in MCF-7 cells.
Our results suggest that CCN5 could antagonize TGF-β signaling. To examine the effects of CCN5 on TGF-β signal transduction, we performed a transcription reporter assay. Reporter plasmids containing either a TGF-β/Smad3 response element, (9XCAGA)-Lux (11), or TGF-β/Smad2 response element, (ARE)-Lux (10), were transfected into either MCF-7-sh-scrambled or MCF-7-sh-CCN5 cell lines. As shown in Fig. 5C, TGF-β treatment enhanced transcription from the reporters (9XCAGA)-Lux (27-fold) and (ARE)-Lux (14-fold) transfected into MCF-7-sh-CCN5 cells compared to MCF-7-sh-scrambled cells. Furthermore, the loss of CCN5 induces the TGF-β-dependent expression of endogenous PAI1, which is a target of the Smad signaling pathway (Fig. 5D). These findings suggest that CCN5 inhibits the Smad signal transducers of TGF-β signaling.
CCN5 is recruited to the TGF-βRII promoter.
Having shown that depletion of CCN5 in MCF-7 cells can lead to increased expression of TGF-βRII, we then sought out to determine the mechanism by which CCN5 exerts its suppressive function. For this, we used the cloned region of the TGF-βRII gene (nucleotides −1800 to +50) known to confer maximal transcriptional activity in the context of a gene reporter (54). When TGF-βRII (−1800 to + 50)-Luc was cotransfected in the 293T cell line with increasing amounts of the expression plasmid coding for CCN5, the promoter activity decreased in a dose-dependent manner (Fig. 6 A). Truncation of the promoter to leave only the −291 to +50 segment led to significant loss of the basal promoter activity, although this construct was also repressed by CCN5 and the factor of repression was the same as that for the full-length construct (Fig. 6A). These results show that CCN5 represses transcription from the proximal promoter, which includes sites for potential transcription factors, such as Sp1, CCAAT-binding protein and AP1, or CRE/ATF.
Fig. 6.

CCN5 binds and inhibits the TGF-βRII promoter. (A) 5′-flanking region deletion mutants of the TGF-βRII promoter-luciferase constructs were transiently transfected into 293T cells with 0, 0.5, and 1 μg of plasmid expressing hCCN5 and 0.1 μg of RSV-β-galactosidase construct as an internal control. At 48 h posttransfection, cells were harvested and analyzed for luciferase and β-galactosidase activities. Luciferase was expressed as mean ± SD of triplicates from a representative experiment performed at least three times. Significant differences: *, P < 0.05 versus control. (B) Cross-linked sheared chromatin from the MCF-7-sh-scrambled cells or from the MCF-7-sh-CCN5 and MDA-MB-231 cells were immunoprecipitated with the indicated specific antibodies. DNA was analyzed by PCR using primers to amplify the hTGF-βRII promoter region. Results shown are representative of three independent experiments.
Next, we wished to determine whether endogenous CCN5 is recruited to the TGF-βRII gene promoter in vivo, using a chromatin immunoprecipitation assay. Antibodies specific for acetylated histones H3 and H4 and for CCN5 were used to immunoprecipitate chromatin from MCF-7-sh-scrambled cells as well as from MCF-7-sh-CCN5 stably depleted of CCN5. As a control, we used MDA-MB-231 cells that do not express endogenous CCN5. The resulting partitioned genomic DNA was then analyzed by PCR, using primers spanning the bp −294 to +27-bp region of the TGF-βRII promoter for changes in the relative level of DNA associated with acetylated histones and CCN5. As shown in Fig. 6B, CCN5 was recruited to the TGF-βRII promoter in MCF-7-sh-scrambled cells. Of note, we did not detect any significant increase in CCN5 at the distal region of the TGF-βRII promoter (Fig. 6B). During the same time, ChIP analysis with antibodies to acetylated H4 and acetylated H3 revealed no acetylated H3 and a weak acetylated H4 signal at the TGF-βRII promoter. As expected, in MDA-MB-231 and MCF-7-sh-CCN5 cells antibodies against CCN5 did not precipitate the promoter. However, there was a clear association of the TGF-βRII promoter with acetylated histones H3 and H4 in these cells, suggesting that CCN5 might recruit HDACs to the TGF-βRII promoter, leading to its deacetylation and, consequently, silencing.
As HDAC1 and Sp1 have been shown to interact with and mediate the repression of the TGF-βRII promoter (54), we investigated the requirement of CCN5 in this process using MCF-7 cells. As shown in Fig. 6B, we observed an increase in the association of HDAC1 and Sp1 in MCF-7-sh-scrambled cells, a pattern that is similar to that seen for CCN5. Crucially, we detected very little or no association of HDAC1 and Sp1 with the TGF-βRII promoter in MCF-7-sh-CCN5 cells. We also used the CCN5-deficient MDA-MB-231 cell line and found no association of HDAC1 with the TGF-βRII promoter. These results suggest that CCN5 may be recruited to the TGF-βRII gene promoter through interaction with HDAC1, resulting in transcriptional repression.
Inhibition of TGF-β signaling reduced the invasiveness and restored E-cadherin expression of CCN5-downregulated cells.
We previously demonstrated that knockdown of CCN5 induces an invasive phenotype in MCF-7 cells (18). TGF-β signaling can increase the invasive potential of cancer cells and promote EMT that are implicated in cancer dissemination (53). We therefore tested whether activation of TGF-β signaling in MCF-7-sh-CCN5 cells was associated with the invasive phenotype. Control MCF-7-sh-scrambled cells form a monolayer on collagen type I gels and were unable to invade the underlying collagen matrix. In contrast, MCF-7-sh-CCN5 cells cultured for 14 days on type I collagen gels massively infiltrate the matrix structure. A superficial invasion front and several deeply penetrating single cells are observed. Quantification of deep single-cell infiltration revealed that persistent SB431542 treatment, which inhibits the TGF-β type 1 receptor Alk5, significantly reduces the deep cell infiltration of MCF-7-sh-CCN5 cells (Fig. 7 A and B) but had no effect on viability (trypan blue exclusion) or the Ki67 proliferation index (data not shown). We previously demonstrated that knockdown of CCN5 decreases the epithelial marker E-cadherin. This cell-cell adhesion molecule was recognized as an invasion suppressor molecule through in vitro genetic manipulation of human cell lines (47). Having demonstrated an anti-invasive effect of the TGF-β inhibitor SB431542, we sought to determine whether inhibition of TGF-β signaling would reinduce expression of E-cadherin in MCF-7-sh-CCN5 cells. We observed that TGF-β inhibitor treatment restored E-cadherin expression in MCF-7-sh-CCN5 cells, and this may, at least in part, be responsible for decreased deep cell infiltration (Fig. 7C).
Fig. 7.

Pharmacological inhibition of TGF-β signaling reduced the invasiveness and restored E-cadherin expression of CCN5-downregulated cells. (A) MCF-7-sh-scrambled and MCF-7-sh-CCN5 cells were seeded on collagen type I gel in the presence or absence of SB431542 (10 μM). Representative H&E cross-sections of the collagen gel after a 14-day culture period. White double arrow shows superficial invasion front. Black double arrow shows region of deeply infiltrated cancer cells. Individual deeply penetrated cancer cells are indicated by arrows. Scale bar = 50 μm. (B) Data indicate mean percentage of deep invasive cells counted from 6 cross-sections, with error bars indicating the standard error of mean of three experiments. Significant differences: **, P < 0.01 versus control; ***, P < 0.001 versus control. (C) Paraffin sections of 14-day collagen invasion experiments with MCF-7-sh-scrambled or MCF-7sh-CCN5 with or without SB431542 were probed with anti-E-cadherin monoclonal antibody. Scale bar = 50 μm.
DISCUSSION
Although CCN5 expression has been shown to have a protective effect on breast cancer progression, the role of CCN5 in mammalian carcinogenesis has not been well defined. In this work, we show that in MCF-7 cells the loss of CCN5 leads to an increased abundance of key components of the TGF-β signaling pathway.
We find that endogenous CCN5 localizes to the nucleus and cytoplasm of MCF-7 breast cancer cells and in noninvasive breast tumors. In considering potential functions of CCN5 in this newly identified nuclear localization, we observed that the distribution of nuclear CCN5 was reminiscent of the speckled pattern characteristic of components of the transcriptional machinery (44). Consistent with the detection of CCN5 in the nucleus, a repressor function of CCN5 is observed upon directing it to a reporter gene via a heterologous DNA-binding domain, as well as to a reporter containing a natural TGF-βRII promoter element. We have shown that CCN5 interacts with HDAC1 and the histone deacetylase inhibitor TSA can partially block the action of CCN5, suggesting that the CCN5 repression activity is mediated, in part, by histone deacetylation-dependent mechanisms and, in part, by other mechanisms in addition to HDAC recruitment. CCN5 might recruit one of the multiprotein repressor complexes that contain HDACs (25) and acts as a linker between specific factors and general repression complexes. It can be hypothesized that localization of CCN5 concomitant to that of the interacting factor could be a way to activate and render CCN5 available for its targets. Together these data establish a novel role for CCN5 as a transcriptional repressor. The mechanisms of transcriptional repression are important for maintaining the epithelial cell phenotype and during cell differentiation.
One of the pathways that we found specifically activated in CCN5-negative breast cancer cells is the TGF-β signaling pathway, known to play an important role in human embryonic stem cells as well as tumorigenesis (24, 42). TGF-β plays a dual role in tumor progression: it is one of the most potent inhibitors of cell proliferation, but it promotes invasion, angiogenesis, EMT, and metastasis (6, 42). Our pathway analysis and TGF-βRI inhibitor treatment experiment implied that the TGF-β pathway is activated in MCF-7-sh-CCN5 cells and that it regulates, at least in part, their more mesenchymal and invasive phenotype.
We have shown that CCN5 inhibits the Smad signal transducers of TGF-β signaling. Smad3 has also been shown to cross-communicate with ERα, as TGF-β-induced activation of Smad3 responsive genes is significantly suppressed in the presence of activated ERα (32). There exists evidence that a direct physical interaction between Smad3 and estrogen receptor α occurs (32). Previously, we found that CCN5 knockdown in MCF-7 cells induced estrogen-independent growth of these cells linked to a loss of ERα expression and promoted EMT. Although the exact mechanisms leading to the loss of ERα expression are currently uncertain, recent works suggest that activation of growth factor receptor pathways, such as epidermal growth factor (EGF), IGF-1, TGF-β, and heregulin, may contribute to ERα loss (15). It has been suggested that ERα-positive breast cancer cells lose responsiveness to the TGF-β signaling cascade due to the loss or decreasing expression of TGF-βRII (45). In addition, TGF-βRII is repressed in ERα-positive breast cancer cells due to the Sp1/Sp3 family of transcription factors (1). Therefore, understanding the transcription-regulating mechanisms that control TGF-βRII expression is of great importance. However, surprisingly little is known about the transcriptional regulation of the TGF-βRII gene. The TGF-βRII promoter lacks the TATA box and, similar to other promoters that lack the TATA box, Sp1 is required for the transcription of the TGF-βRII gene (3). In this report, we show that CCN5 negatively regulates the transcription of the TGF-βRII gene. ChIP assays revealed that CCN5 is recruited to the TGF-βRII promoter region that contains several Sp1 binding sites. We found that CCN5 is bound together with HDAC1 and Sp1 at the promoter. As HDAC1 and Sp1 have been shown to interact with and mediate the repression of the TGF-βRII promoter in ductal adenocarcinoma cells (54) and p21 WAF1/CIP1 transcription in breast cancer cells (21), it is possible that CCN5, HDAC1, and Sp1 may cooperatively regulate TGF-βRII transcription.
Estrogens are decisive actors responsible for the proliferation and differentiation of normal mammary epithelial cells as well as the development and progression of breast cancer. On the other hand, TGF-β acts like a cell growth inhibitor. Although a number of studies of the cross talk between estrogen and TGF-β signaling have been made, molecular mechanisms remain to be determined. We have previously shown that CCN5 is an estrogen-regulated gene and its loss induces loss of ERα expression and estrogen-dependent growth in MCF-7 breast cancer cells. Here we demonstrate a novel CCN5 function, by which TGF-β signaling is inhibited. In summary, our molecular analysis reveals that the CCN5-negative cells are CD44+, TGF-β1+, TGF-βRII+, and ER−, suggesting that these cells exhibited a breast cancer stem cell-like profile.
ACKNOWLEDGMENTS
We thank Azeddine Atfi and Jan Mester for the critical review of the manuscript and Pauline Sabbah for technical assistance.
This work was supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, the Ligue Nationale contre le Cancer (Comité de Paris), and the Groupement d'Entreprises Françaises dans la Lutte contre le Cancer. Olivier De Wever is supported by a postdoctoral grant from Fund for Scientific Research-Flanders.
Footnotes
Published ahead of print on 24 January 2011.
REFERENCES
- 1. Ammanamanchi S., Kim S. J., Sun L. Z., Brattain M. G. 1998. Induction of transforming growth factor-beta receptor type II expression in estrogen receptor-positive breast cancer cells through SP1 activation by 5-aza-2′-deoxycytidine. J. Biol. Chem. 273:16527–16534 [DOI] [PubMed] [Google Scholar]
- 2. Anonymous 2001. Proposal for a unified CCN nomenclature. Mol. Pathol. 54:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bae H. W., et al. 1995. Characterization of the promoter region of the human transforming growth factor-beta type II receptor gene. J. Biol. Chem. 270:29460–29468 [DOI] [PubMed] [Google Scholar]
- 4. Banerjee S., et al. 2008. CCN5/WISP-2 expression in breast adenocarcinoma is associated with less frequent progression of the disease and suppresses the invasive phenotypes of tumor cells. Cancer Res. 68:7606–7612 [DOI] [PubMed] [Google Scholar]
- 5. Banerjee S., et al. 2003. WISP-2 gene in human breast cancer: estrogen and progesterone inducible expression and regulation of tumor cell proliferation. Neoplasia 5:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bates R. C., Mercurio A. M. 2005. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol. Ther. 4:365–370 [DOI] [PubMed] [Google Scholar]
- 7. Brigstock D. R. 2003. The CCN family: a new stimulus package. J. Endocrinol. 178:169–175 [DOI] [PubMed] [Google Scholar]
- 8. Brigstock D. R. 1999. The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr. Rev. 20:189–206 [DOI] [PubMed] [Google Scholar]
- 9. Brigstock D. R., et al. 2003. Proposal for a unified CCN nomenclature. Mol. Pathol. 56:127–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen X., et al. 1997. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature 389:85–89 [DOI] [PubMed] [Google Scholar]
- 11. Dennler S., Prunier C., Ferrand N., Gauthier J. M., Atfi A. 2000. c-Jun inhibits transforming growth factor beta-mediated transcription by repressing Smad3 transcriptional activity. J. Biol. Chem. 275:28858–28865 [DOI] [PubMed] [Google Scholar]
- 12. De Wever O., et al. 2010. Modeling and quantification of cancer cell invasion through collagen type I matrices. Int. J. Dev. Biol. 54:887–896 [DOI] [PubMed] [Google Scholar]
- 13. De Wever O., et al. 2008. Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front. Histochem. Cell Biol. 130:481–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhar G., et al. 2007. Loss of WISP-2/CCN5 signaling in human pancreatic cancer: a potential mechanism for epithelial-mesenchymal-transition. Cancer Lett. 254:63–70 [DOI] [PubMed] [Google Scholar]
- 15. Dhasarathy A., Kajita M., Wade P. A. 2007. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol. Endocrinol. 21:2907–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fèvre-Montange M., et al. 2006. Microarray analysis reveals differential gene expression patterns in tumors of the pineal region. J. Neuropathol. Exp. Neurol. 65:675–684 [DOI] [PubMed] [Google Scholar]
- 17. Fritah A., Redeuilh G., Sabbah M. 2006. Molecular cloning and characterization of the human WISP-2/CCN5 gene promoter reveal its upregulation by oestrogens. J. Endocrinol. 191:613–624 [DOI] [PubMed] [Google Scholar]
- 18. Fritah A., et al. 2008. Role of WISP-2/CCN5 in the maintenance of a differentiated and noninvasive phenotype in human breast cancer cells. Mol. Cell. Biol. 28:1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fritah A., Saucier C., Mester J., Redeuilh G., Sabbah M. 2005. p21WAF1/CIP1 selectively controls the transcriptional activity of estrogen receptor alpha. Mol. Cell. Biol. 25:2419–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gray M. R., Malmquist J. A., Sullivan M., Blea M., Castellot J. J., Jr 2007. CCN5 expression in mammals. II. Adult rodent tissues. J. Cell Commun. Signal. 1:145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang L., Sowa Y., Sakai T., Pardee A. B. 2000. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene 19:5712–5719 [DOI] [PubMed] [Google Scholar]
- 22. Huang W., et al. 2008. Inhibition of CCN6 (Wnt-1-induced signaling protein 3) down-regulates E-cadherin in the breast epithelium through induction of snail and ZEB1. Am. J. Pathol. 172:893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inadera H., et al. 2000. WISP-2 as a novel estrogen-responsive gene in human breast cancer cells. Biochem. Biophys. Res. Commun. 275:108–114 [DOI] [PubMed] [Google Scholar]
- 24. James D., Levine A. J., Besser D., Hemmati-Brivanlou A. 2005. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132:1273–1282 [DOI] [PubMed] [Google Scholar]
- 25. Jepsen K., Rosenfeld M. G. 2002. Biological roles and mechanistic actions of co-repressor complexes. J. Cell Sci. 115:689–698 [DOI] [PubMed] [Google Scholar]
- 26. Johnson H. M., Subramaniam P. S., Olsnes S., Jans D. A. 2004. Trafficking and signaling pathways of nuclear localizing protein ligands and their receptors. Bioessays 26:993–1004 [DOI] [PubMed] [Google Scholar]
- 27. Jones J. A., Gray M. R., Oliveira B. E., Koch M., Castellot J. J., Jr 2007. CCN5 expression in mammals. I. Embryonic and fetal tissues of mouse and human. J. Cell Commun. Signal. 1:127–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kouzarides T. 1999. Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev. 9:40–48 [DOI] [PubMed] [Google Scholar]
- 29. Leask A., Abraham D. J. 2006. All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 119:4803–4810 [DOI] [PubMed] [Google Scholar]
- 30. Lemercier C., et al. 2000. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J. Biol. Chem. 275:15594–15599 [DOI] [PubMed] [Google Scholar]
- 31. Mason H. R., Lake A. C., Wubben J. E., Nowak R. A., Castellot J. J., Jr 2004. The growth arrest-specific gene CCN5 is deficient in human leiomyomas and inhibits the proliferation and motility of cultured human uterine smooth muscle cells. Mol. Hum. Reprod. 10:181–187 [DOI] [PubMed] [Google Scholar]
- 32. Matsuda T., Yamamoto T., Muraguchi A., Saatcioglu F. 2001. Cross-talk between transforming growth factor-beta and estrogen receptor signaling through Smad3. J. Biol. Chem. 276:42908–42914 [DOI] [PubMed] [Google Scholar]
- 33. Ng H. H., Bird A. 2000. Histone deacetylases: silencers for hire. Trends Biochem. Sci. 25:121–126 [DOI] [PubMed] [Google Scholar]
- 34. Olsnes S., Klingenberg O., Wiedlocha A. 2003. Transport of exogenous growth factors and cytokines to the cytosol and to the nucleus. Physiol. Rev. 83:163–182 [DOI] [PubMed] [Google Scholar]
- 35. Pennica D., et al. 1998. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc. Natl. Acad. Sci. U. S. A. 95:14717–14722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pessah M., et al. 2001. c-Jun interacts with the corepressor TG-interacting factor (TGIF) to suppress Smad2 transcriptional activity. Proc. Natl. Acad. Sci. U. S. A. 98:6198–6203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Planque N. 2006. Nuclear trafficking of secreted factors and cell-surface receptors: new pathways to regulate cell proliferation and differentiation, and involvement in cancers. Cell Commun. Signal. 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Planque N., Long Li C., Saule S., Bleau A. M., Perbal B. 2006. Nuclear addressing provides a clue for the transforming activity of amino-truncated CCN3 proteins. J. Cell. Biochem. 99:105–116 [DOI] [PubMed] [Google Scholar]
- 39. Planque N., Perbal B. 2003. A structural approach to the role of CCN (CYR61/CTGF/NOV) proteins in tumourigenesis. Cancer Cell Int. 3:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prunier C., Ferrand N., Frottier B., Pessah M., Atfi A. 2001. Mechanism for mutational inactivation of the tumor suppressor Smad2. Mol. Cell. Biol. 21:3302–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rachfal A. W., Brigstock D. R. 2005. Structural and functional properties of CCN proteins. Vitam. Horm. 70:69–103 [DOI] [PubMed] [Google Scholar]
- 42. Roberts A. B., Wakefield L. M. 2003. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 100:8621–8623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sabbah M., et al. 2008. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist. Updat. 11:123–151 [DOI] [PubMed] [Google Scholar]
- 44. Sexton T., Schober H., Fraser P., Gasser S. M. 2007. Gene regulation through nuclear organization. Nat. Struct. Mol. Biol. 14:1049–1055 [DOI] [PubMed] [Google Scholar]
- 45. Sun L., et al. 1994. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J. Biol. Chem. 269:26449–26455 [PubMed] [Google Scholar]
- 46. Thiery J. P. 2002. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2:442–454 [DOI] [PubMed] [Google Scholar]
- 47. Vleminckx K., Vakaet L., Jr., Mareel M., Fiers W., van Roy F. 1991. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 66:107–119 [DOI] [PubMed] [Google Scholar]
- 48. Wahab N. A., Brinkman H., Mason R. M. 2001. Uptake and intracellular transport of the connective tissue growth factor: a potential mode of action. Biochem. J. 359:89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wei L. N., Hu X., Chandra D., Seto E., Farooqui M. 2000. Receptor-interacting protein 140 directly recruits histone deacetylases for gene silencing. J. Biol. Chem. 275:40782–40787 [DOI] [PubMed] [Google Scholar]
- 50. Wiesman K. C., et al. 2010. CCN5, a secreted protein, localizes to the nucleus. J. Cell Commun. Signal. 4:91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang J., Weinberg R. A. 2008. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell 14:818–829 [DOI] [PubMed] [Google Scholar]
- 52. Yang S. H., Vickers E., Brehm A., Kouzarides T., Sharrocks A. D. 2001. Temporal recruitment of the mSin3A-histone deacetylase corepressor complex to the ETS domain transcription factor Elk-1. Mol. Cell. Biol. 21:2802–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zavadil J., Bottinger E. P. 2005. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24:5764–5774 [DOI] [PubMed] [Google Scholar]
- 54. Zhao S., Venkatasubbarao K., Li S., Freeman J. W. 2003. Requirement of a specific Sp1 site for histone deacetylase-mediated repression of transforming growth factor beta type II receptor expression in human pancreatic cancer cells. Cancer Res. 63:2624–2630 [PubMed] [Google Scholar]